

Abstract
Astrocytes are the gatekeepers of neuronal energy supply. In neurodegenerative diseases, bio-energetics demand increases and becomes reliant upon fatty acid oxidation as a source of energy. Defective fatty acid oxidation and mitochondrial dysfunctions correlate with hippocampal neurodegeneration and memory deficits in Alzheimer’s disease (AD), but it is unclear whether energy metabolism can be targeted to prevent or treat the disease. Here we show for the first time an impairment in fatty acid oxidation in human astrocytes derived from induced pluripotent stem cells of AD patients. The impairment was corrected by treatment with a synthetic peroxisome proliferator activated receptor delta (PPARβ/δ) agonist GW0742 which acts to regulate an array of genes governing cellular metabolism. GW0742 enhanced the expression of CPT1a, the gene encoding for a rate-limiting enzyme of fatty acid oxidation. Similarly, treatment of a mouse model of AD, the APP/PS1-mice, with GW0742 increased the expression of Cpt1a and concomitantly reversed memory deficits in a fear conditioning test. Although the GW0742-treated mice did not show altered astrocytic glial fibrillary acidic protein-immunoreactivity or reduction in amyloid beta (Aβ) load, GW0742 treatment increased hippo-campal neurogenesis and enhanced neuronal differentiation of neuronal progenitor cells. Furthermore, GW0742 prevented Aβ-induced impairment of long-term potentiation in hippocampal slices. Collectively, these data suggest that PPARβ/δ-agonism alleviates AD related deficits through increasing fatty acid oxidation in astrocytes and improves cognition in a transgenic mouse model of AD.
Keywords: Alzheimer’s disease, astrocyte, fatty acid oxidation, GW0742, neurodegenerative diseases, PPARβ/δ
1. INTRODUCTION
Astrocytes have a fundamental role in the brain energy metabolism and provide energy substrates to support neuronal transmission. Although astrocytes are commonly known to utilize glucose for energy, they exhibit a preference for fatty acids (FAs) and ketones as their primary metabolic fuel and produce ketone bodies when FAs are available (Edmond, 1992). Approximately 20% of total adult brain energy comes from fatty acid oxidation (FAO) (Ebert, Haller, & Walton, 2003) occurring mainly in astrocytes (Edmond, Higa, Korsak, Bergner, & Lee, 1998). FAO is driven by a metabolic pathway, in which carnitine palmitoyltransferase 1A (CPT1A) is a rate-limiting enzyme, catalyzing transfer of FAs into the mitochondria, where the β-oxidation occurs.
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly (Grimm, Mett, Grimm, & Hartmann, 2017). In AD, as in many other neurodegenerative diseases, normal astrocytic processes are compromised, leading to altered neuronal functions and memory deficits (Acosta, Anderson, & Anderson, 2017). Whereas the majority of AD cases are late-onset (LOAD) without evidence of genetic inheritance, 1–6% of the cases manifest at an early age (early-onset, EOAD) and are caused by mutations in genes, such as presenilin-1 (PSEN1), responsible for the accumulation of toxic amyloid beta (Aβ) in the brain, representing the main pathological hallmark of AD (Grimm, Rothhaar, & Hartmann, 2012). PSEN1 is a catalytic subunit of the γ-secretase complex that participates in the amyloidogenic cleavage of amyloid precursor protein (APP) releasing the 1–42 fragment of Aβ in the extracellular space where it further aggregates and deposits as senile Aβ plaques (Grimm et al., 2012). APP cleavage products play an essential role in regulating lipid homeostasis, and vice versa, FA metabolism is intertwined with APP processing and Aβ generation (Grimm et al., 2017).
Defects in metabolism correlate with the progression of AD. Glycolysis is impaired (Bigl, Apelt, Eschrich, & Schliebs, 2003), but also the metabolic products of FAO are reduced in the brains of AD patients (Ciavardelli et al., 2016). Furthermore, inhibition of FAO increases oxidative stress and induces the accumulation of Aβ in vitro and in vivo (Shi et al., 2016). Administration of substrates for FAO rescues Aβ-induced memory impairment by facilitating astrocytic oxidative metabolism (Gibbs, Gibbs, & Hertz, 2009). Similarly, facilitation of FAO has been linked to memory improvement (Gibbs et al., 2009) and an increase in neurogenesis (Stoll et al., 2015). Metabolism of FAs and Aβ is influenced by peroxisome proliferator receptor beta/delta (PPARβ/δ) target genes which regulate the lipid transport and processing in other cell types (Silva-Veiga et al., 2018). Therefore, activation of PPARβ/δ may emerge as a relevant approach to manage also brain metabolism, especially through astrocytic functions, since astrocytes are the major site for FAO in the brain.
