

Several cationic amphiphilic drugs (CADs) have been found to inhibit cell entry of filoviruses and other enveloped viruses. Structurally unrelated CADs may have antiviral activity, yet the underlying common mechanism and structure-activity relationship are incompletely understood. We aimed to understand how widespread antiviral activity is among CADs and which structural and physico-chemical properties are linked to entry inhibition. We measured inhibition of Marburg virus pseudoparticle (MARVpp) cell entry by 45 heterogeneous and mostly FDA-approved CADs and cytotoxicity in EA.
KEYWORDS: Marburg virus, amiodarone, antiviral therapy, dronedarone, entry inhibitors, structure-activity relationships
ABSTRACT
Several cationic amphiphilic drugs (CADs) have been found to inhibit cell entry of filoviruses and other enveloped viruses. Structurally unrelated CADs may have antiviral activity, yet the underlying common mechanism and structure-activity relationship are incompletely understood. We aimed to understand how widespread antiviral activity is among CADs and which structural and physico-chemical properties are linked to entry inhibition. We measured inhibition of Marburg virus pseudoparticle (MARVpp) cell entry by 45 heterogeneous and mostly FDA-approved CADs and cytotoxicity in EA.hy926 cells. We analyzed correlation of antiviral activity with four chemical properties: pKa, hydrophobicity (octanol/water partitioning coefficient; ClogP), molecular weight, and distance between the basic group and hydrophobic ring structures. Additionally, we quantified drug-induced phospholipidosis (DIPL) of a CAD subset by flow cytometry. Structurally similar compounds (derivatives) and those with similar chemical properties but unrelated structures (analogues) to those of strong inhibitors were obtained by two in silico similarity search approaches and tested for antiviral activity. Overall, 11 out of 45 (24%) CADs inhibited MARVpp by 40% or more. The strongest antiviral compounds were dronedarone, triparanol, and quinacrine. Structure-activity relationship studies revealed highly significant correlations between antiviral activity, hydrophobicity (ClogP > 4), and DIPL. Moreover, pKa and intramolecular distance between hydrophobic and hydrophilic moieties correlated with antiviral activity but to a lesser extent. We also showed that in contrast to analogues, derivatives had antiviral activity similar to that of the seed compound dronedarone. Overall, one-quarter of CADs inhibit MARVpp entry in vitro, and antiviral activity of CADs mostly relies on their hydrophobicity yet is promoted by the individual structure.
INTRODUCTION
Outbreaks of emerging viral diseases have repeatedly posed major challenges to global health in recent years (https://www.who.int/csr/don/en/) (1). Important examples include the Filoviridae members Ebola virus (EBOV) and Marburg virus (MARV), the Arenaviridae member Lassa virus (LASV), Coronaviridae members like Middle East respiratory syndrome coronavirus (MERS-CoV), or the Flaviviridae member Zika virus (ZIKV). Some of these infections are associated with high case fatality rates, e.g., 50% for EBOV and MARV, 35% for MERS-CoV (2), and up to 30% for hospitalized LASV patients (3). Thus, according to the WHO list of Blueprint priority diseases of 2018, the aforementioned pathogens share priority status concerning the need for research and development, given that effective vaccines or directly acting antivirals are unavailable in most cases (4). It would be of great value to have drugs that are active against a broad range of viral pathogens (5, 6). Such agents would likely not target specific components of individual viruses but, rather, cellular structures or processes that are utilized by various unrelated viruses. An attractive target pathway concerns cellular endosomal processes since these are used by numerous pathogens, including members of the families Filoviridae, Arenaviridae, Rhabdoviridae, Coronaviridae, Togaviridae, Flaviviridae, and Bunyaviridae.
Cationic amphiphilic drugs (CADs) are a diverse group of compounds, many of which are in medical use for various clinical indications. CADs are defined by a hydrophilic group marked by a basic amine with high pKa (the negative logarithm of the acid dissociation constant Ka) and by hydrophobic group(s) characterized by several aromatic or aliphatic structures (7). Due to their amphiphilic nature, CADs transmigrate membranes, which can be enhanced by different substituents like halogen residues. Upon reaching acidic compartments like late endosomes (LE) and endolysosomes, the basic amine groups get protonated, which leads to lysosomal trapping of the compounds (8). Consequently, CADs primarily accumulate in acidified compartments (9, 10).
Several studies, most of which analyzed sets of approved drugs, have found that various CADs have antiviral activity. The antiarrhythmics amiodarone, dronedarone, and verapamil (11), as well as the estrogen receptor antagonists clomifene and toremifene (12), have been shown to inhibit filoviral entry in different cell lines. Moreover, clomifene and toremifene were shown to elevate survival rates of mice infected with mouse-adapted EBOV up to 90% (12). Of note, they could show that the antiviral activity of these two CADs was unrelated to their clinical mechanism of action as estrogen receptor modulators. Beyond filoviruses, CADs have also been found to inhibit the Arenaviridae members LASV and Guanarito virus (GTOV) (13), hepatitis C virus (HCV) (14–18), Japanese encephalitis virus (JEV) (19), severe acute respiratory syndrome-related coronavirus (SARS-CoV) (20), and Epstein-Barr virus (EBV) (21). Moreover, it was found that the CADs amiodarone, bepridil, raloxifene, and amodiaquine, in addition to several non-CAD compounds, have broad antiviral effects against all tested alpha- and flaviviruses (with the exception of Zika virus), as well as various other viruses, but none inhibited human immunodeficiency virus (HIV) (22). Highest antiviral activity for all four CADs was found against EBOV virus-like particles (VLPs). Despite these observations, it is not yet known how widespread antiviral activity really is among CADs. Similarly, the mechanism of action behind the antiviral activity is unclear. For this reason, we undertook a comprehensive study of antiviral activity among 45 heterogeneous CAD compounds and correlated antiviral activity with various physicochemical properties.
RESULTS
Side-by-side assessment of MARV GP-driven cell entry inhibition and cytotoxicity of 45 CADs.
We first attempted to evaluate how common antiviral activity is among CADs. Therefore, we selected an initial set of 45 CADs (Table 1) based on the following criteria: (i) coverage of multiple drug classes, (ii) diverse chemical structures aside from being CADs, (iii) preferentially FDA- or European Medicines Agency (EMA)-approved compounds/no known major toxicity, and (iv) commercial availability. We included CADs that have either previously been reported to have antiviral activity or are known to cause drug-induced phospholipidosis (DIPL).
TABLE 1.
