

Resistance to polymyxin antibiotics is increasing. Without new antibiotic classes, combination therapy is often required. We systematically investigated bacterial killing with polymyxin-based combinations against multidrug-resistant (including polymyxin-resistant), carbapenemase-producing Klebsiella pneumoniae. Monotherapies and double- and triple-combination therapies were compared to identify the most efficacious treatment using static time-kill studies (24 h, six isolates), an in vitro pharmacokinetic/pharmacodynamic model (IVM; 48 h, two isolates), and the mouse thigh infection model (24 h, six isolates).
KEYWORDS: Klebsiella pneumoniae, polymyxin resistance, combination, amikacin, rifampin
ABSTRACT
Resistance to polymyxin antibiotics is increasing. Without new antibiotic classes, combination therapy is often required. We systematically investigated bacterial killing with polymyxin-based combinations against multidrug-resistant (including polymyxin-resistant), carbapenemase-producing Klebsiella pneumoniae. Monotherapies and double- and triple-combination therapies were compared to identify the most efficacious treatment using static time-kill studies (24 h, six isolates), an in vitro pharmacokinetic/pharmacodynamic model (IVM; 48 h, two isolates), and the mouse thigh infection model (24 h, six isolates). In static time-kill studies, all monotherapies (polymyxin B, rifampin, amikacin, meropenem, or minocycline) were ineffective. Initial bacterial killing was enhanced with various polymyxin B-containing double combinations; however, substantial regrowth occurred in most cases by 24 h. Most polymyxin B-containing triple combinations provided greater and more sustained killing than double combinations. Standard dosage regimens of polymyxin B (2.5 mg/kg of body weight/day), rifampin (600 mg every 12 h), and amikacin (7.5 mg/kg every 12 h) were simulated in the IVM. Against isolate ATH 16, no viable bacteria were detected across 5 to 25 h with triple therapy, with regrowth to ∼2-log10 CFU/ml occurring at 48 h. Against isolate BD 32, rapid initial killing of ∼3.5-log10 CFU/ml at 5 h was followed by a slow decline to ∼2-log10 CFU/ml at 48 h. In infected mice, polymyxin B monotherapy (60 mg/kg/day) generally was ineffective. With triple therapy (polymyxin B at 60 mg/kg/day, rifampin at 120 mg/kg/day, and amikacin at 300 mg/kg/day), at 24 h there was an ∼1.7-log10 CFU/thigh reduction compared to the starting inoculum for all six isolates. Our results demonstrate that the polymyxin B-rifampin-amikacin combination significantly enhanced in vitro and in vivo bacterial killing, providing important information for the optimization of polymyxin-based combinations in patients.
TEXT
Klebsiella pneumoniae is a major Gram-negative opportunistic pathogen responsible for nosocomial respiratory tract, bloodstream, and urinary tract infections (1–4). Infections caused by multidrug-resistant (MDR) K. pneumoniae have been increasingly reported over the past few years (3, 5, 6), with a mortality rate of up to 40% in patients infected with carbapenemase-producing (KPC) K. pneumoniae (7–11). The World Health Organization (WHO) has designated carbapenem-resistant K. pneumoniae one of three critical priority pathogens on the list for the research and development of new antibiotics (12). Given the critical shortage of effective treatment options for use against infections caused by MDR K. pneumoniae, including KPC-producing K. pneumoniae strains (13, 14), the old polymyxins (i.e., polymyxin B [PMB] and colistin) are increasingly used in the clinic. Polymyxins are naturally occurring cyclic lipopeptides (15, 16) whose use declined in the 1980s due to reports of nephro- and neurotoxicity (17–19). However, they have attracted significant interest over the last decade, given their activity against many MDR Gram-negative organisms (20, 21). Worryingly, polymyxin-resistant K. pneumoniae isolates have been reported both in vitro and in vivo (21–23). Although a number of in vitro studies have demonstrated that polymyxin-based double combinations can exhibit rapid initial killing against polymyxin-susceptible and -resistant MDR K. pneumoniae isolates, bacterial regrowth commonly occurs soon after (24, 25). Thus, there is an urgent need to develop novel, rational combinations for the treatment of polymyxin-resistant, MDR K. pneumoniae isolates. This in vitro and in vivo study aimed to evaluate bacterial killing and resistance suppression of polymyxin-based double and triple combinations against polymyxin-resistant, MDR K. pneumoniae clinical isolates and to identify the most active triple combination in vitro and in vivo.
(This study was presented at ASM Microbe, 7 to 11 June 2018.)
RESULTS
MICs and static-concentration time-kill experiments.
MICs to each antibiotic are shown in Table 1. All six K. pneumoniae isolates were susceptible or intermediately resistant to minocycline (MIN) and resistant to all remaining antibiotics tested, except ATH 8, which was not resistant to meropenem (MER). The results of the static-concentration time-kill experiments are shown in Table 2 (log changes in viable cell counts) and Fig. S1 to S3 in the supplemental material (time-kill curves). All antibiotic monotherapies were ineffective against all isolates across 24 h (maximum killing of 0.7 log10 CFU/ml, with subsequent regrowth; Table 2 and Fig. S1). Polymyxin B with either minocycline or rifampin (RIF) were additive or synergistic against most isolates across 24 h (except polymyxin B plus minocycline against BD 32), with substantially enhanced initial killing (at 1 h) of up to ∼4 log10 CFU/ml compared to monotherapy (Table 2 and Fig. S2). Polymyxin B plus either amikacin (AMI) or meropenem showed additivity or synergy against fewer isolates and generally not at 24 h. Other double combinations against all isolates, with the exception of BD 46 at 1 and 4 h, typically were ineffective. Despite the enhanced killing, regrowth was observed with all isolates treated with double antibiotic combinations such that by 24 h, even synergistic combinations had exceeded, or were close to, the initial inoculum.
TABLE 1.
MICs of KPC-producing K. pneumoniae isolates used in this study
| Isolatea | MIC (μg/ml) |
||||
|---|---|---|---|---|---|
| PMBb | RIFd | AMIb | MINc | MERb | |
| ATH 8 | 64 | 16 | 16 | 4 | 4 |
| ATH 16 | 32 | 16 | 16 | 4 | 64 |
| ATH 18 | 64 | 16 | 16 | 4 | 128 |
| ATH 24 | 32 | 16 | 16 | 4 | 64 |
| BD 32 | 32 | 128 | 128 | 8 | 64 |
| BD 46 | 128 | 32 | 16 | 4 | 64 |
All isolates were multidrug resistant, defined as nonsusceptible to at least one agent in three or more antimicrobial categories (67). BD 32 additionally was pandrug resistant, defined as nonsusceptible to all agents in all antimicrobial classes (66).
EUCAST breakpoints (S, susceptible; R, resistant) were S ≤ 8 μg/ml and R > 8 μg/ml for amikacin and S ≤ 2 μg/ml and R > 8 μg/ml for meropenem; the EUCAST breakpoints for colistin of S ≤ 2 and R > 2 were applied to polymyxin B (69, 70).
CLSI breakpoints (S, susceptible; R, resistant) were S ≤ 4 μg/ml and R ≥ 16 μg/ml for minocycline (68).