Here, we show for the first time that human astrocytes, differentiated from induced pluripotent stem cells (iPSCs) of AD patients with an amyloidogenic mutation in delta 9 exon of PSEN1 (PSEN1ΔE9), show impaired FAO capacity. The impairment is corrected by treatment with GW0742, a synthetic ligand agonist of PPARβ/δ, which subsequently increases astrocytic expression of CPT1A. Similarly, treatment of APPswe/PSEN1dE9 (APP/PS1) mice with GW0742 increases the expression of Cpt1a, concomitantly reverses memory deficits in fear conditioning test and reduces the expression of pro-inflammatory mediators. This study is the first to address the AD-related defect in FAO, specifically in astrocytes, and suggests that astrocytic lipid metabolism provides a potential target for AD therapy.
2. MATERIALS AND METHODS
2.1. Human iPSC lines
Skin biopsies or blood samples of participants were collected upon approval from the committee on Research Ethics of Northern Savo Hospital district (license no. 123/2016). Written informed consent was obtained from the participating patients and healthy volunteers. All procedures were conducted in accordance with Finnish directives including the Act on Medical Research (488/1999), Decree on Medical Research (986/1999), and The Act on the Medical Use of Organs and Tissues (2001/101).
The iPSC-lines used in this study were generated from three healthy volunteers and from three EOAD patients carrying a deletion in the exon 9 of the PSEN1 gene (4.6 kb mutation; the Finnish PSEN1ΔE9) (Crook et al., 1998; Oksanen et al., 2017). Two patients were diagnosed with AD and one was pre-symptomatic with no clinical diagnosis at the time of sample collection. To examine the correlation between ΔE9 mutation and AD phenotype, we also used corrected isogenic control lines created by CRISPR/Cas9 system from the pre-symptomatic and one symptomatic patients (Oksanen et al., 2017). In addition, we used commercial embryonic stem cell line H9 (NIH approved WA09, WiCell, Madison, WI). The absence of the most common genetic determinant of LOAD, namely APOE ε4 allele, was confirmed, and all cell lines carried neutral APOE ε3/ε3 alleles. All used iPSC lines had undergone detailed karyotype and phenotype evaluations described in Oksanen et al. (2017), and expressed high levels of pluripotent markers, formed embryoid bodies, had the ability to form all three germ layers and presented normal euploid karyotypes (Oksanen et al., 2017). The genders, phenotype, and origin of each line used in this study are presented in Table 1.
TABLE 1.
Panel of used human induced pluripotent stem cell lines and their features
| Line | Sex | Health status |
Age/years | APOE type | PSEN1 genotype | Sample origin | Reprogramming method | Karyotype | References | Used in figure |
|---|---|---|---|---|---|---|---|---|---|---|
| Ctrl1 | F | Healthy | Adult | ε3/ε3 | - | Skin biopsy | SeV 1.0 | 46 XX Normal | Holmqvist et al. (2016) | Figure 1a–d |
| Cltr2 | M | Healthy | 62 years | ε3/ε3 | - | Skin biopsy | SeV 1.0 | 46 XY Normal | Holmqvist et al. (2016) | Figure 1a–d |
| Ctrl3 | F | Healthy | 44 years | ε3/ε3 | - | Skin biopsy | SeV 1.0 | 46 XX Normal | Oksanen et al. (2017) | Figure 1a–d |
| AD1 | F | EOAD | 64 years | ε3/ε3 | ΔE9 deletion | Blood sample | SeV 1.0 | 46 XX Normal | Oksanen et al. (2017) | Figure 1a–d |
| AD2 | M | EOAD | 48 years | ε3/ε3 | ΔE9 deletion | Skin biopsy | SeV 2.0 | 46 XY Normal | Oksanen et al. (2017) | Figure 1a–i |
| AD2 iso | M | EOAD corrected | 48 years | ε3/ε3 | ΔE9 corrected | Skin biopsy | SeV 2.0 CRISPR/Cas9 | 46 XYNormal | Oksanen et al. (2017) | Figure 1e–i |
| AD3 | F | Pre-symptomatic | 47 years | ε3/ε3 | ΔE9 deletion | Skin biopsy | SeV 2.0 | 46 XX Normal | Oksanen et al. (2017) | Figure 1a–i |
| AD3 iso | F | EOAD corrected | 47 years | ε3/ε3 | ΔE9 corrected | Skin biopsy | SeV 2.0 CRISPR/Cas9 | 46 XX Normal | Oksanen et al. (2017) | Figure 1f–h, e, i |
| H9 | F | Healthy | Embryo | ε3/ε3 | - | ESC blastocyst | - | 46 XX Normal | Thomson et al. (1998) | Figure 1a–f, g; i |
Iso = isogenic.