Cationic amphiphilic drugs included in single-point assay and their properties
| Compound name | Drug class | pKa | ClogP | Mol wt (g/mol) |
|---|---|---|---|---|
| Fenfluramine HCl | Anorexic | 10.22 | 3.36 | 267.72 |
| Clemastine fumarate salt | Antiallergic | 9.55 | 5.2 | 459.96 |
| Perhexiline maleate salt | Antianginal | 10.58 | 6.2 | 393.56 |
| Bepridil HCl | Antianginal | 9.16 | 5.49 | 403 |
| Dronedarone HCl | Antiarrhythmic | 9.08 | 5.28 | 593.22 |
| Propranolol HCl | Antiarrhythmic | 9.67 | 3.48 | 295.8 |
| Verapamil HCl | Antiarrhythmic | 9.68 | 5.04 | 491.06 |
| Amiodarone HCl | Antiarrhythmic | 8.47 | 7.64 | 681.77 |
| Gentamicin sulfate | Antibiotic | 10.18 | −3.1 | 516.604 |
| Teicoplanin | Antibiotic | 7.1 | −3.71 | 1879.7 |
| Sertraline HCl | Antidepressant | 9.85 | 5.1 | 342.69 |
| N-Desethylamiodarone HCl solution | Antidepressant | 9.4 | 6.27 | 653.72 |
| Clomipramine HCl | Antidepressant | 9.2 | 5.19 | 351.31 |
| Fluoxetine HCl | Antidepressant | 9.8 | 4.09 | 345.79 |
| Imipramine HCl | Antidepressant | 9.2 | 4.8 | 316.87 |
| Maprotiline HCl | Antidepressant | 10.54 | 5.1 | 313.86 |
| Mianserin HCl | Antidepressant | 6.92 | 3.52 | 300.83 |
| Zimelidine dihydrochloride | Antidepressant | 8.62 | 3.39 | 390.15 |
| Cyclizine HCl | Antiemetic | 8.51 | 3 | 302.84 |
| Amorolfine HCl | Antifungal | 7.1 | 6.4 | 353.97 |
| Terconazole | Antifungal | 8.8 | 4.8 | 532.46 |
| Thioridazine HCl | Antifungal | 8.8 | 4.8 | 407.04 |
| Promethazine HCl | Antihistaminic | 9.05 | 4.81 | 320.88 |
| Tripelennamine HCl | Antihistaminic | 8.76 | 3.3 | 291.82 |
| Chlorcyclizine | Antihistaminic | 8 | 4.15 | 300.83 |
| Quinacrine dihydrochloride | Antiprotozoal | 10.33 | 5.5 | 472.88 |
| Chloroquine diphosphate salt | Antiprotozoal | 10.32 | 4.63 | 515.86 |
| Promazine HCl | Antipsychotic | 9.4 | 4.55 | 320.88 |
| Chlorpromazine HCl | Antipsychotic | 9.35 | 5.41 | 355.33 |
| Prochlorperazine dimaleate salt | Antipsychotic | 8.39 | 4.88 | 606.09 |
| Flupentixol dihydrochloride | Antipsychotic | 8.51 | 4.51 | 507.44 |
| Trifluoperazine HCl | Antipsychotic | 8.39 | 5.03 | 480.42 |
| Clozapine | Antipsychotic | 7.35 | 3.23 | 326.82 |
| Fluphenazine dihydrochloride | Antipsychotic | 8.21 | 4.36 | 510.44 |
| Amantadine HCl | Antiviral | 10.71 | 2.53 | 187.71 |
| Clomiphene citrate salt | Hormone analogue | 9.31 | 6.47 | 598.08 |
| Enclomiphene HCl | Hormone analogue | 9.31 | 6.47 | 442.42 |
| Ro 48-8071 | Lipid lowering | 8.8 | 5.7 | 448.4 |
| Triparanol | Lipid lowering | 9.6 | 6.7 | 438 |
| W-5 HCl | Nonclinical | 10.3 | 2.9 | 342.88 |
| U18666A | Nonclinical | 9.7 | 5.1 | 424.06 |
| Benzylamine | Nonclinical | 9.51 | 1.1 | 107.15 |
| W-7 | Nonclinical | 10.3 | 3.8 | 377.33 |
| Dasatinib | Antineoplastic | 8.49 | 1.8 | 488.01 |
| Toremifene citrate salt | Antineoplastic | 8.76 | 6.8 | 598.08 |
In order to test many drugs at the same time, a single-point, 96-well assay was established that enabled testing the entire set of compounds at the same micromolar concentrations against a high-titer preparation of lentiviral particles pseudotyped with the MARV GP (MARV pseudoparticles, MARVpp) in endothelium-derived EA.hy926 cells. Beforehand, we conducted concentration-response and -toxicity curves of the prototypical, antivirally active CAD amiodarone in EA.hy926 cells against MARVpp. The half-maximal inhibitory (IC50) and cytotoxic (CC50) concentrations were 1.6 μM and 54 μM, respectively (see Fig. S1 in the supplemental material). Consequently, we chose a test concentration of 5 μM for all CADs, i.e., 2.8-fold above the IC50 and almost 10-fold below the CC50 of amiodarone.
The ability to inhibit MARVpp entry in EA.hy926 cells differed widely within the set of CADs tested (Fig. 1a). Approximately 24% of CADs showed antiviral activity (>40% inhibition), with strongest effects by dronedarone and triparanol (>80% inhibition), followed by >60% inhibition by quinacrine, sertraline, amiodarone, perhexiline, clomifene and N-desethylamiodarone. Notably, cytotoxicity measurement in EA.hy926-NLuc (EA.hy cells expressing the nano-luciferase gene) cells revealed that most CADs do not have major toxic effects on these cells. However, treatment with dasatinib, quinacrine, and sertraline showed reductions in viability of 42%, 22%, and 19%, respectively (Fig. 1b). Our results demonstrate that antiviral activity is not a common feature of all CADs and independent of the drug class.
FIG 1.

Antiviral activity against MARVpp GP-driven cell entry and cytotoxicity of 45 CADs. (a) EA.hy926 target cells were transduced with MARVpp expressing an NLuc reporter gene in a single-point approach in the presence of various CADs at 5 μM. Results were normalized to the levels for the solvent control. (b) Cytotoxic and antiproliferative effects were evaluated for CADs at 5 μM in EA.hy926 cells stably expressing NLuc. Box-whisker plots show the median, interquartile range, and standard deviation of 3 independent experiments. Asterisks indicate statistical significance of differences between results for the compounds and those for the solvent control that was calculated by one-way ANOVA with a correction for multiple comparisons (*, P < 0.01; **, P < 0.001; ***, P < 0.0001). RLU, relative light units.
Determination of antiviral potency and safety of top and bottom CADs.
Next, we generated concentration-response curves for the six CADs that showed the highest antiviral activity (dronedarone, triparanol, sertraline, amiodarone, perhexiline, and clomifene) (Fig. S1) and for the six least antivirally active CADs (propranolol, tripelennamin, amantadine, promazine, zimelidine, and benzylamine) (Fig. S2). Quinacrine and dasatinib were excluded from further analyses due to their high cytotoxicity (Fig. 1). Similarly, teicoplanin was excluded because its high molecular size and structural complexity would hamper the structure-function analysis. The determined IC50 and CC50 values as well as the selective indices (SIs) are summarized in Table 2. We observed that strong antiviral CADs act in micromolar concentrations, with IC50 values between 1.3 μM (dronedarone) and 2.9 μM (sertraline) and CC50 values between 13.2 μM (dronedarone) and 54 μM (amiodarone). In contrast, weakly active CADs in most cases did not allow for IC50 and CC50 calculation except for propranolol (IC50 of 64.6 μM) and promazine (IC50 of 29.3 μM and CC50 of 51.6 μM). Notably, we observed relatively narrow nontoxic activity windows, with the broadest one for amiodarone (SI of 34), followed by clomifene (SI of 17), triparanol (SI of 13), and dronedarone (SI of 10).
TABLE 2.
Antiviral activity and toxicity as well as selective indices of top and bottom antiviral CADsa
| Compound group and name | IC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|
| Most active antiviral CADs | |||
| Dronedarone | 1.3 | 13.2 | 10 |
| Triparanol | 2.0 | 25.1 | 13 |
| Sertraline | 2.9 | 18.7 | 6 |
| Amiodarone | 1.6 | 54.0 | 34 |
| Perhexiline | 2.4 | 13.8 | 6 |
| Clomifene | 2.7 | 44.8 | 17 |
| Least active antiviral CAD | |||
| Promazine | 29.3 | 51.6 | 2 |
Activity and toxicity indicated as IC50 and CC50 values, respectively.