Rifampin as monotherapy is normally inactive against the Enterobacteriaceae, and breakpoints have not been established.
TABLE 2.
Log changes in viable cell counts at 1, 4, and 24 h with clinically relevant concentrations of PMB, RIF, AMI, MIN, and MER against six MDR K. pneumoniae isolates using static time-kill experimentsa

PMB, polymyxin B (2 μg/ml); RIF, rifampin (5 μg/ml); AMI, amikacin (20 μg/ml); MIN, minocycline (4 μg/ml); MER, meropenem (50 μg/ml). A green background indicates synergy (a ≥2-log10 decrease in the number of CFU/ml with combination therapy compared with its most active component at the specified time and with the number of surviving bacteria in the presence of the combination being ≥2 log10 CFU/ml below the starting inoculum); a red background indicates additivity (a ≥1.0-log10 decrease in number of CFU/ml with the combination compared with its most active component, without being synergistic).
With the exception of BD 32, where only the combinations of polymyxin B with rifampin plus either amikacin or meropenem enhanced bacterial killing, the polymyxin B-based triple combinations were highly synergistic (up to ∼5 log10 CFU/ml greater killing than the most active monotherapy) or additive against all isolates, including at 24 h in the majority of cases (Table 2 and Fig. S3). However, only the polymyxin B-rifampin-amikacin combination was additive or synergistic against all isolates at 24 h. Despite the observed additivity/synergy at 24 h, in most cases bacterial growth was trending upwards at this time.
One-compartment IVM.
The most active triple combination from the static-concentration time-kill experiments, namely, polymyxin B-rifampin-amikacin, was further examined in an in vitro dynamic infection model (IVM) against isolates ATH 16 and BD 32. The results of IVM experiments are shown in Fig. 1 (time-kill curves) and 2 (population analysis profiles) and Table S1 (log changes in viable cell counts). The growth control grew steadily to ∼8.5 log10 CFU/ml throughout the experiment. Against ATH 16, monotherapy and the double combination of rifampin plus amikacin were ineffective, with growth mirroring that of the growth control. The combination of polymyxin B plus amikacin produced a maximum killing of ∼3 log10 CFU/ml at 5 h, but regrowth to control values occurred by 24 h. The combination of polymyxin B plus rifampin produced maximal killing of ∼5 log10 CFU/ml at 5 h, with slow regrowth (to ∼5 log10 CFU/ml at 48 h) occurring thereafter. The polymyxin B-rifampin-amikacin triple combination was the most effective, with no viable bacteria detected from 5 to 25 h and regrowth to only ∼2 log10 CFU/ml at 48 h. For the latter two combinations (polymyxin B-rifampin double and polymyxin B-rifampin-amikacin triple combinations), compared to the level of the most active monotherapy, additivity or synergy was observed at all times across 48 h (Table S1); on many occasions and including at 48 h, the triple combination was synergistic compared to all mono- and double-combination therapies. Against BD 32, only polymyxin B-rifampin and the triple combination showed substantial bacterial killing. With both combinations against BD 32, killing of ∼3 log10 CFU/ml had occurred at 5 h; however, slow regrowth to ∼6.5 log10 CFU/ml at 48 h occurred with polymyxin B-rifampin. Bacterial numbers continued to decline slowly with the triple combination, with growth of ∼2 log10 CFU/ml at 48 h. With these two combinations, additivity and synergy patterns were similar to those observed with ATH 16.
FIG 1.

Time-kill curves with clinically relevant concentrations of polymyxin B (constant concentration of 2 μg/ml), rifampin (Cmax, 5 μg/ml; t1/2, 2.5 h), and amikacin (Cmax, 20 μg/ml; t1/2, 2.5 h) alone, in double combinations, and in a triple combination with an inoculum of ∼107 CFU/ml against two isolates of polymyxin-resistant, MDR K. pneumoniae (ATH 16 [A] and BD 32 [B]) using a dynamic one-compartment infection model. The y axis starts from the limit of detection (∼1.3 log10 CFU/ml), and the dashed horizontal line represents the limit of quantification. PMB, polymyxin B; RIF, rifampin; AMI, amikacin.
Baseline population analysis profiles (PAPs) for both isolates showed that the entire population was highly resistant to polymyxin B (growing in the presence of 32 μg/ml polymyxin B) (Fig. 2). Subsequent PAPs at 24 and 48 h revealed that irrespective of the treatment regimen (polymyxin B monotherapy or polymyxin B double or triple therapy), virtually all bacteria detected remained highly resistant to polymyxin B.
FIG 2.

Population analysis profiles of polymyxin B at baseline (0 h; inoculum of ∼107 CFU/ml) or 24 h and 48 h after exposure to no antibiotics (control) or clinically relevant concentrations of polymyxin B (constant concentration of 2 μg/ml) alone or combined with rifampin (Cmax, 5 μg/ml; t1/2, 2.5 h), and/or amikacin (Cmax, 20 μg/ml; t1/2, 2.5 h). (A to C) ATH 16. (D to F) BD 32. The y axis starts from the limit of detection (∼1.3 log10 CFU/ml), and the dashed horizontal line represents the limit of quantification. PMB, polymyxin B; RIF, rifampin; AMI, amikacin.
Mouse thigh infection model.
Log10 CFU/thigh counts in the mouse thigh infection model are shown in Fig. 3 and 4. In the control mice, values ranged from ∼9 to 10 log10 CFU/thigh at 24 h. With polymyxin B monotherapy, little or no reduction in log10 CFU/thigh compared to that of the controls occurred at 24 h with any of the 6 isolates. Larger reductions (4 to 4.6 log10 CFU/thigh) occurred with the triple therapy, with a log10 CFU/thigh of ∼5 observed at 24 h for all isolates; the latter value was at least 2.5 log10 CFU/thigh below that observed with polymyxin B monotherapy at this time and ∼1 log10 CFU/thigh lower than that of the starting inoculum (∼107 log10 CFU/thigh). Viable bacterial counts with triple therapy were significantly different from those of the control and polymyxin B monotherapy groups at 24 h, while no significant differences were observed between the polymyxin B monotherapy and untreated control groups at this time (P < 0.05). The in vivo efficacy of all treatments (polymyxin B, rifampin, or amikacin alone and in double and triple combinations) was further evaluated against BD 32 (Fig. 4). For all monotherapies, there was only a small reduction in bacterial counts at 24 h compared to the level for the control. Similarly, there was no improvement in bacterial killing at this time with the rifampin-amikacin combination, although larger reductions were observed with the polymyxin B-amikacin, polymyxin B-rifampin, and polymyxin B-rifampin-amikacin combinations. Only the polymyxin B-rifampin and triple combinations reduced the bacterial burden to below that of the initial inoculum.
FIG 3.

Log10 CFU/thigh counts in the mouse thigh infection model at baseline (0 h; inoculum of ∼107 CFU/thigh) and 24 h after exposure to no antibiotics (control) or polymyxin B (60 mg/kg/day) alone or combined with rifampin (120 mg/kg/day) and amikacin (300 mg/kg/day) against six isolates of multidrug-resistant (including polymyxin-resistant) K. pneumoniae. PMB, polymyxin B; RIF, rifampin; AMI, amikacin. ***, Approximately 5 log10 CFU/thigh lower than that of the control group at 24 h (P < 0.05).
