

Long-acting (LA) administration using a subcutaneous (s.c.) implant presents opportunities to simplify administration of antiretroviral drugs, improve pharmacological profiles, and overcome suboptimal adherence associated with daily oral formulations. Tenofovir alafenamide (TAF) is a highly potent nucleoside reverse transcriptase inhibitor (NRTI) and an attractive agent for LA delivery, with a high potency and long intracellular half-life. The aim of this study was to predict minimum TAF doses required to achieve concentrations effective for HIV preexposure prophylaxis (PrEP).
KEYWORDS: PBPK, tenofovir alafenamide, implanted devices, long acting, subcutaneous
ABSTRACT
Long-acting (LA) administration using a subcutaneous (s.c.) implant presents opportunities to simplify administration of antiretroviral drugs, improve pharmacological profiles, and overcome suboptimal adherence associated with daily oral formulations. Tenofovir alafenamide (TAF) is a highly potent nucleoside reverse transcriptase inhibitor (NRTI) and an attractive agent for LA delivery, with a high potency and long intracellular half-life. The aim of this study was to predict minimum TAF doses required to achieve concentrations effective for HIV preexposure prophylaxis (PrEP). Daily drug release requirements were then ascertained by averaging across the dosing interval. A TAF physiologically based pharmacokinetic (PBPK) model was developed and partially qualified against available oral single- and multiple-dose pharmacokinetics. The models were assumed to be qualified when simulated values were within 2-fold of the observed mean. TAF s.c. implants were simulated in five hundred individuals, reporting predicted TAF plasma and tenofovir (TFV) plasma concentrations for various release rates. Intracellular TFV diphosphate (TFV-DP) concentrations were also simulated in peripheral blood cells and cervical and rectal tissues. The minimum dose predicted to achieve intracellular TFV-DP levels above a target concentration of 48 fmol/106 cells for a month was identified. TAF, TFV, and TFV-DP concentrations for release rates between 1.0 and 1.6 mg/day were simulated. The PBPK model indicated that a minimum release of 1.4 mg/day TAF is necessary to achieve TFV-DP concentrations above the identified target in peripheral blood mononuclear cells (PBMCs). TFV-DP cervical and rectal tissue concentrations were predicted to be between 1.5 and 2.0 fmol/106 cells and 0.9 and 1.1 fmol/106 cells, respectively, for release rates between 1.3 and 1.6 mg/day. These simulations provide target minimum doses for LA TAF PrEP in humans. Based on the generated results, multiple implants delivering a total of 1.4 mg/day of TAF subcutaneously could provide protection levels for approximately 6 months to 1 year. This modeling may inform future design of s.c. implants to mitigate adherence issues for effective PrEP applications.
Human immunodeficiency virus (HIV) is a global epidemic, with an estimated 37.9 million people currently living with the virus (1). Although existing antiretroviral (ARV) regimes for treatment and preexposure prophylaxis (PrEP) have dramatically reduced the incidence of new infections annually over the last decade, an estimated 1.7 million people became newly infected in 2018; sex workers and clients of sex workers, men who have sex with men (MSM), people who inject drugs (PWIDs), transgender women (TGW), and their partners accounted for over half of these new infections globally (1). In sub-Saharan Africa, young women and adolescent girls accounted for two out of three new HIV infections that occurred in 2017 in the region (2). Development and implementation of more HIV prevention options for these high-risk populations are key to decreasing the incidence of new infections and overall prevalence of the virus (3, 4).
Tenofovir disoproxil fumarate (TDF), a prodrug of tenofovir, and emtricitabine (FTC [F]) were FDA approved for treatment of HIV in 2004 and for PrEP in 2012 as the once-daily oral combination FTC/TDF (Truvada) (5). Oral PrEP has been successful among serodiscordant heterosexual couples, MSM, and TGW when they are able to adhere to daily pill-taking regimens (6, 7). However, several social, behavioral, and biological factors can contribute to reduced user adherence and effectiveness of oral PrEP (e.g., poor accessibility to health care clinics for monthly refills, dosing fatigue, social stigma of taking ARVs in public, and lower drug sequestration in vaginal tissues than in rectal tissues) (8–11). Studies in women have shown that suboptimal adherence to oral PrEP yielded no protection (12, 13) and that nearly perfect adherence (up to 6 to 7 doses per week) was needed to achieve complete protection via the vaginal route of exposure (10, 14).