Anti-MARVpp activity of CADs correlates with specific physicochemical properties and DIPL induction.
Next, we addressed the question of whether strong antiviral activity of CADs was associated with physicochemical properties. Specifically, we correlated the CADs’ antiviral activities with their acidity (pKa), hydrophobicity (ClogP), molecular weight (MW), and the distance between hydrophilic amine and hydrophobic ring structure of the molecules (linker length) (Fig. 2a to d). The parameters pKa, ClogP, and MW were obtained mainly from databases, whereas the linker length was calculated according to the three-dimensional (3D) structure of the molecules. Although we could not observe any correlation between antiviral activity and linker length (Fig. 2d), we noticed a modest yet nonsignificant (R = −0.24; P = 0.11) association of antiviral activity with pKa, showing that more-alkaline CADs have higher antiviral activity (Fig. 2a). We observed a similar correlation with molecular weight (R = −0.33; P = 0.02) (Fig. 2c). Most strikingly, we found a highly significant correlation between antiviral activity and hydrophobicity (R = −0.78; P < 0.0001) (Fig. 2b). Although the majority of CADs are rather hydrophobic (1 < ClogP < 8), all strong antiviral CADs had an octanol/water partitioning coefficient larger than 4 (ClogP > 4). Taken together, CADs with ClogP values of >4 and pKa values of >8 are more likely to be antiviral.
FIG 2.

Correlation of MARVpp antiviral activity of 45 CADs and their physicochemical properties. Residual transduction of 45 CADs against MARVpp GP-driven cell entry of single-point assays was normalized, and the average of 3 independent experiments was correlated with pKa (a), ClogP (b), molecular weight (c), and linker length (d) of all tested CADs. The prototypical CAD amiodarone is labeled. Nonparametric Spearman correlation was computed, and the correlation coefficient R and P value represent statistical significance (ns, not significant [P > 0.12]; *, P < 0.03; **, P < 0.002; ***, P < 0.0002; ****, P < 0.0001).
Next, we analyzed to what extent antiviral activity is associated with induction of DIPL (Fig. 3). Therefore, we treated EA.hy926 cells with 16 of the CADs at 5 μM or with their solvents, both in the presence of LipidTox. LipidTox is a fluorophore-conjugated phospholipid which permeates cellular membranes but is believed to be complexed in endosomes upon DIPL induction. The selected CADs were chosen for their various degrees of antiviral activity against MARVpp, and the group was comprised of 10 CADs that were more or less active and 6 inactive CADs; thus, we aimed to represent the full spectrum of antiviral activity seen with CADs. We detected green fluorescence as a measure for lysosomal accumulation of LipidTox, representing CAD-induced DIPL. Here, we could show not only a highly significant correlation of MARVpp antiviral activity with the degree of DIPL induction (R = −0.81; P = 0.0001) (Fig. 3a) but also significant association of DIPL induction and hydrophobicity of these compounds (R = 0.73; P = 0.0011) (Fig. 3b). These findings underline the importance of hydrophobicity, mediating both antiviral activity and DIPL of CADs. Furthermore, they hint that DIPL and antiviral activity might be more closely related than previously suggested.
FIG 3.

Correlation of MARVpp antiviral activity, DIPL induction, and hydrophobicity. (a) EA.hy926 cells were treated in triplicate with 16 different CADs at a 5 μM concentration and the fluorescent phospholipid LipidTox green (1:1). After 6 h of incubation, medium was exchanged, and compounds (without LipidTox) were added again for an extra 18 h. Fluorescence measured by FACS represents DIPL induction by CADs. The change in mean fluorescence intensity (ΔMFI) was calculated by first subtracting the mean value of the solvent control from each measured value. Averages of 3 independent stainings were calculated and plotted against average antiviral activity. (b) DIPL induction was correlated with ClogP of the investigated 16 CADs. Correlation coefficients (R values) were determined via Spearman correlation (ns, not significant [P > 0.12]; *, P < 0.03; **, P < 0.002; ***, P < 0.0002; ****, P < 0.0001).
Selection of additional CADs based on our previous findings and validation of their antiviral activity and cytotoxicity.
We observed that strong antiviral activity in CADs is associated with certain physicochemical features, yet antiviral CADs can vary widely in their scaffold structures. In order to gain an insight into how far the chemical structure and/or chemical properties influence antiviral activity, we validated additional CADs that were identified by two distinct approaches.
First, we tested whether compounds with comparable molecular structures (derivatives) would have similar or even stronger antiviral effects than antiviral CADs. Dronedarone derivatives (N-mesyldronedarone [D-1] and S-desmethyl S-chloromethyl dronedarone [D-2]) were obtained by a similarity search using SciFinder, a curated chemistry information database (23). We thus analyzed their antiviral activity against MARVpp GP-driven cell entry as well as their cytotoxicity in comparison to levels for dronedarone and solvent (Fig. 4a and b). We found 94% and 91% inhibition for 5 μM D-1 and D-2, respectively. This is comparable to the antiviral effect seen with dronedarone itself and suggests that the CAD structure may be important for antiviral activity. Furthermore, we could show that these two compounds induce DIPL at levels similar to those of dronedarone (Fig. S4). In line with our previous observations, D-1 and D-2 have basic pKa values and are highly hydrophobic, with ClogP values of 8.6 and 9.2, respectively.
FIG 4.
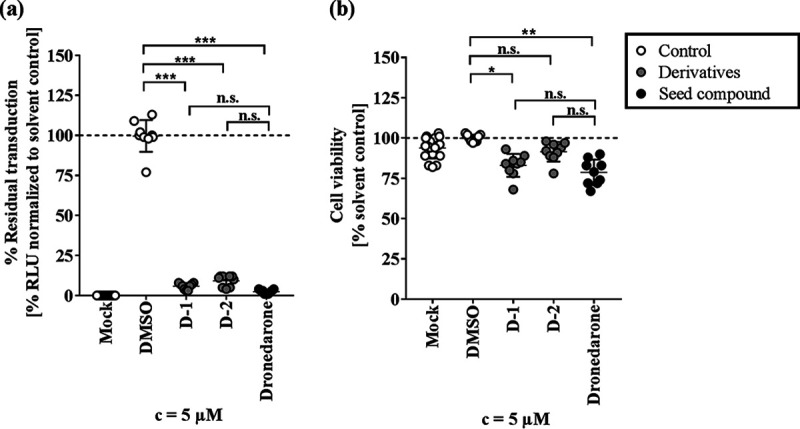
Antiviral activity and cytotoxicity of dronedarone structural derivatives. Dronedarone derivatives D-1 and D-2 were found by a SciFinder similarity search and applied in a 5 μM concentration together with MARVpp on EA.hy926 cells (a) or on EA.hy-NLuc cells (b) for 6 h. Luciferase activity was determined at 72 hpt, and triplicate values of 3 independent experiments were normalized to those of the solvent control. Statistical difference was calculated by one-way ANOVA corrected for multiple comparisons and is indicated by asterisks (*, P < 0.01; **, P < 0.001; ***, P < 0.0001; ns, not significant). c, concentration.