2.2. IPSC-derived astrocyte cultures
Generation of iPSCs and their differentiation to astrocytes was carried out as described earlier (Oksanen et al., 2017). Briefly, somatic cells were reprogrammed into iPSCs using CytoTune™ -iPS 1.0 or CytoTune™ -iPS 2.0 Sendai Reprogramming Kits (Invitrogen, Eugene, OR). The resulting iPSCs were grown in Essential 8 Medium (E8; Life Technologies) on Matrigel-coated (Corning, NY) dishes and maintained by passaging with 0.5 mM ethylenediaminetetraacetic acid (EDTA) and keeping additional 5 μM Y-27632 ROCK-inhibitor (Selleckchem, Munich, Germany) in medium for 24 hr after passaging.
Astrocytic differentiation was initiated by changing the medium into neural differentiation medium (NDM) consisting of DMEM/F12 and Neurobasal (1:1), 1% B27 w/o vitamin A, 0.5% N2, 1% Glutamax, and 0.5% penicillin/streptomycin (P/S, 50 IU/50 μg/ml) (all from Invitrogen), 10 μM SB431542 (Sigma-Aldrich Co., St. Louis, MO) and 200 nM LDN193189 (Selleckchem). The resulting rosettes were expanded in NDM supplemented with 20 ng/ml basic fibroblastic growth factor (bFGF, Peprotech, Rocky Hill, NJ) for 2–3 days and then mechanically lifted onto ultra-low attachment plates (Corning). The resulting spheres were grown in astrocyte differentiation medium (DMEM/F12, 1% N2, 1% Glutamax, 1% nonessential amino acids, 0.5% P/S (50 IU/50 μg/ml) and 0.5 IU/ml heparin (Leo Pharma, Ballerup, Denmark), 10 ng/ml bFGF, and 10 ng/ml epidermal growth factor (EGF, Peprotech). Medium was changed every 2–3 days and the spheres maintained in astrocytic differentiation medium (ADM) for 6–8 months allowing the formation of pure astrocytic cultures. For experiments, spheres between 180 and 240 days in vitro (DIV) were dissociated with Accutase (Stem Cell Technologies, Cambridge, United Kingdom) to single cells and plated on Matrigel (BD Matrigel matrix, growth factor reduced; BD Biosciences, San Jose, CA) coated well plates or on Seahorse microplates (Seahorse Bioscience, North Billerica, MA) in ADM supplemented with 10 ng/ml ciliary neurotrophic factor and 10 ng/ml bone morphogenetic protein 4 (both from Peprotech).
2.3. Adult mouse astrocyte cultures
Adult astrocytes were isolated as previously published (Iram et al., 2016) from 4.5-, 9-, and 20-month-old APP/PS1 mice. Briefly, mice were sacrificed by overdose with tribromoethanol (Avertin, Sigma-Aldrich) and transcardially perfused with ice-cold heparinized saline. The brains excluding cerebellum and brain stem were dissected out and minced in Hank’s balanced salt solution (HBSS) followed by dissociation into single cell suspension using the Neural dissociation kit (Miltenyi Biotech) according to manufacturer’s instructions. After washing with phosphate-buffered saline (PBS), the cells were suspended into culture medium containing DMEM/F-12, 1× Glutamax, 10% iFBS, and 1% P/S, and plated on Poly-D-Lysine coated 6-well plates. The medium was changed every day for the first 7 days. The cells were detached with 0.25% Trypsin-EDTA and split every 7 days to expand the cell amount and used for experiments on 21–27 DIV.