FIG 4.
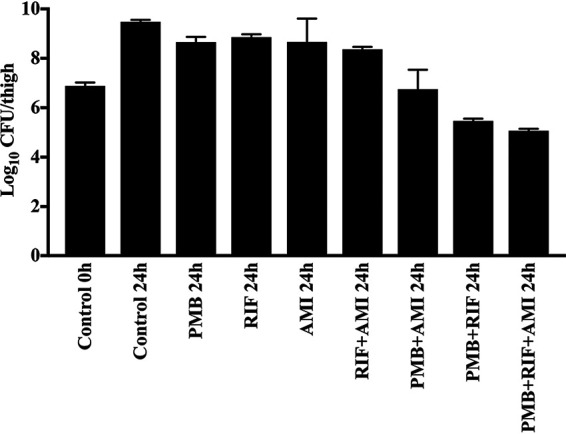
Log10 CFU/thigh counts in the mouse thigh infection model against polymyxin-resistant, MDR K. pneumoniae BD 32 at baseline (0 h; inoculum of ∼107 CFU/thigh) and 24 h after exposure to no antibiotics (control) or polymyxin B (60 mg/kg/day), rifampin (120 mg/kg/day), and amikacin (300 mg/kg/day) alone, in double combinations, and in the triple combination (n = 4). PMB, polymyxin B; RIF, rifampin; AMI, amikacin.
DISCUSSION
As MDR K. pneumoniae spreads globally (26, 27), reports of strains resistant to the last-line polymyxins have become increasingly more frequent (22, 23). Such a dire situation leaves clinicians with virtually no therapeutic options to treat this problematic human pathogen. Polymyxin combination therapy is considered a viable clinical strategy to salvage bacterial killing efficacy against pathogens resistant to the individual monotherapies (28, 29). In vitro and in vivo studies provide considerable support for polymyxin use as part of combination therapies, including when combined with antibiotics (e.g., rifampin) that typically are inactive against Gram-negative organisms (29). Although polymyxin-based combinations are increasingly used clinically (30, 31), the choice of antimicrobial agents in these combinations often are based on trial and error or anecdotal experiences. Thus, investigating rational, scientifically based polymyxin-based combinations is critical for the optimization of antimicrobial therapy. Our study systematically examined polymyxin combination therapy with other antibiotics (rifampin, amikacin, minocycline, and meropenem) against six polymyxin-resistant, MDR clinical isolates of K. pneumoniae. The time-kill studies revealed good initial killing (∼3 to 7 log10 CFU/ml) against many of the isolates with the various combinations, although slightly greater initial bacterial killing and suppression of regrowth was observed with the triple combinations (polymyxin B-rifampin-amikacin) compared to that of the double combinations. Nevertheless, substantial regrowth occurred in most cases with these static-concentration experiments.
The antibiotics forming the most active triple combination from the static time-kill experiments (polymyxin B-rifampin-amikacin) subsequently were evaluated in the one-compartment pharmacokinetic/pharmacodynamic (PK/PD) model; notably, both K. pneumoniae isolates tested in this experiment were resistant to each antibiotic in this combination when used as monotherapy. Our group has previously shown the importance of using dynamic models that mimic the antibiotic concentration-time profiles in patients when assessing the efficacy of antibiotic combinations (32, 33). The simulated plasma concentration-time profiles reflected free (unbound) concentrations achieved in patients using standard dosage regimens: polymyxin B at 2.5 mg/kg/day, giving an unbound average steady-state concentration (fCss,avg) of up to approximately 2 μg/ml (34, 35); rifampin at 600 mg every 12 h to achieve a peak unbound concentration (fCmax) of 5 μg/ml and half-life (t1/2) of 2.5 h (36, 37); and amikacin at 7.5 mg/kg/12 h to achieve an fCmax of 20 μg/ml and t1/2 of 2.5 h (38). The combination of polymyxin B plus rifampin and the triple combination were most effective against both extremely drug-resistant isolates tested, each causing substantial initial killing (>3 log10 CFU/ml), and, for the triple therapy, continued killing across 48 h (BD 32) or with no detectable viable bacteria for a prolonged period, followed by minimal regrowth (ATH 16).
While a number of studies have examined polymyxin-based combinations against K. pneumoniae, very few have utilized polymyxin-resistant KPC-producing isolates (39–42). Using checkerboard and time-kill studies, Tascini et al. (43) and Gaibani et al. (44) found the combination of colistin plus rifampin was consistently synergistic against colistin-resistant, KPC-producing isolates of K. pneumoniae. Similarly, our results support the effectiveness of colistin/rifampin double combinations on initial bacterial killing (across the first 4 to 8 h). However, our static concentration and IVM studies with this double combination (the latter utilizing dynamic concentrations over 48 h; inoculum, ∼107 CFU/ml) indicated that substantial regrowth occurs following the initial killing phase (Fig. 1; also see Fig. S2 in the supplemental material). Jernigan et al. (40) used time-kill studies (24 h, inoculum of 1 × 106 CFU/ml) to examine colistin combined with either doripenem, gentamicin, or doxycycline against 12 KPC-producing isolates of K. pneumoniae (10 were polymyxin-resistant isolates). Synergy at 24 h was observed with six isolates (colistin-doripenem), three isolates (colistin-gentamicin), and one isolate (colistin-doxycycline). In our time-kill study, the analogous combinations containing a polymyxin (polymyxin B) and a carbapenem (meropenem) or aminoglycoside (amikacin) were even less effective, with no synergy observed for any K. pneumoniae isolate at 24 h; apart from initial killing against ATH 16, the latter combination was similarly ineffective in the IVM.
Diep et al. (25) used an in vitro dynamic model (48 h, inoculum of ∼107 CFU/ml) to examine the effectiveness of various triple combinations containing polymyxin B (constant concentration of 0.5, 1, or 2 μg/ml), rifampin (Cmax, 5 μg/ml; t1/2, 2 h; dosing every 8 and 12 h), and meropenem (Cmax, 40 or 80 μg/ml; t1/2, 2 h; dosing every 8 h) against one polymyxin-susceptible and one polymyxin-resistant KPC-producing K. pneumoniae isolate; both isolates were resistant to meropenem and rifampin. Irrespective of the individual regimen used for each antibiotic, the triple combination produced substantial bacterial killing against each isolate, with bacterial counts at 48 h at least ∼3 to 4 log10 CFU/ml lower than that of the starting inoculum. In our static time-kill experiments, the same triple combination similarly enhanced bacterial killing, although the combination of polymyxin B-rifampin-amikacin was slightly more effective across all six isolates. To the best of our knowledge, ours is the first study to examine the triple combination of polymyxin B, rifampin, and amikacin in an IVM or in vivo against polymyxin-resistant, KPC-producing K. pneumoniae. The triple combination of polymyxin B, rifampin, and amikacin substantially enhanced bacterial killing in the IVM compared to the double combinations and was superior to polymyxin B monotherapy in the thigh infection model. Given a lack of therapeutic options and the absence of evidence-based treatment guidelines for treating patients with infections caused by KPC-producing K. pneumoniae, combination therapy with three or more antibiotics is increasingly used (40). In these patients, triple-combination therapies, of which a polymyxin is considered the backbone antibiotic, have been shown to significantly reduce overall mortality compared to monotherapy (45, 46).