As a second approach, we looked for chemically similar, but structurally diverse, CADs (analogues) by chemically advanced template search (CATS) analysis (24). CATS analysis largely disregards structural similarities and considers only similar chemical properties of compounds. As query substances, we submitted the seven most potent CADs: amiodarone, dronedarone, triparanol, sertraline, quinacrine, perhexiline, and clomifene. We found 33 new compounds that were identified as double hits for showing functional similarity to two unrelated seed CADs. As we were aiming for putative antiviral candidates, and in accordance with the observed importance of hydrophobicity for antiviral effective CADs, we further narrowed the number of hits down to compounds with ClogP values of >4. By further selecting for diverse functional groups, we obtained a set of eight compounds and named them according to the two seed compounds they derive from (CT1, clomifene and triparanol; CS1 to CS4, clomifene and sertraline; AC1, amiodarone and clomifene; QT1, quinacrine and triparanol; DQ1, dronedarone and quinacrine). We tested their antiviral activity against MARVpp as well as their cytotoxicity (Fig. 5a and b) and found that the compounds CT1, CS3, AC1, CS1, CS2, and QT1 decreased MARVpp entry (inhibition ranged from 17% to 45%). Still, the newly identified compounds did not reach the level of inhibition seen with their respective seed compounds.
FIG 5.

Inhibition of MARVpp GP-mediated cell entry and cytotoxicity of additional CADs identified by CATS analysis. New CADs were identified in a CATS screen for compounds with chemical properties similar to those of seed compounds. Eight double hits were chosen which fitted the requirements of being a CAD, having diverse functional groups, and having a ClogP value of >4. In order to simplify compound nomenclature, new CADs were abbreviated according to the initials of their two seed compounds. (a) For analysis of viral entry inhibition, 5 μM CADs were applied together with MARVpp on EA.hy926 cells, and luciferase activity was measured in transduced cells at 72 hpt. (b) Cytotoxicity of new CADs, seed compounds, and solvent controls was measured in EA.hy-NLuc cells treated with 5 μM CADs for 6 h. Normalized results of 3 experiments with three technical replicates are shown, and asterisks indicate statistical significance relative to results with the solvent control calculated by one-way ANOVA including a multiple-comparison test (*, P < 0.01; **, P < 0.001; ***, P < 0.0001).
DISCUSSION
This study is to date the most comprehensive evaluation of both the distribution of antiviral activity among different types of CADs and their antivirally relevant physicochemical properties. It reveals the following (i) that antiviral activity is not universal among CADs but is limited to a minority; (ii) that antiviral CADs come from a broad range of pharmacological classes and have diverse molecular structures; (iii) that certain physicochemical features are associated with antiviral activity, most notably hydrophobicity; (iv) that there is a strong association between antiviral activity, the ability to induce cellular phospholipidosis, and hydrophobicity.
Previous studies showed that amiodarone, as well as its close relative dronedarone, acts in low-micromolar concentrations against pseudoparticles of different filovirus species, GTOV, and influenza H5N1 virus. Furthermore, amiodarone was shown to inhibit authentic EBOV, HCV, SARS, and recently Semliki forest virus, dengue virus, Sindbis virus, Ross river virus, herpes simplex virus, and the attenuated vaccine strain of yellow fever virus. Conversely, ZIKV, HIV, or vaccinia virus was not affected (14, 15, 20, 22, 25–27). Triparanol was found to inhibit EBOV VLPs as well as EBOV infection and HCV replication (17, 28). Among a variety of other CADs, quinacrine, sertraline, and clomifene inhibited mainly EBOV VLPs, mouse-adapted EBOV, and other filovirus strains, including MARV (12, 29). To our knowledge, perhexiline has not been investigated in the context of antivirals yet and was included in our study due to reported association with DIPL (8). Furthermore, the amiodarone metabolite mono-N-desethylamiodarone (MDEA) was shown to have additive effects with amiodarone (26). With our results, we partially confirmed and extended the previous studies. Notably, we could show that the drugs dronedarone and triparanol, followed by quinacrine, sertraline, amiodarone, perhexiline, clomifene, and N-desethylamiodarone, have higher antiviral potential within the broad field of described CADs. Furthermore, we show that candidates previously not associated with antiviral effects but with only DIPL (e.g., perhexiline) may also be potent antivirals or templates for further drug development. Experiments were performed in the endothelium/lung hybrid cell line EA.hy926 that is easy to cultivate and gives low background. In addition to monocytes, macrophages, endothelial cells, hepatocytes, and fibroblasts, this cell line was shown to be susceptible to several enveloped viruses including filoviruses (11, 13, 30, 31).
Concentration-response analyses showed that the six strongest CADs act at low-micromolar concentrations. The IC50 values for amiodarone and sertraline are comparable to the reported maximum concentration (Cmax) values in plasma (Cmax(amiodarone) = 1.5 to 2.5 mg/liter [2.2 to 3.7 μmol/liter] and Cmax(sertraline) = 0.352 μmol/liter) which can accumulate up to 20-fold in the liver (32–34). In the case of dronedarone, the determined IC50 value is 10-fold higher than the plasma concentration achieved in treatment of arrhythmia (Cmax(dronedarone) = 84 – 147 μg/liter [0.14 to 0.25 μmol/liter]) (35). The selectivity index values of these CADs are relatively narrow in vitro. However, it may be worth probing whether a therapeutic window exists in vivo, which might be acceptable when dealing with a severe or even life-threating disease or in an outbreak setting triggered by emerging enveloped viruses, given that they act against a range of viruses.
In a previous study, Shoemaker et al. identified six highly antiviral CADs among various sterol pathway inhibitors. These CADs were characterized with a positively charged amine group, alkaline pKa (>8.8), small MW, and high hydrophobicity, and they caused cholesterol accumulation (28). However, it is not known whether the previous observations apply necessarily to all antivirally active CADs and which set of structural features define antiviral activity. To understand the relationship between CAD structure and antiviral activity, we initially focused on a limited number of properties, including pKa, ClogP, MW, and linker length. Hydrophobicity is an important determinant of membrane permeability and bioavailability of a molecule and has been described as one of the key factors for ligand binding, together with molar refractivity and formal charge density (8, 36, 37). We could show that strongly hydrophobic compounds have significantly stronger capacity to inhibit viral entry. The reason for this observation is so far unclear, but it may suggest that CADs need to be able to traverse membranes in order to exert their antiviral effects. Apart from the CAD-determining amine and hydrophobic groups, we were not able to identify a set of functional groups that are required for antiviral activity.
We further analyzed CADs with either structural (derivatives) or functional (analogues) similarity to strong antiviral CADs of our screen (24, 38). The fact that only the derivatives, but none of the analogues, reached the level of inhibition seen with their respective seed compounds suggests that structural architecture seemed to have more impact on the CAD activity than chemical properties only. Nevertheless, the analogues’ activity rate is far higher than the rate obtained by screening random collections. However, these data show that hydrophobicity alone is not a certain predictor for antiviral activity but, rather, a combination of chemical properties is required, which is also suggested for DIPL induction (39, 40). An extended multiparametric analysis would be useful to identify a larger set of (interdependent) parameters that enable and enhance activity (24).
In addition, our studies showed a strong association between CAD antiviral activity and the ability to induce DIPL in target cells, i.e., lysosomal accumulation of charged phospholipids, an effect that has been reported for many CADs (8, 41). DIPL shows morphological resemblance of Niemann-Pick type C disease (NPC-1), an inherited and generally fatal lipid storage disorder, and similar diseases (8, 42). Amiodarone preferentially accumulates in the lung, causes DIPL, and is associated with lung dysfunction (43). However, the CADs in clinical use have not been linked to Niemann-Pick-like symptoms, and a clear general link between cellular DIPL and organ toxicity has not been demonstrated. Moreover, tissue accumulation seems to be reversible within a few days (44). Nevertheless, DIPL is considered a concern in drug development. In our study, we found that DIPL strongly correlates with both antiviral activity and with hydrophobicity. However, visible DIPL itself is not required for antiviral activity of CADs since effects like EBOV VLP inhibition (28) or lysosomal calcium flux changes (45) precede DIPL induction for hours. In conclusion, DIPL induction seems to be more prominent among CADs with antiviral activity. In fact, in our series, all CADs with antiviral activity also induce DIPL to some extent. Yet the ability to induce DIPL is not the only determinant of antiviral activity as some CADs with similar DIPL values have quite different degrees of antiviral activity.