Long-acting (LA) methods (e.g., lasting longer than 2 to 3 months between dosing intervals) offer a promising strategy for users to overcome some of the documented adherence challenges. Two ARV drugs, cabotegravir and rilpivirine, are currently in late-stage clinical development as an intramuscular LA injection for HIV treatment. The latest results show that the combination of cabotegravir and rilpivirine LA as maintenance therapy provided viral suppression equivalent to that with existing daily oral therapy with FTC/TDF (15–17). Cabotegravir LA as a stand-alone agent is also being compared to FTC/TDF for prevention in two phase 3 studies among a population of healthy MSM and TGW who have sex with men (ClinicalTrials registration no. NCT02720094) and healthy women (ClinicalTrials registration no. NCT03164564). Evidence suggests that persistent subtherapeutic levels of cabotegravir in plasma occur long after secession, requiring a “tailing” regimen of oral PrEP to prevent risk of antiviral-resistant infection (18). Implant systems containing highly potent ARVs are also in development as LA methods. These systems are inserted subcutaneously (s.c.) in the upper arm during a minimally invasive surgical procedure and can provide efficacious concentrations for months to years with no clinical follow-up until implant removal or replacement. They can also be removable in case of an adverse event, an advantage over LA-injectable formulations. Some implants are made of bioabsorbable material designed to degrade after the therapeutic use window, thus eliminating the need for an additional clinic visits to remove the depleted implant (19). Several groups are developing implants with the nucleoside reverse transcriptase inhibitor (NRTI) tenofovir alafenamide (TAF) (19–22). TAF is approved for oral treatment as the combination F/TAF (Descovy) and more recently for PrEP in at-risk adult and adolescent males. Like TDF, TAF is a tenofovir prodrug, but it is about 10 times more potent and has an improved safety profile and longer intracellular half-life of the active metabolite tenofovir diphosphate (TFV-DP) (23). Unlike TDF, TAF is not approved for HIV PrEP in women at risk of infection from the vaginal route of exposure, but plans to evaluate the efficacy for HIV PrEP among this population are ongoing (24). TAF is an attractive single agent for LA administration due to its superior potency (lower target plasma concentrations) and low-dose long intracellular half-life.
Physiologically based pharmacokinetic (PBPK) modeling is a computational approach to simulate pharmacokinetics in humans. PBPK models mimic human anatomy and physiology through anthropometric equations and combine drug physicochemical data (e.g., log P, pKa, and molecular weight) and in vitro data (protein binding, apparent permeability, blood-to-plasma ratio, and intrinsic clearance) to describe drug disposition kinetics. PBPK models are increasingly used to support candidate selection during drug discovery and dose selection for clinical development, facilitating regulatory submissions and optimizing therapy after market approval across different subpopulations (25).
The aim of this study was to develop a TAF PBPK model and simulate the minimum dose suitable as a subcutaneous implant in virtual healthy women. The developed model was qualified against observed data from oral administration and then used to simulate theoretical LA options. The minimum daily dose for the subcutaneous implant was evaluated such that the intracellular concentration of TFV-DP was above the target concentration of 48 fmol/106 cells (6, 21) at the end of a 4-week interval. Given the unanswered scientific question of whether mucosal drug concentrations play an important role in PrEP, in addition to systemic drug levels (26), cervical and rectal tissue concentrations of TFV-DP were also simulated.
RESULTS
Model qualification.
The observed and predicted pharmacokinetic parameters and tables comparing the area under the concentration-time curve (AUC) and maximum concentration in serum (Cmax) of TAF, TFV, and TFV-DP for different dosing regimens are shown in the supplemental material. The simulated pharmacokinetic parameters are in the agreeable 2-fold limit from the mean observed values except for the TFV Cmax of single 10-mg TAF oral dose, which exceeds the limit by 6%. The difference in the simulated pharmacokinetic parameters AUC and Cmax of plasma TAF and TFV on day 14 for 8-mg and 25-mg oral doses of TAF was less than 50% from the observed mean; however, the difference between observed and simulated AUCs for single oral doses of 5 mg and 10 mg was between 10 and 50%, and that for Cmax was between 90 and 107%, for plasma TFV. The difference in the intracellular TFV-DP AUC and Cmax for the single TAF doses of 5 mg and 10 mg was between 10% and 35%.
Model prediction.
The pharmacokinetics of TAF, TFV, and TFV-DP for various release amounts from the implant per day are presented in Table 1, and the intracellular TFV-DP concentrations in cervical and rectal tissues for various release rates are reported in Table 2. The model indicated that a minimum release of 1.4 mg TAF per day is necessary to achieve a TFV-DP intracellular concentration above the target concentration of 48 fmol/106 cells. The simulated TAF implant plasma concentrations were predicted to reach a steady level within half a day (assuming constant uninterrupted release from the implant). However, TFV and TFV-DP concentrations required a longer interval of time (up to 14 days), as shown in Fig. 1 and 2.
TABLE 1.
TAF subcutaneous implant pharmacokinetic predictions at different zero-order release rates for 28 consecutive days
| Release rate (mg/day) | Compound and location | Mean ± SD |
|
|---|---|---|---|
| AUC (ng · h/ml)a | Css (ng/ml)b | ||
| 1.6 | TAF, plasma | 899 ± 193 | 1.341 ± 0.287 |
| TFV, plasma | 1,064 ± 414 | 1.683 ± 0.682 | |
| TFV-DP, PBMCs | 56.59 ± 15.84 | ||
| 1.5 | TAF, plasma | 806 ± 134 | 1.202 ± 0.199 |
| TFV, plasma | 950 ± 369 | 1.497 ± 0.498 | |
| TFV-DP, PBMCs | 51.72 ± 13.54 | ||
| 1.4 | TAF, plasma | 769 ± 148 | 1.146 ± 0.221 |
| TFV, plasma | 899 ± 296 | 1.418 ± 0.482 | |
| TFV-DP, PBMCs | 49.26 ± 10.32 | ||
| 1.3 | TAF, plasma | 715 ± 135 | 1.067 ± 0.202 |
| TFV, plasma | 811 ± 307 | 1.249 ± 0.492 | |
| TFV-DP, PBMCs | 46.26 ± 11.09 | ||
| 1.2 | TAF, plasma | 678 ± 109 | 1.011 ± 0.163 |
| TFV, plasma | 768 ± 274 | 1.212 ± 0.451 | |
| TFV-DP, PBMCs | 42.95 ± 10.16 | ||
| 1.0 | TAF, plasma | 556 ± 96.3 | 0.831 ± 0.167 |
| TFV, plasma | 609 ± 196 | 0.986 ± 0.238 | |
| TFV-DP, PBMCs | 35.53 ± 8.797 | ||
AUC was measured for 28 days (672 h) after implant administration.