2.4. FA oxidation measurement
The oxidation of endogenous FAs was measured with the Seahorse XF24 analyzer and FA oxidation assay according to the manufacturer’s instructions (Seahorse Bioscience). Cells were seeded at high density (100,000–125,000 per well) and cultured on Matrigel-coated XF24 cell culture microplate (Seahorse Bioscience) and were primed to utilize endogenous FAs as an energy source by replacing the maturation medium with substrate limited medium 24 hr before the measurement. Limited medium for adult mouse astrocytes consisted of DMEM/F12 without glucose (Biowest, Nuaille, France), 0.5% Glutamax, 0.5% P/S (50 IU/50 (μg/ml), supplemented with 2.5 mM glucose (Sigma-Aldrich), and 0.5 mM acyl-carnitine (Sigma-Aldrich). For iPSC-astrocytes, limited medium contained also additional 0.5% N2 and 0.5% nonessential amino acids. Limited medium was supplemented either with vehicle (dimethyl sulfoxide [DMSO]) or 1 μM or 10 μM GW0742.
On the day of the assay, limited medium was replaced with FAO assay medium (Seahorse Bioscience) containing MgS04 0.8 mM, CaCI2 1.8 mM, NaCI 143 mM, KCI 5.4 mM, NaH2P04 0.91 mM, L-Ala-Gln (GlutaMAX) 2 mM, supplemented with additional glucose 1 mM, acyl-carnitine 0.5 mM, and HEPES 5.0 mM, and adjusted to pH 7.4. The plate was transferred to non-CO2 incubator at +37 °C 1 hr before the assay. A CPT1A inhibitor, etomoxir (Sigma-Aldrich), was added to half of the wells in the final concentration of 40 μM 15 min prior to starting the assay to allow it to be taken up by the cells and to bind to CPT1A. Assay test compounds, oligomycin, carbonyl cyanide-4-(tri-fluoromethoxy)phenylhydrazone (FCCP), and rotenone/antimycin A (all from Sigma-Aldrich) were added sequentially through automated injection during the assay at 1 μM final concentrations each, and measurements were repeated three times every 10 min.
The results were analyzed with Wave Desktop 2.4 (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions. The parameters of mitochondrial function, including basal respiration, ATP production, proton leak, spare respiratory capacity, and nonmitochondrial respiration were calculated from oxygen consumption rate (OCR) for basal and maximal energy demands under the limited conditions described above. Basal respiration was derived by subtracting nonmitochondrial respiration from baseline OCR measured before addition of assay test compounds. The ATP-linked respiration was calculated by subtracting the OCR after addition of oligomycin, a complex V inhibitor, from baseline cellular OCR. The proton leak was derived by subtracting nonmitochondrial respiration from the oligomycin rate. Maximal respiratory capacity was calculated by subtracting nonmitochondrial respiration from the FCCP rate, since FCCP is a protonophore that collapses the inner membrane gradient allowing the electron transport chain to function at its maximal rate. Nonmitochondrial respiration was measured directly from OCR after addition of last assay rest compound mix, antimycin A, and rotenone, which are inhibitors of complex III and I and shut down electron transport chain function. Mitochondrial reserve capacity was calculated by subtracting basal respiration from maximal respiratory capacity. Furthermore, the basal and maximal endogenous FA utilization was determined by subtracting the OCR of vehicle or GW0742 treated group exposed to etomoxir that inhibits the FA oxidation, from the corresponding group that was not exposed to etomoxir. The wells that did not show adequate basal respiration or response for FCCP were excluded: altogether 3 of 72 wells from 3 experiments. The experiments were repeated three times with astrocytes derived from six different iPSC-lines and adult astrocytes from APP/PS1 mice of three different ages. The results from individual experiments were pooled together by normalizing the values to vehicle.
2.5. Mitochondrial respiration, glycolysis, and lactate analysis
Mitochondrial metabolism in presence of glucose was analyzed with the Seahorse XF24 analyzer and Mito Stress Test according to the manufacturer’s instructions (Agilent Technologies). Briefly, the cells were similarly seeded, cultured, and measured on XF24 cell culture microplate (Seahorse Bioscience) as for FAO assay, except, instead of changing limited medium 24 hr before the assay, the treatment compounds, GW0742 or vehicle, were added directly to the normal culture medium. In addition, assay medium was supplemented with 1× GlutaMAX, 0.5 mM sodium pyruvate, and 10 mM glucose instead of glucose 1 mM, acyl-carnitine 0.5 mM, and HEPES 5.0 mM. L(+)-lactate was measured from the normal cell culture medium 24 hr after treatment using an enzymatic Lactate Assay kit (Sigma) according to the manufacturer’s instructions. Results were normalized to total protein amount (Pierce BCA protein assay).