Filoviruses are late-penetrating viruses that are taken up into endosomes following low-specificity interactions with one or more of various cell surface attachment factors (30). Within the endosome, they undergo GP priming by low-pH-dependent cathepsin and interact with their endolysosomal receptor NPC-1, a cholesterol transporter (46). This enables membrane fusion, thereby releasing the nucleocapsid into the cytosol (47, 48). It is conceivable that CADs modulate endosomal or endolysosomal membranes or membrane-attached proteins in a manner that perturbs viral penetration. Direct evidence or a clear concept of how this might occur is as yet lacking. So far, it has not even been fully established whether CADs exert their antiviral effects on the host cell or on the viral particle. It has been reported for a group of structurally diverse EBOV-inhibiting CADs—including toremifen, bepridil, and sertraline that were tested in our study—that these compounds decrease the overall stability of the GP1-GP2 dimers, thus inhibiting fusion (49, 50). Another recent study reported that the CAD flunarizine as well as the structurally similar fluphenazine, trifluoperazine, chlorcyclizine, and chlorpromazine seems to inhibit membrane fusion of HCV particles by targeting a specific hydrophobic region in the E1 protein (16, 51). Moreover, the antiviral spectrum of amiodarone, bepridil, amodiaquine, and raloxifene together with lipid mixing assays suggests that mostly late-penetrating viruses and a late entry step, most probably membrane fusion, are affected by these CADs (22).
However, given that we and others have described a range of structurally diverse CADs as inhibitors of cell entry by different unrelated virus species, it seems likely that in addition to these virus-specific effects, there is a mechanism shared by diverse CADs affecting a range of enveloped viruses. The correlation of antiviral effects and early events of DIPL points toward a cell-based effect of CADs that takes place in the endolysosomal compartment. Thus, we consider that the perturbation of late endosomal homeostasis might be the underlying antiviral mechanism of CADs. The clearer elucidation of the antiviral mechanism of some CADs is part of an ongoing follow-up study.
In summary, we describe the most comprehensive evaluation to date of the antiviral properties among CADs and show how antiviral activity is linked to structural features. Most notably, we show that antiviral activity is strongly associated with hydrophobicity and DIPL induction in target cells, corroborating previous studies (28). Overall, our findings help clarify the structure-activity relationship of antiviral CADs. Furthermore, using an in silico screening approach for identification of structurally and functionally related compounds, we identified novel CADs with antiviral properties. By showing that CAD antiviral activity is partially fingerprinted on the CAD molecular architecture, we set a basis for the structure-based design of potent antiviral CADs.
MATERIALS AND METHODS
Pseudoparticle production and transduction.
Pseudoparticles were produced as described before (52, 53). In brief, producer HEK-293T cells were seeded at 8 × 105 cells/well in a poly-l-lysine coated six-well plate. After 24 h cells were cotransfected with 1 μg of DNA using polyethyleneimine (PEI) to produce HIV-based pseudoparticles bearing individual viral envelope GPs. The plasmids used were (i) a packaging plasmid containing HIV-based gag-pol genes (54), (ii) a plasmid encoding the env gene of MARV (pCAGGS-MARVGP) (55) or an empty vector pcDNA3.1, and (iii) a gutted, yet packaging-competent, HIV-based transfer plasmid encoding an NLuc reporter gene (pWPI_NanoLuc_BLR) (in a ratio of 1:1:4). At 6 h posttransfection, medium was exchanged for 3% fetal calf serum (FCS)-supplemented Dulbecco’s modified Eagle’s medium (DMEM) containing 5% penicillin-streptomycin, and pseudoparticles were harvested, pooled, and filtered after 48 h and 72 h. For lentiviral transduction, target EA.hy926 cells were seeded at 1 × 104 cells/well in a 96-well plate. After incubation for 24 h, 50 μl of MARVpp or 50 μl of complete DMEM was mixed with Polybrene (1:1,000) and CADs to a final concentration of 5 μM and applied in triplicates. As a solvent control, sterile water, ethanol (EtOH), or dimethyl sulfoxide (DMSO) was diluted in complete DMEM in the same ratio as the drugs. At 6 h postransduction (hpt), cells were washed once with phosphate-buffered saline (PBS), and 100 μl of complete DMEM was applied. At 72 hpt, cells were washed with PBS and lysed. Next, 20 μl of cell lysate was transferred to a 96-well luminometer plate, mixed with 80 μl of the luciferase substrate coelenterazine, and incubated briefly in the dark before NLuc activity quantification in a GloMax plate luminometer (Promega, Madison, WI, USA), representing GP-driven cell entry.
Cytotoxicity test.
EA.hy926-NLuc cells were seeded at 1 × 104 cells/well in a 96-well plate. The next day, cells were treated in triplicate with either complete DMEM, solvent control (complete DMEM plus solvent), or diluted CADs at a final concentration of 5 μM. After 6 h, cells were washed with PBS and incubated with 100 μl of complete DMEM for another 72 h at 37°C. Then cells were lysed, and luciferase activity was determined as an aggregate measure of cell viability and proliferation.
Compounds and similarity search.
The compounds amiodarone HCl (catalog no. A8423), amorolfine HCl (SML0283), bepridil HCl (B5016), clemastine fumarate salt (SML0445), clomiphene citrate salt (C6272), dasatinib (CDS023389), dronedarone HCl (D9696), perhexiline maleate salt (SML0120), promazine HCl (P6656), sertraline HCl (S6319), terconazole (32355), toremifene citrate salt (T7204), triparanol (T5200), chlorcyclizine (SML1473), clozapine (C6305), cyclizine HCl (C3090000), N-desethylamiodarone HCl solution (D-055), verapamil HCl (V4629), W-7 [N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride] (A3281), amantadine HCl (A1260), benzylamine (185701), chloroquine diphosphate salt (C6628), clomipramine HCl (C7291), chlorpromazine HCl (C0982), enclomiphene HCl (SML0719), fenfluramine HCl (F112), flupentixol dihydrochloride (Y0000064), fluphenazine dihydrochloride (F4765), gentamicin sulfate (G1914), imipramine HCl (I0899), maprotiline HCl (M9651), mianserin HCl (M2525), prochlorperazine dimaleate salt (P9178), promethazine HCl (P4651), propranolol HCl (P0884), quinacrine dihydrochloride (Q3251), teicoplanin (T0578), thioridazine HCl (T9025), trifluoperazine HCl (T6062), tripelennamine HCl (T7511), U18666A (U3633), W-5 [N-(6-aminohexyl)-1-naphthalenesulfonamide hydrochloride] (SML0657), and zimelidine dihydrochloride (Z101) were purchased from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany). The compound Ro 48-8071 was purchased from Biomol GmbH (Hamburg, Germany).