Css, steady-state concentration. Concentrations in PBMCs are in fmol/106 cells.
TABLE 2.
TFV-DP cervical and rectal PKs for a subcutaneous implant at different zero-order release rates for 28 consecutive days
| Release rate (mg/day) | Simulated TFV-DP concn (fmol/106 cells; mean ± SD) ina: |
||
|---|---|---|---|
| PBMCs | Cervical tissue | Rectal tissue | |
| 1.6 | 56.6 ± 15.8 | 1.72 ± 0.38 | 1.11 ± 0.25 |
| 1.5 | 51.7 ± 13.5 | 1.63 ± 0.42 | 1.05 ± 0.27 |
| 1.4 | 49.3 ± 10.3 | 1.52 ± 0.32 | 0.98 ± 0.21 |
| 1.3 | 46.3 ± 11.1 | 1.45 ± 0.36 | 0.94 ± 0.23 |
Average ratios of 0.031 and 0.02 for TFV-DP cervical/TFV-DP PBMC and TFV-DP rectal/TFV-DP PBMC were used for computation of TFV-DP concentrations in cervical and rectal tissues, respectively.
FIG 1.
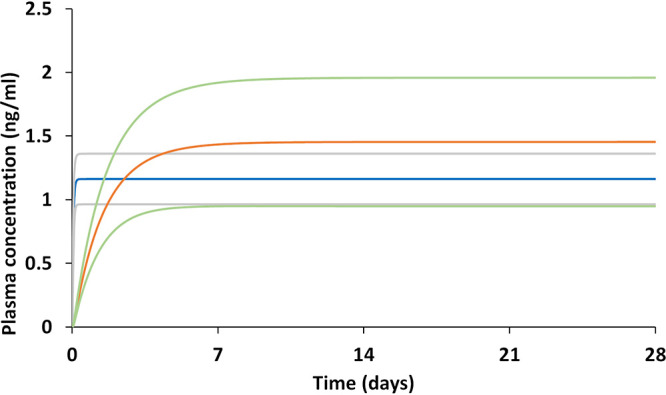
TAF and TFV pharmacokinetics at a constant release of 1.4 mg/day TAF implant through the subcutaneous tissue for 28 days are illustrated, but duration of exposure would ultimately be defined by the total amount of TAF contained within an implant. Blue line, simulated TAF mean; orange line, simulated TFV mean; gray lines, simulated TAF mean ± 1 standard deviation (SD); green lines, simulated TFV mean ± 1 SD.
FIG 2.
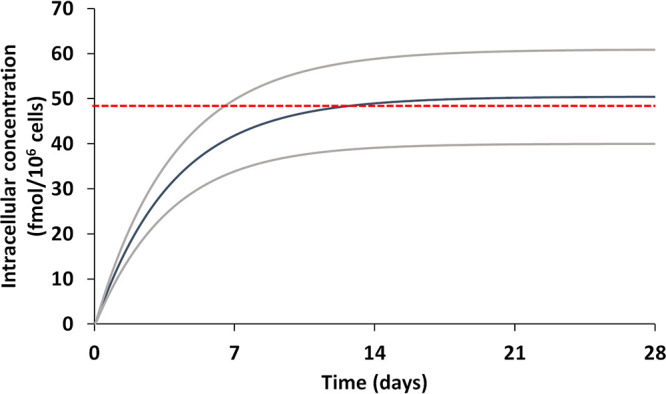
TFV-DP pharmacokinetics at a constant release of 1.4 mg/day TAF implant through the subcutaneous tissue for 28 consecutive days. Red line, target intracellular concentration of 48 fmol/106 cells; blue line, simulated TFV-DP mean; gray line, simulated TFV-DP mean ± 1 SD.