2.6. Cytometric bead arrays
Cytokine concentrations were detected with the cytometric bead array (CBA) multiplex flex sets (BD Biosciences). Human Soluble Protein flex sets were used for detecting CCL5/RANTES, granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein-1 (MCP-1) and IL-6, and Human Enhanced Sensitivity flex sets for IL-2 and IL-10. The iPSC-astrocytes were maturated on Matrigel-coated 48-well plates, with each well receiving 100,000 cells, and stimulated for 24 hr with a combination of pro-inflammatory cytokines TNFα (50 ng/ml, Peprotech) and IL-1β (10 ng/ml, Pepro-tech) and then treated with 1 μM GW0742 or vehicle at the same time. Medium was collected and centrifuged at 300g for 5 min to remove cell debris, and 20 or 25 μl of supernatant was used for analysis. Analysis was run with FACSAriaIII flow cytometer (BD Biosciences) and analyzed with FCAP Array™ v2.0.2 software (BD Biosciences).
2.7. Animals and drug administration
For behavioral testing and immunohistochemistry, we used 21 C57BL/6J wild type (WT) and 19 transgenic (TG) APPswe/PSEN1dE9 mice (Jankowsky et al., 2004) (Jackson Laboratories, Bar Harbor, ME) carrying human APP (K595N and M596L) and PSEN1dE9 mutations maintained in C57BL/6J background. These mice are referred to as APP/PS1 mice. Mice were randomized into vehicle or GW0742 treatment groups. Starting at the age of 12 months, the mice were administered with 30 mg/kg GW0742 in water suspension, or only water with the same volume, every 24 hr by oral gavage for 14 days. The mice were subjected to behavioral testing while under treatment and were killed for sample collection 12 hr after the final drug administration. For electrophysiology measurements, we used slices from 31 adult male WT mice. For cell culture assays, also WT mice with C57BL/6JRccHsd background were used.
All animals were group-housed in a controlled environment (temperature, 21 ± 1 °C; humidity, 50 ± 10%; light period, 7:00 a.m. to 7:00 p.m.) and had ad libitum access to food and water. Experiments were conducted according to the European Communities Directive (86/609/EEC) and approved by the Animal Experiment Board in Finland; or according to the Case Western Reserve University Institutional Animal Care and Use Committee guidelines.
2.8. Reagents
Powdered Ap 1–42 peptide (American Peptide Company, Sunnyvale, CA) and diluted in dH2O to the concentration of 500 μM, and then, by addition of Ca2+-free artificial cerebrospinal fluid (aCSF) to concentration of 50 μM, then aliquoted and frozen. Single aliquot was sufficient for one experiment, which was then immediately diluted to aCSF containing 10 μM DMSO to the final concentration of 1.0 μM before use. GW0742 for the animal experimentation was kindly provided by GlaxoSmithKline (Research Triangle, NC). For the cell culture studies, GW0742 was purchased from Sigma (Sigma-Aldrich).
2.9. Contextual fear conditioning
We chose to examine memory functions of APP/PS1-mice with the hippocampus-dependent memory test 1 day after contextual fear conditioning as described before (Kimura & Ohno, 2009; Mariani et al., 2017) . The mice were 12 months old at the time of experiment to ensure that they have fully developed amyloid pathology (Garcia-Alloza et al., 2006; Jackson et al., 2016; Jankowsky et al., 2004; Kam-phuis et al., 2012) and memory impairments typically observed in the strain (Kilgore et al., 2010; Lalonde, Kim, Maxwell, & Fukuchi, 2005; Minkeviciene et al., 2008; Volianskis, Kostner, Molgaard, Hass, & Jensen, 2010). The measurements were performed in standard conditioning chambers equipped with a stainless-steel grid floor connected to a solid-state shock scrambler. Two mice were measured simultaneously in two chambers, each of which was housed in a soundproof isolation cubicle. Each scrambler was connected to an electronic constant-current shock source that was controlled via an interface on a Windows XP computer running FreezeFrame software (Coulbourn Instruments, Allentown, PA). On the side of each chamber was mounted a digital camera and video signals were sent to the same computer for analysis. During training, one mouse at a time was placed in a conditioning chamber for 12 min and then received four footshocks (1.0 mA, 2 s) in 1-min interval to ensure initial hippocampal memory formation. After the last shock delivery, mice were left in the chamber for another 30 s.
Hippocampus-dependent contextual fear memory formation was evaluated by scoring freezing behavior for 5 min when the mice were placed back into the same conditioning chamber 24 hr after training.