Dronedarone derivatives were confirmed using SciFinder software (23). D-1 (N-mesyldronedarone; TRC-M225785) and D-2 (S-desmethyl S-chloromethyl dronedarone; TRC-D291470) were purchased from Biozol Diagnostica Vertrieb GmbH (Eching, Germany). To obtain analogous CADs, chemical advanced template search (CATS) software was used (24). The compounds AC1 (1-(4-ethyl-1-piperazinyl)-4-[1-(4-methylbenzyl)-1H-indol-3-yl]-1-butanone; E230-0651), CS3 (2-[(E)-2-{4-[ethyl(4-methoxyphenyl)amino]phenyl}vinyl]-3-methyl-1.3-benzothiazol-3-ium; 7165-0012), and QT1 (G119-1593; G119-1593) were obtained by ChemDiv (San Diego, CA, USA). CS1 (2-{2-[(3.4-dichlorophenyl)thio]ethyl}-1-(2-oxo-2-phenylethyl)pyridinium bromide; 5841320) and CS2 (1-{2-[4-(1-methyl-1-phenylethyl)phenoxy]ethyl}pyrrolidine; 7020606) were ordered from ChemBridge Corporation (San Diego, CA, USA). DQ1 (2-(4-(sec-butyl)phenyl)-N′(1-phenethylpiperidin-4-ylidine)quinolone-4-carbohydrazide; IVK 9315735) was purchased from SRC Alinda (Moscow, Russia), and CS4 (5-bromo-2-{[1-(3-fluorophenyl)ethyl]sulfanyl}pyridine; PB742999078) was from Chemspace (Riga, Latvia). CT1 ((E)-1-(5-chlorothiophen-2-yl)-3-(4-(diphenylamino)phenyl)prop-2-en-1-one; Z46049784) was purchased from Enamine (Monmouth Junction, NJ, USA). As solvents, water, ethanol (EtOH) (Carl Roth, Karlsruhe, Germany), or dimethyl sulfoxide (DMSO) (Carl Roth, Karlsruhe, Germany) was used.
Cell culture.
Immortalized epithelial HEK-293T and EA.hy926 cells, a fusion product of A549 and human umbilical vein endothelial cells, were cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal calf serum (FCS), 5% penicillin-streptomycin, 5% nonessential amino acids (NEAA), and 5% l-glutamine (complete DMEM) at 37°C with 5% CO2. Luciferase-expressing EA.hy926-NLuc cells were obtained by transduction with vesicular stomatitis virus glycoprotein pseudoparticles (VSV-Gpp) harboring the nano luciferase (NLuc) gene and cultured as previously described.
Phospholipidosis assay.
EA.hy926 cells were seeded at 1 × 104 cells/well in a 96-well plate. The next day, CADs and their solvents (DMSO and water) were diluted in culture medium. Phospholipidosis was quantified using LipidTOX green phospholipidosis detection reagent (H34350; Thermo Fisher Scientific, Waltham, MA, USA) according to a modified version of a degradation assay (56). In brief, LipidTOX was diluted 1:500 in complete DMEM and sterile filtered. Then 50 μl of either CAD mixture or controls was applied to the cells together with 50 μl of diluted LipidTox, resulting in a final CAD concentration of 5 μM and a final LipidTox dilution of 1:1,000. After incubation for 6 h at 37°C, cells were washed once with PBS. Medium was exchanged for 100 μl of culture medium, and only the drugs (5 μM) but no LipidTOX were added a second time. After 24 h in total, cells were washed with PBS, trypsinized, and transferred to fluorescence-activated cell sorting (FACS) tubes. Cells were washed with 1 ml of FACS buffer (2% FCS in PBS) and centrifuged for 5 min at 1,000 rpm, and the supernatant was discarded. In some replicates, cells were fixed with 100 μl of 3 to 6% paraformaldehyde (PFA) with no influence on the results. Finally, CAD-induced lysosomal accumulation of LipidTOX was quantified as intracellular green fluorescence by flow cytometry in a BD FACSCanto II instrument (BD, Franklin Lakes, NJ, USA) or for derivates in Spectral Cell Analyzer SA3800 (Sony Biotechnology, San Jose, CA, USA) and analyzed with FlowJo, versions 9 and 10.
Chemical properties/parameters.
The chemical parameters of CADs, including molecular weight, pKa, and octanol/water partitioning coefficient (ClogP), were obtained from either the respective data sheets or the databases PubChem (57), DrugBank (58), and ChemDraw Professional 18 (PerkinElmer, Hamburg, Germany). The parameter “linker length” was defined as the distance between the amine group and the first hydrophobic ring structure. We determined linker length by compiling the 3D structure of each CAD in Chem3D Pro, version 18 (PerkinElmer, Hamburg, Germany), performed MM2 energy minimization, and then used coordinates x, y, and z of the amine and hydrophobic groups to calculate the distance.
Statistical analysis.
Unless stated otherwise, experiments were conducted three times in triplicate. Statistical analysis was performed in GraphPad Prism, version 7 (La Jolla, CA, USA). The statistical significance between multiple groups and the control groups was evaluated by ordinary one-way analysis of variance (ANOVA), followed by a multiple-comparison test (Dunnett), and for correlation analyses a nonparametric Spearman correlation was run. Concentration-response curves were fitted by nonlinear regression ([inhibitor] versus response-variable slope [four parameters]), and IC50 and CC50 values were interpolated from the resulting standard curve.
Supplementary Material
ACKNOWLEDGMENTS
We thank Petra Schneider (ETH, Pharmaceutical Sciences, Zürich, Switzerland) for help with CATS software, Barbara Hertel (University of Potsdam, Department for Food Chemistry, Potsdam, Germany) for excellent experimental support, and Beate Sodeik (MHH, Institute of Virology, Hannover, Germany) for helpful discussions.
This work was supported by Deutsches Zentrum für Infektionsforschung (DZIF; German Center for Infection Research; grant number 05807) and the German Research Foundation (DFG) via grant SFB738 (Research Project B2). A.P.G. is a Ph.D. student of The Center for Infection Biology (ZIB) and Hannover Biomedical Research School (HBRS).
We have no conflicts of interest to report.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.WHO. 2019. Disease outbreak news. https://www.who.int/csr/don/en/. Accessed 2 September 2019.