DISCUSSION
This study developed a PBPK model for TAF to inform the minimum dose required for a subcutaneous implant to achieve protection against HIV infection in healthy adult women. The PBPK model was qualified against TAF oral formulations, and the model simulations were in agreement with the clinically observed pharmacokinetic data. The observed data considered for this study comprised both single- and multiple-dose studies of TAF in a healthy population. Qualification against both single- and multiple-dose scenarios improved the performance and confidence of the TAF PBPK model for long-term simulations. This study focused on the use of TAF as a single agent, and therefore clinical studies during which no other concomitant drugs were administered were considered for model qualification, but coadministration with other drugs may affect TAF pharmacokinetics. The model qualification resulted in simulations well within 2-fold of the mean observed values. The difference in the Cmax of plasma TFV for both 5-mg and 10-mg doses was on the higher side (98 to 107%), which may be due to the low values of Cmax for these doses (0.8 and 1.5 ng/ml for 5 mg and 10 mg, respectively). However, since the simulated AUC and Cmax values of plasma TAF and TFV for multiple doses of 8 mg and 25 mg were within 50% of the mean observed values, the model was considered qualified, and the long-term performance was better than for a single dose. The simulated TFV-DP intracellular concentration in healthy individuals was also compared with observed data, and the mean values were within the 2-fold limit (data not shown) (27).
Studies have indicated that a 90% effective concentration (EC90) for TFV-DP is between 26 and 48 fmol/106 cells, and therefore, a concentration of 48 fmol/106 cells was considered the conservative target concentration (6, 21), since no standardized target concentration for prophylactic use of TAF has been identified. For the model predictions, simulations starting with 0.1 mg/day were gradually increased to identify the minimal release from the implant per day to achieve a TFV-DP concentration above the target concentration. Only data for simulations conducted with release over 1.0 mg/day are presented in Table 1. A declining trend in the plasma concentration-time curve is observed for conventional implants due to the physical degradation and declining amount of drug and the surface area of the implant. However, the PBPK model assumes 100% bioavailability from the subcutaneous environment and a constant drug release from the implant. Figures 1 and 2 show a steady curve at the end of the 4-week period without a decline in TAF plasma concentrations. Figures 1 and 2 show that longer time scales are likely to be needed to reach a steady concentration for TFV-DP compared to TFV and TAF. The mean simulated intracellular TFV-DP concentrations for 1.4 mg/day are over the target concentration of 48 fmol/106 cells (Fig. 1). However, the implant is predicted to need at least 14 days to reach that level of protection if this is experimentally confirmed. This may warrant additional oral doses to compensate for the low TFV-DP levels immediately following implantation. The variability observed in the simulated pharmacokinetics means that a higher dose per day (>1.6 mg per day) might be necessary to protect all individuals (TFV-DP levels of >48 fmol/106 cells). A minimum dose of 1.4 mg/day would result in 42 mg monthly, 126 mg quarterly, and 252 mg every 6 months. Recent efforts with a biodegradable implant containing up to 115 mg TAF demonstrated a tuneable release rate between approximately 0.2 and 0.9 mg/day and a sustained zero-order release of TAF at approximately 0.3 mg/day for over 6 months in vitro (19). Multiple implants may thus be needed to achieve protection for a target duration of 6 months to 1 year in humans. Notably, a recent study among reproductive-age women in sub-Saharan Africa suggested that multiple implants were acceptable for PrEP, provided that an increased number of rods afforded a longer duration of protection (28).
Although the presented PBPK model was successfully qualified against available data and a minimum daily implant dose was identified, there are some important limitations. The effect of transporters such as P-gp have not been directly accounted for due to current data limitations, and this may affect TAF pharmacokinetics (29). Also, there is evidence of granuloma formation for injectable formulations (30), which can change the pharmacokinetics, and this may also occur for subcutaneous implants when the normal foreign-body response results in a thick fibrous capsule formed around the implant (30). The model does not account for potential toxicity of TAF delivered subcutaneously. Local reactivity was noted with TAF implants releasing from 0.15 mg/day to 1.8 mg/day in different animal species (20, 31, 32) and with doses greater than 1.0 mg/day specifically in beagles (32). Recently, Su et al. reported marked inflammation with their TAF implant in New Zealand White (NZW) rabbits and rhesus macaques with doses from 0.13 mg/day up to 0.78 mg/day after 4- and 12-week durations (22). It is thus critical for a minimum protective dose of TAF to exist below the threshold of toxicity and be delivered safely in humans via the subcutaneous route of administration over long durations.
Conclusion.
A PBPK model for a theoretical subcutaneous TAF implant is presented along with the minimum dose needed to provide protection against HIV. The PBPK model was qualified against available data from existing oral formulations, and the predictions suggest that a dose of at least 1.4 mg/day is needed to sustain mean intracellular TFV-DP concentrations over 48 fmol/106 cells. This approach may be valuable to support the design of future LA s.c. implants, addressing problems such as suboptimal adherence and pill fatigue associated with oral drug delivery that are known to impact the success of PrEP.
MATERIALS AND METHODS
A whole-body PBPK model was described using Simbiology (MATLAB v.2018b; MathWorks, Natick, MA, USA), and a virtual population of 500 healthy adult women was used in this study considering a previously published model framework (33). The PBPK model assumed (i) a well-stirred model (i.e., the drug distribution across organs and tissues is instant and uniform), (ii) blood flow limited first-order kinetics to describe drug distribution (34), and (iii) no drug reabsorption from the colon. This study is based on computational data generated by the model, so no ethics approval was needed.
Anatomy and physiology.