2.10. Open field (OF) test
The mice were placed in the center of an open arena area of 40 × 40 × 40 cm with transparent walls and the movement was recorded for 10 min. The total distance moved, speed in 30-s bins, and the percentage of time spent in the center of the area were calculated.
2.11. Immunohistochemistry
At the end of the behavioral testing, the mice were terminally anesthetized using tribromoethanol (Avertin, Sigma-Aldrich) and perfused transcardially with 0.01 M PBS, pH 7.4. After perfusion, the brains were dissected out, cut midsagittal and the left hemisphere was immersion fixed in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.4) overnight after which the hemibrains were cryoprotected in 30% sucrose in 0.1 M phosphate buffer (pH 7.4) for 48 hr. The brains were frozen and cut in 20 μm thick sagittal sections using a cryostat. Total of six sections per mouse, spanning in 400 μm intervals through the hippocampus, were stained with antibodies against ionized calcium binding protein (Iba-1, 1:200 dilution, Wako Chemicals, Richmond, VA), glial fibrillary acidic protein (GFAP; 1:500 dilution, Dako, Carpinteria, CA), doublecortin (DCX; 1:500 dilution, Cell Signaling, Danvers, MA), or anti-human Aβ antibody 6E10 (1:1,000 dilution, BioLegend, Dedham, MA), followed by incubation with appropriate Alexafluor 488 or 546 conjugated secondary antibodies (Molecular Probes/Life Technologies, Grand Island, NY). Images of the hippocampi were taken and the immunoreactivities for GFAP, Iba-1, and 6E10 were quantified using Fiji/ImageJ software (freeware). Dense core plaques were imaged by thioflavin-T (ThT) staining carried out as described earlier (Malm et al., 2007). ThT staining was quantified and averaged from three hippocampal sections from each animal. ThT fluorescent tif-images were batch processed using Fiji version 2.0.0-rc-41/1.50d. Percentage ThT immunoreactivity was quantified as the percentage of positive pixels for the pixel intensity range between 60 and 120, which visually correlated with the minimal (core dense plaques) and maximal (halo around plaque) specific ThT staining. ThT images were converted to binary masks using the thresholding algorithm to allow plaque count and total plaque area to be quantified. Data are presented as the mean (+SEM) of n = 7–8 individual animals per group.
2.12. Quantitative RT-PCR
After perfusion, cortices of mice were dissected out from the right hemibrains and were homogenized in 800 μl of tissue homogenization buffer (250 mM sucrose, 20 mM Tris pH 7.4,1 mM EDTA, and 1 mM ethylene glycol tetraacetic acid [EGTA] in diethylpyrocarbonate treated water) containing Protease Inhibitor Cocktail (1:100, Sigma-Aldrich). After homogenization, an equivalent volume of RNABee (TelTest Inc., Friendwood, TX) was added into the samples followed by addition of chloroform. The samples were centrifuged for 15 min at 13,000g at 4 °C after which an equal volume of 70% EtOH was added to the upper aqueous layer. RNA was isolated using RNease Mini Kit (Qiagen, Valencia, CA) including the elimination of genomic DNA. mRNA concentration and purity were determined using a Nano-Drop 2000 (Thermo Scientific, Hudson, NH). About 0.5 μg of RNA was transcribed using a QuantiTect Reverse Transcription kit (Qiagen) according to the manufacturer’s instructions.
For the cell culture samples, RNA was extracted using Qiagen RNA-easy kit and 0.5 μg of RNA was transcribed and used for qPCR as described above.