- 2.Coltart CEM, Lindsey B, Ghinai I, Johnson AM, Heymann DL. 2017. The Ebola outbreak, 2013–2016: old lessons for new epidemics. Philos Trans R Soc Lond B Biol Sci 372:20160297. doi: 10.1098/rstb.2016.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaffer JG, Grant DS, Schieffelin JS, Boisen ML, Goba A, Hartnett JN, Levy DC, Yenni RE, Moses LM, Fullah M, Momoh M, Fonnie M, Fonnie R, Kanneh L, Koroma VJ, Kargbo K, Ottomassathien D, Muncy IJ, Jones AB, Illick MM, Kulakosky PC, Haislip AM, Bishop CM, Elliot DH, Brown BL, Zhu H, Hastie KM, Andersen KG, Gire SK, Tabrizi S, Tariyal R, Stremlau M, Matschiner A, Sampey DB, Spence JS, Cross RW, Geisbert JB, Folarin OA, Happi CT, Pitts KR, Geske FJ, Geisbert TW, Saphire EO, Robinson JE, Wilson RB, Sabeti PC, Henderson LA, Khan SH, Bausch DG, Branco LM, Garry RF, the Viral Hemorrhagic Fever Consortium . 2014. Lassa fever in post-conflict Sierra Leone. PLoS Negl Trop Dis 8:e2748. doi: 10.1371/journal.pntd.0002748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO. 2018. 2018 Annual review of diseases prioritized under the Research and Development Blueprint. WHO, Geneva, Switzerland. [Google Scholar]
- 5.Martinez JP, Sasse F, Brönstrup M, Diez J, Meyerhans A. 2015. Antiviral drug discovery: broad-spectrum drugs from nature. Nat Prod Rep 32:29–48. doi: 10.1039/c4np00085d. [DOI] [PubMed] [Google Scholar]
- 6.Bekerman E, Einav S. 2015. Infectious disease. Combating emerging viral threats. Science 348:282–283. doi: 10.1126/science.aaa3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reasor MJ, Hastings KL, Ulrich RG. 2006. Drug-induced phospholipidosis: issues and future directions. Expert Opin Drug Saf 5:567–583. doi: 10.1517/14740338.5.4.567. [DOI] [PubMed] [Google Scholar]
- 8.Lüllmann H, Lüllmann-Rauch R, Wassermann O. 1978. Lipidosis induced by amphiphilic cationic drugs. Biochem Pharmacol 27:1103–1108. doi: 10.1016/0006-2952(78)90435-5. [DOI] [PubMed] [Google Scholar]
- 9.Morissette G, Ammoury A, Rusu D, Marguery MC, Lodge R, Poubelle PE, Marceau F. 2009. Intracellular sequestration of amiodarone: role of vacuolar ATPase and macroautophagic transition of the resulting vacuolar cytopathology. Br J Pharmacol 157:1531–1540. doi: 10.1111/j.1476-5381.2009.00320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, Parkinson A. 2013. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab Dispos 41:897–905. doi: 10.1124/dmd.112.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gehring G, Rohrmann K, Atenchong N, Mittler E, Becker S, Dahlmann F, Pöhlmann S, Vondran FWR, David S, Manns MP, Ciesek S, von Hahn T. 2014. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J Antimicrob Chemother 69:2123–2131. doi: 10.1093/jac/dku091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, Hoffstrom BG, DeWald LE, Schornberg KL, Scully C, Lehar J, Hensley LE, White JM, Olinger GG. 2013. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med 5:190ra79. doi: 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klintworth A, Nolden T, Westhaus S, Rohrmann K, David S, Manns MP, Finke S, Ciesek S, von Hahn T. 2015. Cationic amphiphilic drugs enhance entry of lentiviral particles pseudotyped with rabies virus glycoprotein into non-neuronal cells. Antiviral Res 124:122–131. doi: 10.1016/j.antiviral.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 14.Cheng Y-L, Lan K-H, Lee W-P, Tseng S-H, Hung L-R, Lin H-C, Lee F-Y, Lee S-D, Lan K-H. 2013. Amiodarone inhibits the entry and assembly steps of hepatitis C virus life cycle. Clin Sci (Lond) 125:439–448. doi: 10.1042/CS20120594. [DOI] [PubMed] [Google Scholar]
- 15.Gastaminza P, Whitten-Bauer C, Chisari FV. 2010. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc Natl Acad Sci U S A 107:291–296. doi: 10.1073/pnas.0912966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perin PM, Haid S, Brown RJP, Doerrbecker J, Schulze K, Zeilinger C, von Schaewen M, Heller B, Vercauteren K, Luxenburger E, Baktash YM, Vondran FWR, Speerstra S, Awadh A, Mukhtarov F, Schang LM, Kirschning A, Müller R, Guzman CA, Kaderali L, Randall G, Meuleman P, Ploss A, Pietschmann T. 2016. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63:49–62. doi: 10.1002/hep.28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owens CM, Mawhinney C, Grenier JM, Altmeyer R, Lee MS, Borisy AA, Leha J, Johansen LM. 2010. Chemical combinations elucidate pathway interactions and regulation relevant to Hepatitis C replication. Mol Syst Biol 6:375–313. doi: 10.1038/msb.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He S, Lin B, Chu V, Hu Z, Hu X, Xiao J, Wang AQ, Schweitzer CJ, Li Q, Imamura M, Hiraga N, Southall N, Ferrer M, Zheng W, Chayama K, Marugan JJ, Liang TJ. 2015. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci Transl Med 7:282ra49. doi: 10.1126/scitranslmed.3010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nawa M, Takasaki T, Yamada KI, Kurane I, Akatsuka T. 2003. Interference in Japanese encephalitis virus infection of Vero cells by a cationic amphiphilic drug, chlorpromazine. J Gen Virol 84:1737–1741. doi: 10.1099/vir.0.18883-0. [DOI] [PubMed] [Google Scholar]
- 20.Stadler K, Ha HR, Ciminale V, Spirli C, Saletti G, Schiavon M, Bruttomesso D, Bigler L, Follath F, Pettenazzo A, Baritussio A. 2008. Amiodarone alters late endosomes and inhibits SARS coronavirus infection at a post-endosomal level. Am J Respir Cell Mol Biol 39:142–149. doi: 10.1165/rcmb.2007-0217OC. [DOI] [PubMed] [Google Scholar]
- 21.Nemerow GR, Cooper NR, Muller-Eberhard HJ. 1984. Infection of B lymphocytes by a human herpesvirus, Epstein-Barr virus, is blocked by calmodulin antagonists. Proc Natl Acad Sci U S A 81:4955–4959. doi: 10.1073/pnas.81.15.4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazzon M, Ortega-Prieto AM, Imrie D, Luft C, Hess L, Czieso S, Grove J, Skelton JK, Farleigh L, Bugert JJ, Wright E, Temperton N, Angell R, Oxenford S, Jacobs M, Ketteler R, Dorner M, Marsh M. 2019. Identification of broad-spectrum antiviral compounds by targeting viral entry. Viruses 11:176–126. doi: 10.3390/v11020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.American Chemical Society. 2017. SciFinder. https://sso.cas.org/as/EZa7f/resume/as/authorization.ping.