Simulations were conducted in females between the ages of 18 and 60 years, weighing between 40 and 120 kg (76.4 ± 30.9 kg) and having a body mass index (BMI) between 18 and 40 kg/m2 (29.2 ± 12.51 kg/m2) (35). Various anthropometric equations that relied on the characteristics of the individual (age, weight, BMI, height, and body surface area) were used to derive the anatomical components, i.e., organ weights and volumes and blood flow rates (36). Means and standard deviations were provided for each of these components, and built-in functions of the model were used to generate a random unique female individual for every simulation, thus generating a population over multiple simulations.
A compartmental absorption and transit (CAT) model was used to describe effective absorption kinetics within the oral TAF model used for qualification (37). The CAT model consisted of seven compartments representing the stomach, and various parts of the small intestine (duodenum, jejunum, and ileum). An absorption rate equivalent to 6.24 h−1, derived from the two-compartmental population pharmacokinetic model, was used (38). The absorption model did not account for drug solubility and assumed that all drug is in solution and readily available for absorption. An apparent systemic clearance of 149 liters/h was used due to the unavailability of in vitro data (38). Drug-specific parameters used in this study are shown in Table 3.
TABLE 3.
Physicochemical properties in vitro and population pharmacokinetic data for tenofovir alafenamide
| Propertya | Value for tenofovir alafenamide (reference) |
|---|---|
| Mol wt | 476.474 (44) |
| Solubility in water | 4.86 mg/ml (44) |
| Log Po:w | 1.6 (44) |
| pKa | 3.96 (44) |
| Blood-to-plasma ratio | 1.5b |
| Protein binding | 80 % (44) |
| Absorption rate | 6.24 h−1 (38) |
| Apparent clearance | 149 liters/h (38) |
| Oral bioavailability | 0.53c (38) |
Log Po:w, partition coefficient between octanol and water; pKa, logarithmic value of the dissociation constant.
Mean of 0.6 and 2.4.
Assumed since 47.2% ± 4.62% is excreted in feces.
First-order kinetics were used to describe drug distribution across the multicompartmental model (39). The tissue-to-plasma partition coefficients were computed by equations obtained from Poulin and Theil (40). A subcutaneous (s.c.) compartment was added to this previously published whole-body PBPK model (41) to describe zero-order TAF release from the implant.
Model qualification.
The TAF PBPK model was qualified against available pharmacokinetic data for TAF, TFV, and TFV-DP from various clinical studies (29, 42, 43). The models were assumed to be qualified if (i) the mean simulated pharmacokinetic parameters (area under the curve [AUC] and maximum concentration [Cmax] were within ±50% from the mean observed values and (ii) the simulated and the observed pharmacokinetic data points versus time had an absolute average fold error (AAFE) of less than two. An AAFE value of 1 is representative of an exact match to the observed data.
The PBPK model was initially qualified against available oral data for 8 mg and 25 mg TAF given once daily on day 1 (29) and day 14 (42). Once the TAF model was qualified, using the observed TAF plasma concentration and the pharmacokinetic curves of TFV on day 1 and day 14 (29, 42) and TFV-DP on day 1 (43), the rate of change of TAF to TFV/TFV-DP (Kin) and the rate of elimination of TFV/TFV-DP (Kout) (as shown in Fig. 3) were estimated by trial and error with the equation dC/dt = Kin × CTAF − Kout × C, where C is the plasma concentration of TFV or intracellular TFV-DP and CTAF is the plasma concentration of TAF at time t, using 25 mg TAF. In this model, TFV-DP concentrations refer to concentrations in peripheral blood mononuclear cells (PBMCs). The estimated Kin and Kout values were then used to further validate available TFV and TFV-DP pharmacokinetics for multiple 5-mg, 8-mg, 10-mg, 25-mg, and 40-mg TAF doses. The estimated values of Kin and Kout were 0.035 ± 0.007 h−1 and 0.03 ± 0.006 h−1 for TFV plasma concentration and 0.465 ± 0.05 h−1 and 0.011 ± 0.001 h−1 for TFV-DP intracellular concentration, respectively.
FIG 3.

Diagrammatic representation of TFV/TFV-DP concentration dependence on Kin, Kout, and TAF plasma concentration.
Model prediction.
The qualified PBPK model was used for dose prediction of implants through the subcutaneous route of administration. A range of zero-order release rates was simulated from 1.0 to 1.6 mg/day, and the minimum amount required per day was identified such that the TFV-DP intracellular concentration in PBMCs was above the target concentration of 48 fmol/106 cells for the entire dose interval (6, 21). Additionally, TFV-DP concentrations in cervical and rectal tissues were also simulated. Average ratios of 0.031 and 0.02 for TFV-DP cervical tissue/TFV-DP PBMCs and TFV-DP rectal tissue/TFV-DP PBMCs, respectively, as described in clinical studies (43), were applied (see the supplemental material). Drug distributions in the cervical and rectal tissues were assumed to be dependent primarily on plasma concentrations and therefore not affected by the route of administration.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R24 AI 118397).