2.13. Ex vivo electrophysiological recording
Acute hippocampal slices were dissected from 36 mice at the age of 3–4 months. From each mouse, 4–6 slices were quickly screened, and the one in the best condition was further recorded. Slices from five animals were excluded due to epileptic activity, four in the Aβ group, one in the Aβ + GW0742 group. The mice were deeply anesthetized with 5% isoflurane (Attane vet®, Piramal Healthcare UK Limited, Morpeth, United Kingdom), the brains dissected out, and 400 μm thick coronal hippocampal slices prepared using a vibratome (“Pelco101,” Redding, CA). The slices were maintained at the interface between an oxygen-rich atmosphere and aCSF containing 124 mM NaCl, 3 mM KCl, 1.25 mM KH2PO4, 2 mM CaCl2,1 mM MgSO4, 26 mM NaHCO3, and 15 mM D-glucose. Calcium precipitation was prevented by equilibrating the slices with 95% O2 and 5% CO2. The slices were incubated in a chamber containing standard aCSF and DMSO (Sigma-Aldrich, the vehicle solvent for GW0742) for 1 hr before exposing them to one of four treatments for 1 hr 30 min. The treatments included: (a) 1.0 μM oligomeric Aβ1–42 (American Peptide Company, Sunnyvale, CA, n = 7 mice) for 1 hr, followed by vehicle (10 μM DMSO in aCSF) for 30 min; (b) vehicle for 1 hr, followed by 10 μM GW0742 for 30 min (n = 9 mice); (c) 1.0 μM Aβ1–42 for 1 hr followed by 10 μM GW0742 (n = 9 mice) for 30 min; and (d) vehicle only for 1 hr 30 min (n = 6 mice). After the treatments, the slices were moved to a recording chamber maintained at 34 ± 1 °C with perfusion rate of 0.8 ml/min and equilibrated for 15 min before placing the electrodes and starting the stimulation. We searched for 1–2 mV amplitude of field excitatory postsynaptic potential (fEPSP) at maximum current and stable baseline for 30 min (changes of fEPSP magnitude <10% with mild stimulation current) before application of the stimulation protocol.
2.13.1. Electrical stimulation
A bipolar nichrome stimulating electrode was placed in CA3 Schaffer collaterals, and two glass microelectrodes filled with aCSF were positioned into the dendritic layer of the hippocampal CA1 to record fEPSP. The distance between the recording and stimulation sites was between 250 and 450 μm. The stimulation, controlled by pClamp system (Digidata-1,200 and Clampex 8.0 software [Axon Instruments, Foster City, CA]), was delivered through a Stimulus Isolator A365 (World Precision Instruments, San Carlos, CA). To obtain the data for the input/output curves, an ascending series of ~10–30 stimulation intensities (20–500 μA, step 20 μA) was applied, covering a range of responses from subthreshold to supramaximal. For baseline recording, the pathway was stimulated (0.1 ms pulse duration) every 30 s and the stimulation intensity was adjusted to obtain 40% of the maximum amplitude of the fEPSP. After establishing 30 min of stable baseline, long-term potentiation (LTP) was elicited by delivering theta-burst stimulation (TBS) (a train of 10 bursts of 4 pulses, 10 ms between pulses, 200 ms between bursts, 0.1 ms pulse duration; repeated twice with a 30 s interval), a protocol that induced LTP lasting ~1 hr in the control group.
2.13.2. Data analysis
The amplified (×1,000) (Differential Amplifier, World Precision Instruments, San Carlos, CA), filtered (0.1 Hz–1 kHz), and digitized (20 kHz; Digidata 1,200 A/D converter) signal was acquired using Clampex 8.0 software and some online analyzes done using Clampfit 8.0 software (both Axon Instruments, Foster City, CA). The main analysis was done offline using a custom-written Matlab® program (Mathworks R2011a, Natick, MA). To import Axon abf file format “abfload” Matlab script was used (Copyright 2004, Harald Hentschke; http://www.mathworks.com/matlabcentral/). The fEPSP amplitude was defined as the absolute voltage of a vertical line running from the min/max point of fEPSP to pre-stimulation baseline.
2.14. Murine neuronal progenitor cell cultures
Neuronal progenitor cells (NPCs) were isolated from the hippocampi of WT C57BL/6JRccHsd mice at embryonic Day 15 (E15) or 18 (E18) and cultured as free-floating neurospheres as described (Gritti et al., 1996; Karkkainen et al., 2014; Reynolds, Tetzlaff, & Weiss, 1992) in the presence of EGF and bFGF. NPCs were maintained and expanded in cell culture medium containing DMEM/F12 (Gibco, Waltham, Massachusetts), 1 M HEPES (Sigma-Aldrich), penicillin streptomycin (100 U/mL/100 μg/mL), B27 supplement with vitamin A (Gibco), 20 ng/ml EGF, and 10 ng/ml bFGF. Fresh medium, supplemented with EGF and bFGF, was added to the cells every 3 days and cells were cultivated at 37 °C in 5% CO2. Neurospheres were dissociated into single cells with TrypLE (Gibco) once a week. Up to 10 passages were used for the experiments.