- 24.Schneider G, Neidhart W, Giller T, Schmid G. 1999. Scaffold-hopping by topological pharmacophore search: a contribution to virtual screening. Angew Chem Int Ed 38:2894–2896. doi:. [DOI] [PubMed] [Google Scholar]
- 25.Madrid PB, Panchal RG, Warren TK, Shurtle AC, Endsley AN, Green CE, Kolokoltsov A, Davey ΔR, Manger ΠID, Gil L, Bavari S, Tanga MJ. 2015. Evaluation of Ebola virus inhibitors for drug repurposing. ACS Infect Dis 1:317–326. doi: 10.1021/acsinfecdis.5b00030. [DOI] [PubMed] [Google Scholar]
- 26.Salata C, Baritussio A, Munegato D, Calistri A, Ha HR, Bigler L, Fabris F, Parolin C, Palù G, Mirazimi A. 2015. Amiodarone and metabolite MDEA inhibit Ebola virus infection by interfering with the viral entry process. Pathog Dis 73:ftv032. doi: 10.1093/femspd/ftv032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chockalingam K, Simeon RL, Rice CM, Chen Z. 2010. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proc Natl Acad Sci U S A 107:3764–3769. doi: 10.1073/pnas.0915117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, White JM. 2013. Multiple cationic amphiphiles induce a Niemann-pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johansen LM, Dewald LE, Shoemaker CJ, Hoffstrom BG, Lear-Rooney CM, Stossel A, Nelson E, Delos SE, Simmons JA, Grenier JM, Pierce LT, Pajouhesh H, Lehár J, Hensley LE, Glass PJ, White JM, Olinger GG. 2015. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci Transl Med 7:290ra89. doi: 10.1126/scitranslmed.aaa5597. [DOI] [PubMed] [Google Scholar]
- 30.Zapatero-Belinchón FJ, Dietzel E, Dolnik O, Döhner K, Costa R, Hertel B, Veselkova B, Kirui J, Klintworth A, Manns MP, Pöhlmann S, Pietschmann T, Krey T, Ciesek S, Gerold G, Sodeik B, Becker S, von Hahn T. 2019. Characterization of the filovirus-resistant cell line SH-SY5Y reveals redundant role of cell surface entry factors. Viruses 11:275–226. doi: 10.3390/v11030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wool-Lewis RJ, Bates P. 1998. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J Virol 72:3155–3160. doi: 10.1128/JVI.72.4.3155-3160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holt DW, Tucker GT, Jackson PR, Storey G. 1983. Amiodarone pharmacokinetics. Am Heart J 106:840–847. doi: 10.1016/0002-8703(83)90006-6. [DOI] [PubMed] [Google Scholar]
- 33.Goldschlager N, Epstein A, Naccarelli G, Olshansky B, Singh B. 2000. Practical guidelines for clinicians who treat patients with amidoarone. Arch Intern Med 160:1741–1748. doi: 10.1001/archinte.160.12.1741. [DOI] [PubMed] [Google Scholar]
- 34.Reis M, Aberg-Wistedt A, Agren H, Höglund P, Akerblad A-C, Bengtsson F. 2004. Serum disposition of sertraline, N-desmethylsertraline and paroxetine: a pharmacokinetic evaluation of repeated drug concentration measurements during 6 months of treatment for major depression. Hum Psychopharmacol 19:283–291. doi: 10.1002/hup.599. [DOI] [PubMed] [Google Scholar]
- 35.Sanofi-Aventis Deutschland GmbH. 2011. MULTAQ package insert. Sanofi-Aventis Deutschland GmbH, Frankfurt, Germany. [Google Scholar]
- 36.Drayer DE. 1984. Clinical consequences of the lipophilicity and plasma protein binding of antiarrhythmic drugs and active metabolites in man. Ann N Y Acad Sci 432:45–56. doi: 10.1111/j.1749-6632.1984.tb14507.x. [DOI] [PubMed] [Google Scholar]
- 37.Ghose AK, Pritchett A, Crippen GM. 1988. Atomic physicochemical parameters for 3D-QSAR III: modeling hydrophobic interactions relationships. J Comput Chem 9:80–90. doi: 10.1002/jcc.540090111. [DOI] [PubMed] [Google Scholar]
- 38.Reutlinger M, Koch CP, Reker D, Todoroff N, Schneider P, Rodrigues T, Schneider G. 2013. Chemically advanced template search (CATS) for scaffold-hopping and prospective target prediction for “orphan” molecules. Mol Inform 32:133–138. doi: 10.1002/minf.201200141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ploemen J-P, Kelder J, Hafmans T, van de Sandt H, van Burgsteden JA, Salemink PJM, van Esch E. 2004. Use of physicochemical calculation of pKa and CLogP to predict phospholipidosis-inducing potential. Exp Toxicol Pathol 55:347–355. doi: 10.1078/0940-2993-00338. [DOI] [PubMed] [Google Scholar]
- 40.Pelletier DJ, Gehlhaar D, Tilloy-Ellul A, Johnson TO, Greene N. 2007. Evaluation of a published in silico model and construction of a novel Bayesian model for predicting phospholipidosis inducing potential. J Chem Inf Model 47:1196–1205. doi: 10.1021/ci6004542. [DOI] [PubMed] [Google Scholar]
- 41.Halliwell WH. 1997. Cationic amphiphilic drug-induced phospholipidosis. Toxicol Pathol 25:53–60. doi: 10.1177/019262339702500111. [DOI] [PubMed] [Google Scholar]
- 42.Adams PC, Holt DW, Storey GCA, Morley AR, Callaghan J, Campbell RW. 1985. Amiodarone and its desethyl metabolite: tissue distribution and morphologic changes during long-term therapy. Circulation 72:1064–1075. doi: 10.1161/01.cir.72.5.1064. [DOI] [PubMed] [Google Scholar]
- 43.Kodavanti U, Mehendale HM. 1990. Cationic amphiphilic drugs and phospholipid storage disorder. Pharmacol Rev 42:327–354. [PubMed] [Google Scholar]
- 44.McCloud CM, Beard TL, Kacew S, Reasor MJ. 1995. In vivo and in vitro reversibility of chlorphentermine-induced phospholipidosis in rat alveolar macrophages. Exp Mol Pathol 62:12–21. doi: 10.1006/exmp.1995.1002. [DOI] [PubMed] [Google Scholar]
- 45.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. 2008. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 46.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simmons JA, D'Souza RS, Ruas M, Galione A, Casanova JE, White JM. 2016. Ebolavirus glycoprotein directs fusion through NPC1+ endolysosomes. J Virol 90:605–610. doi: 10.1128/JVI.01828-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mingo RM, Simmons JA, Shoemaker CJ, Nelson EA, Schornberg KL, D'Souza RS, Casanova JE, White JM. 2015. Ebola virus and severe acute respiratory syndrome coronavirus display late cell entry kinetics: evidence that transport to NPC1+ endolysosomes is a rate-defining step. J Virol 89:2931–2943. doi: 10.1128/JVI.03398-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, Padilla-Parra S, Fry EE, Stuart DI. 2016. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 535:169–172. doi: 10.1038/nature18615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren J, Zhao Y, Fry EE, Stuart DI. 2018. Target identification and mode of action of four chemically divergent drugs against Ebolavirus infection. J Med Chem 61:724–733. doi: 10.1021/acs.jmedchem.7b01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banda DH, Perin PM, Brown RJP, Todt D, Solodenko W, Hoffmeyer P, Sahu KK, Houghton M, Meuleman P, Müller R, Kirschning A, Pietschmann T. 2019. A central hydrophobic E1 region controls the pH range of hepatitis C virus membrane fusion and susceptibility to fusion inhibitors. J Hepatol 70:1082–1092. doi: 10.1016/j.jhep.2019.01.033. [DOI] [PubMed] [Google Scholar]
- 52.Kutner RH, Zhang XY, Reiser J. 2009. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4:495–505. doi: 10.1038/nprot.2009.22. [DOI] [PubMed] [Google Scholar]
- 53.Ciesek S, Westhaus S, Wicht M, Wappler I, Henschen S, Sarrazin C, Hamdi N, Abdelaziz AI, Strassburg CP, Wedemeyer H, Manns MP, Pietschmann T, von Hahn T. 2011. Impact of intra- and interspecies variation of occludin on its function as coreceptor for authentic hepatitis C virus particles. J Virol 85:7613–7621. doi: 10.1128/JVI.00212-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flint M, von Hahn T, Zhang J, Farquhar M, Jones CT, Balfe P, Rice CM, McKeating JA. 2006. Diverse CD81 proteins support hepatitis C virus infection. J Virol 80:11331–11342. doi: 10.1128/JVI.00104-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mittler E, Kolesnikova L, Strecker T, Garten W, Becker S. 2007. Role of the transmembrane domain of Marburg virus surface protein GP in assembly of the viral envelope. J Virol 81:3942–3948. doi: 10.1128/JVI.02263-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mesens N, Steemans M, Hansen E, Peters A, Verheyen G, Vanparys P. 2009. A 96-well flow cytometric screening assay for detecting in vitro phospholipidosis-induction in the drug discovery phase. Toxicol In Vitro 23:217–226. doi: 10.1016/j.tiv.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE. 2019. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 47:D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N, Iynkkaran I, Liu Y, MacIejewski A, Gale N, Wilson A, Chin L, Cummings R, Le Di, Pon A, Knox C, Wilson M. 2018. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.