All authors contributed to the study design. R.K.R.R. designed the model and performed the simulations and analysis. Z.R.D. provided the description of the implant. R.K.R.R., Z.R.D., and M.S. wrote the manuscript with inputs from C.F. and A.O. All authors contributed toward the writing and reviewing of the final manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.UNAIDS. 2019. Global HIV & AIDS statistics—2019 fact sheet. http://www.unaids.org/en/resources/fact-sheet. Accessed 1 February 2018.
- 2.UNAIDS. 2018. The youth bulge and HIV. https://www.unaids.org/sites/default/files/media_asset/the-youth-bulge-and-hiv_en.pdf. Accessed 21 January 2020.
- 3.Eakle R, Venter F, Rees H. 2018. Pre-exposure prophylaxis (PrEP) in an era of stalled HIV prevention: can it change the game? Retrovirology 15:29. doi: 10.1186/s12977-018-0408-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LeVasseur MT, Goldstein ND, Tabb LP, Olivieri-Mui BL, Welles SL. 2018. The effect of PrEP on HIV incidence among men who have sex with men in the context of condom use, treatment as prevention, and seroadaptive practices. J Acquir Immune Defic Syndr 77:31–40. doi: 10.1097/QAI.0000000000001555. [DOI] [PubMed] [Google Scholar]
- 5.Pilkington V, Hill A, Hughes S, Nwokolo N, Pozniak A. 2018. How safe is TDF/FTC as PrEP? A systematic review and meta-analysis of the risk of adverse events in 13 randomised trials of PrEP. J Virus Erad 4:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapía M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernández T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker L-G, Mayer KH, Kallás EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang F, McConnell JJ, Zheng J-H, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV. 2010. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 363:2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baeten JM, Donnell D, Ndase P, Mugo NR, Campbell JD, Wangisi J, Tappero JW, Bukusi EA, Cohen CR, Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, Farquhar C, John-Stewart G, Kakia A, Odoyo J, Mucunguzi A, Nakku-Joloba E, Twesigye R, Ngure K, Apaka C, Tamooh H, Gabona F, Mujugira A, Panteleeff D, Thomas KK, Kidoguchi L, Krows M, Revall J, Morrison S, Haugen H, Emmanuel-Ogier M, Ondrejcek L, Coombs RW, Frenkel L, Hendrix C, Bumpus NN, Bangsberg D, Haberer JE, Stevens WS, Lingappa JR, Celum C. 2012. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med 367:399–410. doi: 10.1056/NEJMoa1108524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyers K, Wu Y, Brill A, Sandfort T, Golub SA. 2018. To switch or not to switch: intentions to switch to injectable PrEP among gay and bisexual men with at least twelve months oral PrEP experience. PLoS One 13:e0200296. doi: 10.1371/journal.pone.0200296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatt NR, Cheu R, Birse K, Zevin AS, Perner M, Noël-Romas L, Grobler A, Westmacott G, Xie IY, Butler J, Mansoor L, McKinnon LR, Passmore J-A, Abdool Karim Q, Abdool Karim SS, Burgener AD. 2017. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 356:938–945. doi: 10.1126/science.aai9383. [DOI] [PubMed] [Google Scholar]
- 10.Cottrell ML, Yang KH, Prince HMA, Sykes C, White N, Malone S, Dellon ES, Madanick RD, Shaheen NJ, Hudgens MG, Wulff J, Patterson KB, Nelson JAE, Kashuba A. 2016. A translational pharmacology approach to predicting outcomes of preexposure prophylaxis against HIV in men and women using tenofovir disoproxil fumarate with or without emtricitabine. J Infect Dis 214:55–64. doi: 10.1093/infdis/jiw077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pacífico de Carvalho N, Mendicino CCP, Cândido RCF, Alecrim DJD, Menezes de Pádua CA. 2019. HIV pre-exposure prophylaxis (PrEP) awareness and acceptability among trans women: a review. AIDS Care 31:1234–1240. doi: 10.1080/09540121.2019.1612014. [DOI] [PubMed] [Google Scholar]
- 12.Van Damme L, Corneli A, Ahmed K, Agot K, Lombaard J, Kapiga S, Malahleha M, Owino F, Manongi R, Onyango J, Temu L, Monedi MC, Mak'Oketch P, Makanda M, Reblin I, Makatu SE, Saylor L, Kiernan H, Kirkendale S, Wong C, Grant R, Kashuba A, Nanda K, Mandala J, Fransen K, Deese J, Crucitti T, Mastro TD, Taylor D. 2012. Preexposure prophylaxis for HIV infection among African women. N Engl J Med 367:411–422. doi: 10.1056/NEJMoa1202614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marrazzo JM, VOICE Study Team, Ramjee G, Richardson BA, Gomez K, Mgodi N, Nair G, Palanee T, Nakabiito C, van der Straten A, Noguchi L, Hendrix CW, Dai JY, Ganesh S, Mkhize B, Taljaard M, Parikh UM, Piper J, Mâsse B, Grossman C, Rooney J, Schwartz JL, Watts H, Marzinke MA, Hillier SL, McGowan IM, Chirenje ZM. 2015. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med 372:509–518. doi: 10.1056/NEJMoa1402269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donnell D, Baeten JM, Bumpus NN, Brantley J, Bangsberg DR, Haberer JE, Mujugira A, Mugo N, Ndase P, Hendrix C, Celum C. 2014. HIV protective efficacy and correlates of tenofovir blood concentrations in a clinical trial of PrEP for HIV prevention. J Acquir Immune Defic Syndr 66:340–348. doi: 10.1097/QAI.0000000000000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chloe Orkin KA, Hernández-Mora MG, Pokrovsky V, Overton ET, Girard P-M, Oka S, Ronald D, Dorey D, Griffith S, Margolis DA, Williams PE, Parys W, Spreen W. 2019. Long-acting cabotegravir + rilpivirine for HIV maintenance: FLAIR week 48 results. Conf Retroviruses Opportunistic Infect, abstr 140.