The enhanced bacterial killing observed in this study with both double- and, especially, triple-combination therapy (polymyxin B-rifampin-amikacin) may be due to mechanistic synergy, whereby two drugs that act on different cellular pathways increase the rate and extent of killing of the other drug(s) (47). Although the exact mechanism(s) by which polymyxins exert their bactericidal action against Gram-negative bacteria remains unknown, electrostatically binding to lipopolysaccharide and permeabilization of the outer membrane, including against polymyxin-resistant strains (48, 49), is thought to be a major contributor. Such an action potentially enables the coadministered antibiotics to achieve greater access to their intracellular target sites, thereby enhancing activity. Bacterial killing by aminoglycosides is due primarily to the inhibition of protein synthesis via interaction with the 30S bacterial ribosome (50, 51). However, aminoglycosides, such as amikacin, at clinically relevant concentrations are known to exert an outer membrane permeabilizing effect (50, 52). This occurs prior to any of their effects on protein synthesis and, thus, may increase the access of other antibacterial agents to their intracellular target sites. Rifampin exerts its action on bacteria by inhibiting bacterial DNA-dependent RNA polymerase (53, 54). Ordinarily, rifampin is inactive against Gram-negative bacteria, as this hydrophobic antibiotic is excluded from its target site by the outer membrane (54). Thus, the permeabilizing effect of both polymyxin B and amikacin may have facilitated the access of rifampin into cells. That said, the amikacin-rifampin combination performed poorly compared to the polymyxin B-rifampin combination in both static and dynamic in vitro models, suggesting polymyxin B facilitated greater entry than did amikacin. Thus, for the polymyxin B-rifampin-amikacin triple combination, the greatly enhanced bacterial killing observed may be due to the disruption of the outer membrane, primarily by polymyxin B, that facilitated the entry of both amikacin and rifampin to their intracellular targets. Although all isolates were resistant to amikacin, the increased intracellular amikacin concentrations might be sufficient to enhance the killing effect. Furthermore, our recent metabolomic study with isolate ATH 16 showed that the triple combination of polymyxin B-rifampin-amikacin negatively affects the lipid A modification pathway, thereby attenuating bacterial resistance to polymyxins (55). In addition, subpopulation synergy, namely, where one drug kills the resistant subpopulations of another drug and vice versa (47), seems unlikely to have significantly contributed to enhanced killing of the triple combination, given that all isolates were resistant (and likely remained resistant) to each drug.
Although antibiotic combination therapy can lead to improved clinical outcomes in patients infected with MDR isolates compared to monotherapy (56), a potential disadvantage of combination therapy is a greater risk of drug toxicity, including nephrotoxicity (57). Polymyxin B and amikacin monotherapy have dose-limiting nephrotoxicity (17, 58–60), while rifampin can cause hepatotoxicity (61). The average steady-state concentration (Css,avg) associated with mild nephrotoxicity for polymyxin B was estimated at ∼4 μg/ml (34, 62), whereas for amikacin peak plasma concentrations (fCmax) greater than 40 μg/ml and trough levels (fCmin) above 10 μg/ml are associated with nephrotoxicity (63, 64). Despite the substantial improvements in in vitro and in vivo bacterial killing with the triple combination of polymyxin B-rifampin-amikacin, the potential of combining nephrotoxic agents might cause clinicians concern. Our studies utilized plasma concentrations of amikacin (fCmax of 20 μg/ml and minimum concentration [fCmin] of 1.02 μg/ml) that in patients would expose the kidneys to concentrations lower than those typically associated with nephrotoxicity. Additionally, inadequately treated Gram-negative infections can lead to sepsis and subsequent kidney injury (65). Thus, greater bacterial killing by the combination may allow for earlier recovery from sepsis and reduced kidney dysfunction.
Conclusions.
The dissemination of MDR K. pneumoniae has created significant health care challenges worldwide and led to growing interest in optimizing antibiotic combination therapies to treat these organisms. We have demonstrated, for the first time, in a one-compartment PK/PD model and in vivo that a polymyxin-based triple combination with rifampin and amikacin significantly enhances bacterial killing against polymyxin-resistant, KPC-producing K. pneumoniae. Future PK/PD modeling and clinical trials are required to optimize dosing of this triple combination against MDR K. pneumoniae.
MATERIALS AND METHODS
Bacterial isolates, antibiotics, MICs, and media.
Six isolates of K. pneumoniae were investigated. Four isolates (ATH 8, ATH 16, ATH 18, and ATH 24) were obtained from the Hygeia General Hospital, Marousi, Greece (2016), and two (BD 32 and BD 46) from SUNY Downstate Medical Center, Brooklyn, New York. All isolates were KPC-producing and multidrug resistant, i.e., nonsusceptible to at least one agent in three or more antimicrobial categories, which, in this case, included the polymyxins (66, 67). MICs to all tested antibiotics were determined in duplicate on separate days in cation-adjusted Muller-Hinton broth (CaMHB; Mg2+ at 12.2 μg/ml and Ca2+ at 23.0 μg/ml [Oxoid, Hampshire, UK]). The MIC breakpoint for minocycline was assigned according to the Clinical and Laboratory Standards Institute guidelines (CLSI) (68), and those of the other drugs (except polymyxin B) were assigned according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (69). Although no breakpoints for polymyxin B against the Enterobacteriaceae have been established, susceptibility and resistance breakpoints for colistin have been set at ≤2 μg/ml and >2 μg/ml, respectively (69). For this study, we applied the colistin breakpoint to polymyxin B given the comparable activity of each polymyxin (70). The same medium was used for static-concentration and in vitro dynamic infection model experiments (discussed below).
Polymyxin B (PMB; lot number 20120204; Sigma-Aldrich, Castle Hill, Australia), amikacin (AMI; lot number 058K0803; Sigma-Aldrich), rifampin (RIF; lot number SLBK5059V; Sigma-Aldrich), minocycline (MIN; lot number 20120424; Sigma-Aldrich), and meropenem (MER; Fresenius Kabi, Mount Kuring-Gai, Australia) were carefully chosen from five major classes of antibiotics. Concentrations were selected to mimic the steady-state average concentrations (fCss,avg) or peak concentrations (fCmax) of free (unbound) drug in human plasma in patients administered standard-dosage regimens. In brief, sterile stock solutions of PMB, AMI, MER, and MIN were freshly prepared in Milli-Q water immediately prior to each experiment. RIF stock solutions were prepared in a minimum amount of dimethyl sulfoxide (DMSO) before dilution with Milli-Q water. All drug solutions were filter sterilized using 0.22-μm filters (Millipore, Bedford, MA).
Static-concentration time-kill experiments.