- 16.Susan Swindells J-F-V, Richmond GJ, Rizzardini G, Baumgarten A, Del Mar Masia M, Latiff G, Pokrovsky V, Mrus JM, Huang JO, Hudson KJ, Margolis DA, Smith K, Williams PE, Spreen W. 2019. Long-acting cabotegravir + rilpivirine as maintenance therapy: ATLAS week 48 results. Conf Retroviruses Opportunistic Infect, abstr 139.
- 17.Margolis DA, Gonzalez-Garcia J, Stellbrink H-J, Eron JJ, Yazdanpanah Y, Podzamczer D, Lutz T, Angel JB, Richmond GJ, Clotet B, Gutierrez F, Sloan L, Clair MS, Murray M, Ford SL, Mrus J, Patel P, Crauwels H, Griffith SK, Sutton KC, Dorey D, Smith KY, Williams PE, Spreen WR. 2017. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 390:1499–1510. doi: 10.1016/S0140-6736(17)31917-7. [DOI] [PubMed] [Google Scholar]
- 18.Markowitz M, Frank I, Grant RM, Mayer KH, Elion R, Goldstein D, Fisher C, Sobieszczyk ME, Gallant JE, Van Tieu H, Weinberg W, Margolis DA, Hudson KJ, Stancil BS, Ford SL, Patel P, Gould E, Rinehart AR, Smith KY, Spreen WR. 2017. Safety and tolerability of long-acting cabotegravir injections in HIV-uninfected men (ECLAIR): a multicentre, double-blind, randomised, placebo-controlled, phase 2a trial. Lancet HIV 4:e331–e340. doi: 10.1016/S2352-3018(17)30068-1. [DOI] [PubMed] [Google Scholar]
- 19.Johnson LM, Krovi SA, Li L, Girouard N, Demkovich ZR, Myers D, Creelman B, van der Straten A. 2019. Characterization of a reservoir-style implant for sustained release of tenofovir alafenamide (TAF) for HIV pre-exposure prophylaxis (PrEP). Pharmaceutics 11:315. doi: 10.3390/pharmaceutics11070315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chua CYX, Jain P, Ballerini A, Bruno G, Hood RL, Gupte M, Gao S, Di Trani N, Susnjar A, Shelton K, Bushman LR, Folci M, Filgueira CS, Marzinke MA, Anderson PL, Hu M, Nehete P, Arduino RC, Sastry JK, Grattoni A. 2018. Transcutaneously refillable nanofluidic implant achieves sustained level of tenofovir diphosphate for HIV pre-exposure prophylaxis. J Control Release 286:315–325. doi: 10.1016/j.jconrel.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunawardana M, Remedios-Chan M, Miller CS, Fanter R, Yang F, Marzinke MA, Hendrix CW, Beliveau M, Moss JA, Smith TJ, Baum MM. 2015. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) subdermal implant for HIV prophylaxis. Antimicrob Agents Chemother 59:3913–3919. doi: 10.1128/AAC.00656-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su JT, Simpson SM, Sung S, Tfaily EB, Veazey R, Marzinke M, Qiu J, Watrous D, Widanapathirana L, Pearson E, Peet MM, Karunakaran D, Grasperge B, Dobek G, Cain CM, Hope T, Kiser PF. 2019. A subcutaneous implant of tenofovir alafenamide fumarate causes local inflammation and tissue necrosis in rabbits and macaques. bioRxiv doi: 10.1101/775452. [DOI] [PMC free article] [PubMed]
- 23.Ray AS, Fordyce MW, Hitchcock M. 2016. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res 125:63–70. doi: 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Gilead Sciences. 2019. Gilead statement on commitment to advancing descovy for PrEP™ study in cisgender women & adolescent females.
- 25.Saeheng T, Na-Bangchang K, Karbwang J. 2018. Utility of physiologically based pharmacokinetic (PBPK) modeling in oncology drug development and its accuracy: a systematic review. Eur J Clin Pharmacol 74:1365–1376. doi: 10.1007/s00228-018-2513-6. [DOI] [PubMed] [Google Scholar]
- 26.Hendrix CW. 2018. HIV antiretroviral pre-exposure prophylaxis: development challenges and pipeline promise. Clin Pharmacol Ther 104:1082–1097. doi: 10.1002/cpt.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenna Yager KB, Castillo-Manchilla J, Morrow M, Ibrahim M, Mchugh C, Bushman L, Kiser J, Mawhinney S, Anderson P Tenofovir-diphosphate in PBMC following increasing TAF vs. TDF dosing under directly observed therapy. http://regist2.virology-education.com/presentations/2019/20AntiviralPK/30_Yager.pdf.