Antibiotics were examined as monotherapy and in double and polymyxin B-containing triple combinations against six isolates (Table 2). Prior to each experiment, isolates were subcultured onto nutrient agar (Media Preparation Unit, Monash University, Clayton, Australia) and incubated at 37°C for 24 h. Single colonies were selected and grown overnight in 10 ml of CaMHB with constant shaking (180 rpm), from which early-log-phase bacterial cultures (∼107 CFU/ml) were obtained (20 ml). Antibiotic solutions were added to achieve the desired drug concentrations (PMB, 2 μg/ml; RIF, 5 μg/ml; AMI, 20 μg/ml; MIN, 4 μg/ml; and MER, 50 μg/ml), whereupon tubes were incubated at 37°C in a shaking water bath (180 rpm) for 24 h. Serial samples (0.2 ml) were collected aseptically at 0, 1, 4, and 24 h, with viable counting conducted immediately via serial dilution (using 0.9% saline) and spiral plating (WASP2 spiral plater; Don Whitley Scientific, Ltd., UK) of 50 μl of undiluted or appropriately diluted suspension onto Mueller-Hinton II agar, followed by incubation at 37°C.
IVM.
A one-compartment in vitro PK/PD model (33) was used to examine bacterial killing and resistance suppression over 48 h against isolates ATH 16 and BD 32. Polymyxin B, rifampin, or amikacin was used as a monotherapy and in double combinations (polymyxin B plus rifampin, polymyxin B plus amikacin, and rifampin plus amikacin) and the triple combination. Single colonies of each isolate were selected and grown overnight as described for static time-kill studies. An early-log-phase bacterial suspension then was prepared by the addition of 200 μl of the overnight culture to 20 ml of CaMHB, with 0.8 ml then inoculated into each reservoir of the model (containing 80 ml of CaMHB) to obtain the desired starting inoculum of ∼107 CFU/ml. The temperature of each reservoir was maintained at 37°C via heating in paraffin oil and with constant stirring via a magnetic stirrer located within each reservoir.
Eight sealed reservoirs (compartments) were used, with one acting as a growth control to define growth dynamics in the absence of antibiotic. As polymyxin B was administered at a constant concentration, it was spiked into one central reservoir connected only to polymyxin B-containing reservoirs immediately prior to starting the experiment, such that all media flowing through these reservoirs contained polymyxin B at a constant concentration of 2 μg/ml; this simulated the upper limit of unbound average steady-state concentration (fCss,avg) observed in critically ill patients (35), with the steady-state concentration present from the beginning of each experiment. Following bacterial inoculation, rifampin and amikacin were administered as 1-h infusions every 12 h using an automatic syringe pump to attain unbound peak concentrations (fCmax) of 5 μg/ml and 20 μg/ml, respectively. Medium was pumped through each reservoir at a rate of 0.37 ml/min to simulate a plasma elimination half-life (t1/2) of 2.5 h for both rifampin and amikacin; the chosen Cmax and t1/2 approximate those achieved in patients administered standard doses intravenously (36, 38, 71, 72).
Serial samples (0.5 ml) were collected from each reservoir aseptically at 0, 5, 12, 13, 24, 25, 36, 37, and 48 h for viable cell counting and at 24 and 48 h for population analysis profiles (PAPs). Viable counting was conducted as described for static time-kills but with an additional washing step to reduce the possibility of antibiotic carryover. Washing involved the centrifugation of the samples (10,000 × g, 4°C, 10 min) with resuspension in 0.9% saline prior to dilution. To evaluate the emergence of polymyxin B resistance, PAPs were performed. Similarly treated samples were plated onto Mueller-Hinton II agar containing polymyxin B at 2, 8, 16, or 32 μg/ml, followed by incubation at 37°C for 24 h. All colonies were counted manually.
Mouse thigh infection model.
A previously described neutropenic mouse thigh infection model was used to evaluate the efficacy of the triple combination across 24 h (73). Mice were rendered neutropenic by intraperitoneal injection of two doses of cyclophosphamide (Endoxan; Baxter Healthcare Pty. Ltd., New South Wales, Australia) administered 4 days (150 mg/kg of body weight) and 1 day (100 mg/kg) prior to experimental infection. Mice were anesthetized briefly with isoflurane by inhalation prior to inoculation. An early logarithmic-phase bacterial suspension of 50 μl (∼106 CFU/ml) was injected intramuscularly into each posterior thigh muscle, with antibiotic treatment commencing 2 h later. At this time, infection was reproducibly established with a starting mean bacteria burden for the six strains of ∼107 CFU per thigh. Experiments were conducted using all six isolates.
Two control mice were sacrificed 2 h postinoculation (t = 0 h) to examine bacterial counts prior to antibiotic treatment. Polymyxin B (60 mg/kg/day), rifampin (120 mg/kg/day), and amikacin (300 mg/kg/day) each was examined as monotherapy and in double and triple combinations against isolate BD 32. Against the remaining five isolates, polymyxin alone and the triple combination were tested at equivalent concentrations. The antibiotic treatments then were commenced via subcutaneous injection every 8 h. The polymyxin B dose was chosen to mimic the exposure of unbound polymyxin B in human plasma based on animal scaling and its different plasma protein binding in humans and mice (35, 74). The doses of rifampin and amikacin were determined based on the literature and consideration of toxicities (36, 38, 71, 72). Each dosage regimen and the remaining controls involved two mice and provided four data points. The pharmacodynamics of control mice (to determine the overall level of bacterial growth in the absence of treatment) and mice receiving treatment were determined at 24 h following humane killing of the mice. Both posterior thigh muscles were aseptically collected, homogenized in sterile saline at 9,000 × g for 30 s (T25 ULTRA-TURRAX; IKA), and filtered using a sterile filter bag (9.5 by 16 cm) with a pore size of 280 μm (Labtek Pty. Ltd.). After this, 1 ml of the filtrate was serially diluted for plating (usually 0.1 ml homogenate in 0.9 ml saline) on nutrient agar as previously described, followed by incubation at 37°C for 24 h. Colonies were counted using an automated colony counter (ProtoCol) and expressed as log10 CFU/thigh.
Pharmacodynamic analysis.
Microbial responses were evaluated using the log change method quantified as follows: log change = log10(CFUt) – log10(CFU0), where log10(CFU0) is the bacterial count at 0 h and log10(CFUt) the bacterial count at a specified time (t). For static and dynamic in vitro studies, synergy was considered to be a ≥2-log10 CFU/ml lower bacterial count with the combination compared to the most active single component at the specified time and with the number of surviving bacteria in the presence of the combination being ≥2 log10 CFU/ml below the starting inoculum (75). Additivity was considered a ≥1.0-log10 CFU/ml lower bacterial count at the specified time than that of the combination without being synergistic (75). Bactericidal activity was defined as a ≥3-log10 decrease in the number of CFU/ml compared to that of the starting inoculum. For the thigh infection studies, the viable bacterial counts between the control groups at 24 h and the treated groups at 24 h were statistically compared (P < 0.05) using Tukey’s multiple-comparison test (GraphPad Prism 8 software; La Jolla, CA, USA).
Supplementary Material
ACKNOWLEDGMENTS
J.L. and T.V. are supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI132681). J.L. and T.V. are also supported by the Australian National Health and Medical Research Council (NHMRC) as Principal Research and Career Development Level 2 Fellows, respectively. S.M.A. thanks the Myanmar Government for providing the Presidential Scholarship.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
We have no conflicts of interest to declare.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Pitout JD, Nordmann P, Poirel L. 2015. Carbapenemase-producing Klebsiella pneumoniae, a key pathogen set for global nosocomial dominance. Antimicrob Agents Chemother 59:5873–5884. doi: 10.1128/AAC.01019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broberg CA, Palacios M, Miller VL. 2014. Klebsiella: a long way to go towards understanding this enigmatic jet-setter. F1000Prime Rep 6:64. doi: 10.12703/P6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (CDC). 2019. Antibiotic resistance threats in the United States 2019. CDC, Atlanta, GA: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf. [Google Scholar]
- 4.Navon VS, Kondratyeva K, Carattoli A. 2017. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev 41:252–275. doi: 10.1093/femsre/fux013. [DOI] [PubMed] [Google Scholar]
- 5.Kirst HA. 2015. Circumventing resistance to anti-infective agents. Expert Opin Pharmacother 16:149–150. doi: 10.1517/14656566.2015.1002669. [DOI] [PubMed] [Google Scholar]
- 6.Limbago BM, Rasheed JK, Anderson KF, Zhu W, Kitchel B, Watz N, Munro S, Gans H, Banaei N, Kallen AJ. 2011. IMP-producing carbapenem-resistant Klebsiella pneumoniae in the United States. J Clin Microbiol 49:4239–4245. doi: 10.1128/JCM.05297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molton JS, Tambyah PA, Ang BS, Ling ML, Fisher DA. 2013. The global spread of healthcare-associated multidrug-resistant bacteria: a perspective from Asia. Clin Infect Dis 56:1310–1318. doi: 10.1093/cid/cit020. [DOI] [PubMed] [Google Scholar]
- 8.Ben-David D, Kordevani R, Keller N, Tal I, Marzel A, Gal-Mor O, Maor Y, Rahav G. 2012. Outcome of carbapenem resistant Klebsiella pneumoniae bloodstream infections. Clin Microbiol Infect 18:54–60. doi: 10.1111/j.1469-0691.2011.03478.x. [DOI] [PubMed] [Google Scholar]
- 9.Anderson DJ, Engemann JJ, Harrell LJ, Carmeli Y, Reller LB, Kaye KS. 2006. Predictors of mortality in patients with bloodstream infection due to ceftazidime-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother 50:1715–1720. doi: 10.1128/AAC.50.5.1715-1720.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borer A, Saidel-Odes L, Riesenberg K, Eskira S, Peled N, Nativ R, Schlaeffer F, Sherf M. 2009. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect Control Hosp Epidemiol 30:972–976. doi: 10.1086/605922. [DOI] [PubMed] [Google Scholar]
- 11.Alotaibi FE, Bukhari EE, Al-Mohizea MM, Hafiz T, Essa EB, AlTokhais YI. 2017. Emergence of carbapenem-resistant Enterobacteriaceae isolated from patients in a university hospital in Saudi Arabia. Epidemiology, clinical profiles and outcomes. J Infect Public Health 10:667–673. doi: 10.1016/j.jiph.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tacconelli E, Magrini N, Kahlmeter G. 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery and development of new antibiotics. World Health Organization, Geneva, Switzerland: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf. [Google Scholar]
- 13.Yamamoto M, Pop-Vicas AE. 2014. Treatment for infections with carbapenem-resistant Enterobacteriaceae: what options do we still have? Crit Care 18:229. doi: 10.1186/cc13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold RS, Thom KA, Sharma S, Phillips M, Kristie Johnson J, Morgan DJ. 2011. Emergence of Klebsiella pneumoniae carbapenemase-producing bacteria. South Med J 104:40–45. doi: 10.1097/SMJ.0b013e3181fd7d5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Velkov T, Thompson PE, Nation RL, Li J. 2010. Structure-activity relationships of polymyxin antibiotics. J Med Chem 53:1898–1916. doi: 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velkov T, Roberts KD, Thompson PE, Li J. 2016. Polymyxins: a new hope in combating Gram-negative superbugs? Future Med Chem 8:1017–1025. doi: 10.4155/fmc-2016-0091. [DOI] [PubMed] [Google Scholar]
- 17.Zavascki AP, Nation RL. 2017. Nephrotoxicity of polymyxins: is there any difference between colistimethate and polymyxin B? Antimicrob Agents Chemother 61:e02319-16. doi: 10.1128/AAC.02319-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pogue JM, Lee J, Marchaim D, Yee V, Zhao JJ, Chopra T, Lephart P, Kaye KS. 2011. Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 53:879–884. doi: 10.1093/cid/cir611. [DOI] [PubMed] [Google Scholar]
- 19.Hartzell JD, Neff R, Ake J, Howard R, Olson S, Paolino K, Vishnepolsky M, Weintrob A, Wortmann G. 2009. Nephrotoxicity associated with intravenous colistin (colistimethate sodium) treatment at a tertiary care medical center. Clin Infect Dis 48:1724–1728. doi: 10.1086/599225. [DOI] [PubMed] [Google Scholar]
- 20.Brown P, Dawson MJ. 2017. Development of new polymyxin derivatives for multi-drug resistant Gram-negative infections. J Antibiot (Tokyo) 70:386–394. doi: 10.1038/ja.2016.146. [DOI] [PubMed] [Google Scholar]
- 21.Sherry N, Howden B. 2018. Emerging Gram negative resistance to last-line antimicrobial agents fosfomycin, colistin and ceftazidime-avibactam-epidemiology, laboratory detection and treatment implications. Expert Rev Anti Infect Ther 16:289–306. doi: 10.1080/14787210.2018.1453807. [DOI] [PubMed] [Google Scholar]
- 22.Macesic N, Nelson B, McConville TH, Giddins MJ, Green DA, Stump S, Gomez-Simmonds A, Annavajhala MK, Uhlemann AC. 2019. Emergence of polymyxin resistance in clinical Klebsiella pneumoniae through diverse genetic adaptations: a genomic, retrospective cohort study. Clin Infect Dis 70:2084–2091. doi: 10.1093/cid/ciz623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Cao Y, Yi L, Liu JH, Yang Q. 2019. Emergent polymyxin resistance: end of an era? Open Forum Infect Dis 6:ofz368. doi: 10.1093/ofid/ofz368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zusman O, Altunin S, Koppel F, Dishon Benattar Y, Gedik H, Paul M. 2017. Polymyxin monotherapy or in combination against carbapenem-resistant bacteria: systematic review and meta-analysis. J Antimicrob Chemother 72:29–39. doi: 10.1093/jac/dkw377. [DOI] [PubMed] [Google Scholar]