- 28.Krogstad EA, Montgomery ET, Atujuna M, Minnis AM, Shannon O, Ahmed K, Bekker L-G, Straten A. 2019. Design of an implant for long-acting HIV pre-exposure prophylaxis: input from South African health care providers. AIDS Patient Care Stds 33:157–166. doi: 10.1089/apc.2018.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Custodio JM, Fordyce M, Garner W, Vimal M, Ling KHJ, Kearney BP, Ramanathan S. 2016. Pharmacokinetics and safety of tenofovir alafenamide in HIV-uninfected subjects with severe renal impairment. Antimicrob Agents Chemother 60:5135–5140. doi: 10.1128/AAC.00005-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darville N, van Heerden M, Vynckier A, De Meulder M, Sterkens P, Annaert P, Van den Mooter G. 2014. Intramuscular administration of paliperidone palmitate extended-release injectable microsuspension induces a subclinical inflammatory reaction modulating the pharmacokinetics in rats. J Pharm Sci 103:2072–2087. doi: 10.1002/jps.24014. [DOI] [PubMed] [Google Scholar]
- 31.Gatto G, Girouard N, Brand RM, Johnson L, Marzinke M, Rowshan S, Engstrom JC, McGowan I, Demkovich Z, Lueke E, van der Straten A. 2018. Pharmacokinetics of tenofovir alafenamide by subcutaneous implant for HIV PREP. Conf Retroviruses Opportunistic Infect, abstr 486.
- 32.Manjula G, Mariana R-C, Debbie S, Moss JA, Paul W, Chris B, Dana W, Udayan C, Angel G, Joseph K, Gallay PA, Massoud M, Vincent KL, Marzinke MA, Hendrix CW, Baum MM. 2018. Multispecies in vivo evaluation of subdermal implants delivering tenofovir alafenamide: of mice, dogs and sheep. AIDS Res Hum Retroviruses 34:107. [Google Scholar]
- 33.Rajoli RKR, Back DJ, Rannard S, Freel Meyers CL, Flexner C, Owen A, Siccardi M. 2015. Physiologically based pharmacokinetic modelling to inform development of intramuscular long-acting nanoformulations for HIV. Clin Pharmacokinet 54:639–650. doi: 10.1007/s40262-014-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nestorov I. 2003. Whole body pharmacokinetic models. Clin Pharmacokinet 42:883–908. doi: 10.2165/00003088-200342100-00002. [DOI] [PubMed] [Google Scholar]
- 35.CDC. 2016. Anthropometric reference data for children and adults: United States, 2011–2014. https://www.cdc.gov/nchs/data/series/sr_03/sr03_039.pdf. Accessed 17 October 2019.
- 36.Bosgra S, Eijkeren J, Bos P, Zeilmaker M, Slob W. 2012. An improved model to predict physiologically based model parameters and their inter-individual variability from anthropometry. Crit Rev Toxicol 42:751–767. doi: 10.3109/10408444.2012.709225. [DOI] [PubMed] [Google Scholar]
- 37.Yu LX, Amidon GL. 1999. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm 186:119–125. doi: 10.1016/s0378-5173(99)00147-7. [DOI] [PubMed] [Google Scholar]
- 38.European Medicines Agency. 2016. Assessment report: Vemlidy. https://www.ema.europa.eu/en/documents/assessment-report/vemlidy-epar-public-assessment-report_en.pdf. Accessed 4 July 2019.
- 39.Peters S. 2008. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin Pharmacokinet 47:261–275. doi: 10.2165/00003088-200847040-00004. [DOI] [PubMed] [Google Scholar]
- 40.Poulin P, Theil FP. 2002. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci 91:129–156. doi: 10.1002/jps.10005. [DOI] [PubMed] [Google Scholar]
- 41.Rajoli RKR, Curley P, Chiong J, Back D, Flexner C, Owen A, Siccardi M. 2019. Predicting drug-drug interactions between rifampicin and long-acting cabotegravir and rilpivirine using physiologically based pharmacokinetic modeling. J Infect Dis 219:1735–1742. doi: 10.1093/infdis/jiy726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Begley R, Das M, Zhong L, Ling J, Kearney BP, Custodio JM. 2018. Pharmacokinetics of tenofovir alafenamide when coadministered with other HIV antiretrovirals. J Acquir Immune Defic Syndr 78:465–472. doi: 10.1097/QAI.0000000000001699. [DOI] [PubMed] [Google Scholar]
- 43.Cottrell ML, Garrett KL, Prince HMA, Sykes C, Schauer A, Emerson CW, Peery A, Rooney JF, McCallister S, Gay C, Kashuba A. 2017. Single-dose pharmacokinetics of tenofovir alafenamide and its active metabolite in the mucosal tissues. J Antimicrob Chemother 72:1731–1740. doi: 10.1093/jac/dkx064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DrugBank. 2019. Tenofovir alafenamide. https://www.drugbank.ca/drugs/DB09299. Accessed 29 July 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.