- 25.Diep JK, Jacobs DM, Sharma R, Covelli J, Bowers DR, Russo TA, Rao GG. 2017. Polymyxin B in combination with rifampin and meropenem against polymyxin B-resistant KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 61:e02121-16. doi: 10.1128/AAC.02121-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, Cornaglia G, Garau J, Gniadkowski M, Hayden MK, Kumarasamy K, Livermore DM, Maya JJ, Nordmann P, Patel JB, Paterson DL, Pitout J, Villegas MV, Wang H, Woodford N, Quinn JP. 2013. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis 13:785–796. doi: 10.1016/S1473-3099(13)70190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canton R, European Network on Carbapenemases, Akova M, Carmeli Y, Giske CG, Glupczynski Y, Gniadkowski M, Livermore DM, Miriagou V, Naas T, Rossolini GM, Samuelsen O, Seifert H, Woodford N, Nordmann P. 2012. Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin Microbiol Infect 18:413–431. doi: 10.1111/j.1469-0691.2012.03821.x. [DOI] [PubMed] [Google Scholar]
- 28.Herrmann G, Yang L, Wu H, Song Z, Wang H, Hoiby N, Ulrich M, Molin S, Riethmuller J, Doring G. 2010. Colistin-tobramycin combinations are superior to monotherapy concerning the killing of biofilm Pseudomonas aeruginosa. J Infect Dis 202:1585–1592. doi: 10.1086/656788. [DOI] [PubMed] [Google Scholar]
- 29.Bergen PJ, Smith NM, Bedard TB, Bulman ZP, Cha R, Tsuji BT. 2019. Rational combinations of polymyxins with other antibiotics, p 251–288. In Li J, Nation RL, Kaye KS (ed), Polymyxin antibiotics: from laboratory bench to bedside, vol 1145 Springer, Cham, Switzerland. [DOI] [PubMed] [Google Scholar]
- 30.Aydemir H, Akduman D, Piskin N, Comert F, Horuz E, Terzi A, Kokturk F, Ornek T, Celebi G. 2013. Colistin vs. the combination of colistin and rifampicin for the treatment of carbapenem-resistant Acinetobacter baumannii ventilator-associated pneumonia. Epidemiol Infect 141:1214–1222. doi: 10.1017/S095026881200194X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Batirel A, Balkan II, Karabay O, Agalar C, Akalin S, Alici O, Alp E, Altay FA, Altin N, Arslan F, Aslan T, Bekiroglu N, Cesur S, Celik AD, Dogan M, Durdu B, Duygu F, Engin A, Engin DO, Gonen I, Guclu E, Guven T, Hatipoglu CA, Hosoglu S, Karahocagil MK, Kilic AU, Ormen B, Ozdemir D, Ozer S, Oztoprak N, Sezak N, Turhan V, Turker N, Yilmaz H. 2014. Comparison of colistin-carbapenem, colistin-sulbactam, and colistin plus other antibacterial agents for the treatment of extremely drug-resistant Acinetobacter baumannii bloodstream infections. Eur J Clin Microbiol Infect Dis 33:1311–1322. doi: 10.1007/s10096-014-2070-6. [DOI] [PubMed] [Google Scholar]
- 32.Ly NS, Bulitta JB, Rao GG, Landersdorfer CB, Holden PN, Forrest A, Bergen PJ, Nation RL, Li J, Tsuji BT. 2015. Colistin and doripenem combinations against Pseudomonas aeruginosa: profiling the time course of synergistic killing and prevention of resistance. J Antimicrob Chemother 70:1434–1442. doi: 10.1093/jac/dku567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergen PJ, Tsuji BT, Bulitta JB, Forrest A, Jacob J, Sidjabat HE, Paterson DL, Nation RL, Li J. 2011. Synergistic killing of multidrug-resistant Pseudomonas aeruginosa at multiple inocula by colistin combined with doripenem in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 55:5685–5695. doi: 10.1128/AAC.05298-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nation RL, Rigatto MHP, Falci DR, Zavascki AP. 2019. Polymyxin acute kidney injury: dosing and other strategies to reduce toxicity. Antibiotics 8:24. doi: 10.3390/antibiotics8010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, Behle TF, Bordinhao RC, Wang J, Forrest A, Nation RL, Li J, Zavascki AP. 2013. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis 57:524–531. doi: 10.1093/cid/cit334. [DOI] [PubMed] [Google Scholar]
- 36.Loos U, Musch E, Jensen JC, Mikus G, Schwabe HK, Eichelbaum M. 1985. Pharmacokinetics of oral and intravenous rifampicin during chronic administration. Klin Wochenschr 63:1205–1211. doi: 10.1007/BF01733779. [DOI] [PubMed] [Google Scholar]
- 37.Lee HJ, Bergen PJ, Bulitta JB, Tsuji B, Forrest A, Nation RL, Li J. 2013. Synergistic activity of colistin and rifampin combination against multidrug-resistant Acinetobacter baumannii in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 57:3738–3745. doi: 10.1128/AAC.00703-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White BP, Lomaestro B, Pai MP. 2015. Optimizing the initial amikacin dosage in adults. Antimicrob Agents Chemother 59:7094–7096. doi: 10.1128/AAC.01032-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tangden T, Hickman RA, Forsberg P, Lagerback P, Giske CG, Cars O. 2014. Evaluation of double- and triple-antibiotic combinations for VIM- and NDM-producing Klebsiella pneumoniae by in vitro time-kill experiments. Antimicrob Agents Chemother 58:1757–1762. doi: 10.1128/AAC.00741-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jernigan MG, Press EG, Nguyen MH, Clancy CJ, Shields RK. 2012. The combination of doripenem and colistin is bactericidal and synergistic against colistin-resistant, carbapenemase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 56:3395–3398. doi: 10.1128/AAC.06364-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim TP, Cai Y, Hong Y, Chan EC, Suranthran S, Teo JQ, Lee WH, Tan TY, Hsu LY, Koh TH, Tan TT, Kwa AL. 2015. In vitro pharmacodynamics of various antibiotics in combination against extensively drug-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother 59:2515–2524. doi: 10.1128/AAC.03639-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deris ZZ, Yu HH, Davis K, Soon RL, Jacob J, Ku CK, Poudyal A, Bergen PJ, Tsuji BT, Bulitta JB, Forrest A, Paterson DL, Velkov T, Li J, Nation RL. 2012. The combination of colistin and doripenem is synergistic against Klebsiella pneumoniae at multiple inocula and suppresses colistin resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 56:5103–5112. doi: 10.1128/AAC.01064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tascini C, Ferranti S, Messina F, Menichetti F. 2000. In vitro and in vivo synergistic activity of colistin, rifampin, and amikacin against a multiresistant Pseudomonas aeruginosa isolate. Clin Microbiol Infect 6:690–691. doi: 10.1046/j.1469-0691.2000.00169.x. [DOI] [PubMed] [Google Scholar]
- 44.Gaibani P, Lombardo D, Lewis RE, Mercuri M, Bonora S, Landini MP, Ambretti S. 2014. In vitro activity and post-antibiotic effects of colistin in combination with other antimicrobials against colistin-resistant KPC-producing Klebsiella pneumoniae bloodstream isolates. J Antimicrob Chemother 69:1856–1865. doi: 10.1093/jac/dku065. [DOI] [PubMed] [Google Scholar]
- 45.Jacobs DM, Safir MC, Huang D, Minhaj F, Parker A, Rao GG. 2017. Triple combination antibiotic therapy for carbapenemase-producing Klebsiella pneumoniae: a systematic review. Ann Clin Microbiol Antimicrob 16:76. doi: 10.1186/s12941-017-0249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tzouvelekis LS, Markogiannakis A, Piperaki E, Souli M, Daikos GL. 2014. Treating infections caused by carbapenemase-producing Enterobacteriaceae. Clin Microbiol Infect 20:862–872. doi: 10.1111/1469-0691.12697. [DOI] [PubMed] [Google Scholar]
- 47.Bulitta JB, Li J, Poudyal A, Yu HH, Owen RJ, Tsuji BT, Nation RL, Forrest A. 2009. Quantifying synergy of colistin combinations against MDR Gram-negatives by mechanism-based models. Abstr 49th Intersci Conf Antimicrob Agents Chemother, abstr A1-571.
- 48.Yu Z, Qin W, Lin J, Fang S, Qiu J. 2015. Antibacterial mechanisms of polymyxin and bacterial resistance. Biomed Res Int 2015:679109–679111. doi: 10.1155/2015/679109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Velkov T, Roberts KD, Nation RL, Thompson PE, Li J. 2013. Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol 8:711–724. doi: 10.2217/fmb.13.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramirez MS, Tolmasky ME. 2017. Amikacin: uses, resistance, and prospects for inhibition. Molecules 22:2267. doi: 10.3390/molecules22122267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis BD. 1987. Mechanism of bactericidal action of aminoglycosides. Microbiol Rev 51:341–350. doi: 10.1128/MMBR.51.3.341-350.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yadav R, Bulitta JB, Schneider EK, Shin BS, Velkov T, Nation RL, Landersdorfer CB. 2017. Aminoglycoside concentrations required for synergy with carbapenems against Pseudomonas aeruginosa determined via mechanistic studies and modeling. Antimicrob Agents Chemother 61:e00722-17. doi: 10.1128/AAC.00722-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wehrli W. 1983. Rifampin: mechanisms of action and resistance. Rev Infect Dis 5(Suppl 3):S407–S411. doi: 10.1093/clinids/5.supplement_3.s407. [DOI] [PubMed] [Google Scholar]
- 55.Aye SM, Galani I, Han M, Karaiskos I, Creek DJ, Zhu Y, Lin Y-W, Velkov T, Giamarellou H, Li J. 2019. Lipid A profiling and metabolomics analysis of paired polymyxin-susceptible and -resistant multidrug-resistant Klebsiella pneumoniae clinical isolates, abstr 00512. Abstr 29th Eur Cong Clin Microbiol Infect Dis, Amsterdam, The Netherlands. [DOI] [PMC free article] [PubMed]
- 56.Schmid A, Wolfensberger A, Nemeth J, Schreiber PW, Sax H, Kuster SP. 2019. Monotherapy versus combination therapy for multidrug-resistant Gram-negative infections: systematic review and meta-analysis. Sci Rep 9:15290. doi: 10.1038/s41598-019-51711-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tamma PD, Cosgrove SE, Maragakis LL. 2012. Combination therapy for treatment of infections with Gram-negative bacteria. Clin Microbiol Rev 25:450–470. doi: 10.1128/CMR.05041-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Temocin F, Erdinc S, Tulek N, Demirelli M, Bulut C, Ertem G. 2015. Incidence and risk factors for colistin-associated nephrotoxicity. Jpn J Infect Dis 68:318–320. doi: 10.7883/yoken.JJID.2014.223. [DOI] [PubMed] [Google Scholar]
- 59.Tuon FF, Rigatto MH, Lopes CK, Kamei LK, Rocha JL, Zavascki AP. 2014. Risk factors for acute kidney injury in patients treated with polymyxin B or colistin methanesulfonate sodium. Int J Antimicrob Agents 43:349–352. doi: 10.1016/j.ijantimicag.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 60.Wargo KA, Edwards JD. 2014. Aminoglycoside-induced nephrotoxicity. J Pharm Pract 27:573–577. doi: 10.1177/0897190014546836. [DOI] [PubMed] [Google Scholar]
- 61.Kim JH, Nam WS, Kim SJ, Kwon OK, Seung EJ, Jo JJ, Shresha R, Lee TH, Jeon TW, Ki SH, Lee HS, Lee S. 2017. Mechanism investigation of rifampicin-induced liver injury using comparative toxicoproteomics in mice. Int J Mol Sci 18:1417. doi: 10.3390/ijms18071417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lakota EA, Landersdorfer CB, Nation RL, Li J, Kaye KS, Rao GG, Forrest A. 2018. Personalizing polymyxin B dosing using an adaptive feedback control algorithm. Antimicrob Agents Chemother 62:e00483-18. doi: 10.1128/AAC.00483-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith CR, Maxwell RR, Edwards CQ, Rogers JF, Lietman PS. 1978. Nephrotoxicity induced by gentamicin and amikacin. Johns Hopkins Med J 142:85–90. [PubMed] [Google Scholar]
- 64.Seshadr P, Climaco AB. 2014. Amikacin level. https://emedicine.medscape.com/article/2089686-overview.
- 65.Zarjou A, Agarwal A. 2011. Sepsis and acute kidney injury. J Am Soc Nephrol 22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 66.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. 2012. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 67.Zhu Y, Galani I, Karaiskos I, Lu J, Aye SM, Huang J, Yu HH, Velkov T, Giamarellou H, Li J. 2019. Multifaceted mechanisms of colistin resistance revealed by genomic analysis of multidrug-resistant Klebsiella pneumoniae isolates from individual patients before and after colistin treatment. J Infect 79:312–321. doi: 10.1016/j.jinf.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clinical and Laboratory Standards Institute (CLSI). 2019. Performance standards for antimicrobial susceptibility testing, 29th ed. CLSI supplement M100-S3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 69.European Committee on Antimicrobial Susceptibility Testing. 2020. Breakpoint tables for interpretation of MICs and zone diameters, version 10.0. https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_10.0_Breakpoint_Tables.pdf.
- 70.Gales AC, Jones RN, Sader HS. 2011. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY antimicrobial surveillance program (2006–09). J Antimicrob Chemother 66:2070–2074. doi: 10.1093/jac/dkr239. [DOI] [PubMed] [Google Scholar]
- 71.Kovacevic T, Avram S, Milakovic D, Spiric N, Kovacevic P. 2016. Therapeutic monitoring of amikacin and gentamicin in critically and noncritically ill patients. J Basic Clin Pharm 7:65–69. doi: 10.4103/0976-0105.183260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruslami R, Nijland HM, Alisjahbana B, Parwati I, van Crevel R, Aarnoutse RE. 2007. Pharmacokinetics and tolerability of a higher rifampin dose versus the standard dose in pulmonary tuberculosis patients. Antimicrob Agents Chemother 51:2546–2551. doi: 10.1128/AAC.01550-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dudhani RV, Turnidge JD, Coulthard K, Milne RW, Rayner CR, Li J, Nation RL. 2010. Elucidation of the pharmacokinetic/pharmacodynamic determinant of colistin activity against Pseudomonas aeruginosa in murine thigh and lung infection models. Antimicrob Agents Chemother 54:1117–1124. doi: 10.1128/AAC.01114-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Landersdorfer CB, Wang J, Wirth V, Chen K, Kaye KS, Tsuji BT, Li J, Nation RL. 2018. Pharmacokinetics/pharmacodynamics of systemically administered polymyxin B against Klebsiella pneumoniae in mouse thigh and lung infection models. J Antimicrob Chemother 73:462–468. doi: 10.1093/jac/dkx409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pillai SK, Moellering RC, Eliopoulos GM. 2005. Antimicrobial combinations, p 365–440. In Lorian V. (ed), Antibiotics in laboratory medicine, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.