To quantify the effect of GW0742 on the differentiation of NPCs, 10–15 neurospheres with 200–400 μm diameter were transferred onto 0.5 mg/ml poly-DL-ornithine (Sigma-Aldrich) coated 48-well plate (Nunc®, Thermo Fisher Scientific, Waltham, Massachusetts), and cultured for 3 or 7 days in the presence of vehicle, 10 nM or 100 nM GW0742, 200 μM etomoxir, or 300 μM acyl-carnitine (all from Sigma-Aldrich). Since GW0742 stock was prepared in DMSO (Sigma-Aldrich), similar volume of DMSO was added to the vehicle wells. To induce differentiation and attachment of neurospheres, the growth factors EGF and bFGF were withdrawn from the medium. For 7-day experiments, half of the medium was changed on the third culture day.
2.15. Immunocytochemistry
After 3 or 7 days of treatment with GW0742, differentiated neuro-spheres were fixed with 4% formaldehyde solution in PBS (pH 7.4) for 20 min, permeabilized with ice-cold methanol (Thermo Fisher Scientific) for 30 min, and blocked to prevent nonspecific binding with 10% normal goat serum (NGS, Chemicon International, Merck Millipore, Billerica, MA) in PBS for 2 hr at room temperature. For immunostaining, the neurospheres were incubated with primary antibody for neuron specific class beta III tubulin (TUJ1; 1:2,000 dilution, Covance, Princeton, New Jersey) diluted in 4% NGS in PBS overnight at 4 °C. Appropriate Alexa Fluor® 488 or 568 conjugated secondary antibodies (Molecular Probes, Invitrogen, Eugene, OR) were added in 5% NGS in PBS and incubated for 2 hr at room temperature protected from light. Finally, cell nuclei were stained with 2.5 μg/ml bisBenzimide (Hoechst 33342, Sigma-Aldrich) for 5 min, washed, and the plates were stored at 4 °C until imaging.
For the evaluation of differentiation, three neurospheres per well from four replicate wells were imaged for each treatment group. Images were taken with 20× objective at the periphery of the neuro-sphere colony area, where the cells had migrated separately to make it possible to identify individual cells. Each experiment was repeated three times. Neurospheres were imaged with an Olympus IX71 microscope with MT10 illumination system attached with the DP70 digital camera, running DP software (all from Olympus, Tokyo, Japan) and quantified using ImagePro Plus (Media Cybernetics, Rockville, MD, and ImageJ (Wayne Rasband, National Institutes of Health, Betheda, MD) software.
2.16. Statistical analysis
The data were expressed as mean ± SEM and analyzed by SPSS® statistical software version 21.0.0.0 (IBM Corp., Armonk, NY) or Graph-Pad Prism software version 7.04 for Windows (La Jolla California). Differences between two groups were analyzed with unpaired two-tailed Student’s t test. Differences between several groups were analyzed with one-way anova followed by Tukey post hoc test. Differences between the two data sets including two genotypes as well as treatment groups were analyzed with two-way ANOVA followed by Bonferroni post hoc test.
2. RESULTS
3.1. AD-patient-derived astrocytes exhibit impaired FA oxidation rescued by PPARβ/δ-agonist, GW0742
To test the hypothesis that FAO contributes to impaired energy metabolism in AD, we examined FAO-related mitochondrial respiration of astrocytes derived from AD-patient (PSEN1ΔE9 astrocytes) and nondemented control subject (control astrocytes) iPSCs (Table 1). All of the used iPSC-astrocytes were previously characterized and have been shown to be functional (Oksanen et al., 2017). PSEN1ΔE9 cells have also been reported to display an AD-specific phenotype (Oksanen et al., 2017). When the astrocytes were deprived from exogenous energy substrates for 24 hr, they failed to show significant differences between the genotypes in FAO Seahorse assay measuring mitochondrial parameters, including basal and maximal respiration, proton leak and spare respiratory capacity, and in nonmitochondrial respiration (t(4) = >0.002, p < 1.00, Figure 1a–c). Additional exposure to etomoxir, an inhibitor of CPT1A, allowed the evaluation of the intrinsic capacity of the cells to oxidize endogenous FAs (Blazquez, Sanchez, Daza, Galve-Roperh, & Guzman, 1999). Indeed, the maximal respiration due to utilization of endogenous FAs was significantly lower in PSEN1ΔE9 astrocytes compared to control astrocytes (t (4) = 3.8, p = .02; Figure 1c). PSEN1ΔE9 also showed decreased basal FAO, yet the difference failed to reach statistical significance (t(4) = 1.9, p = .13).
FIGURE 1.
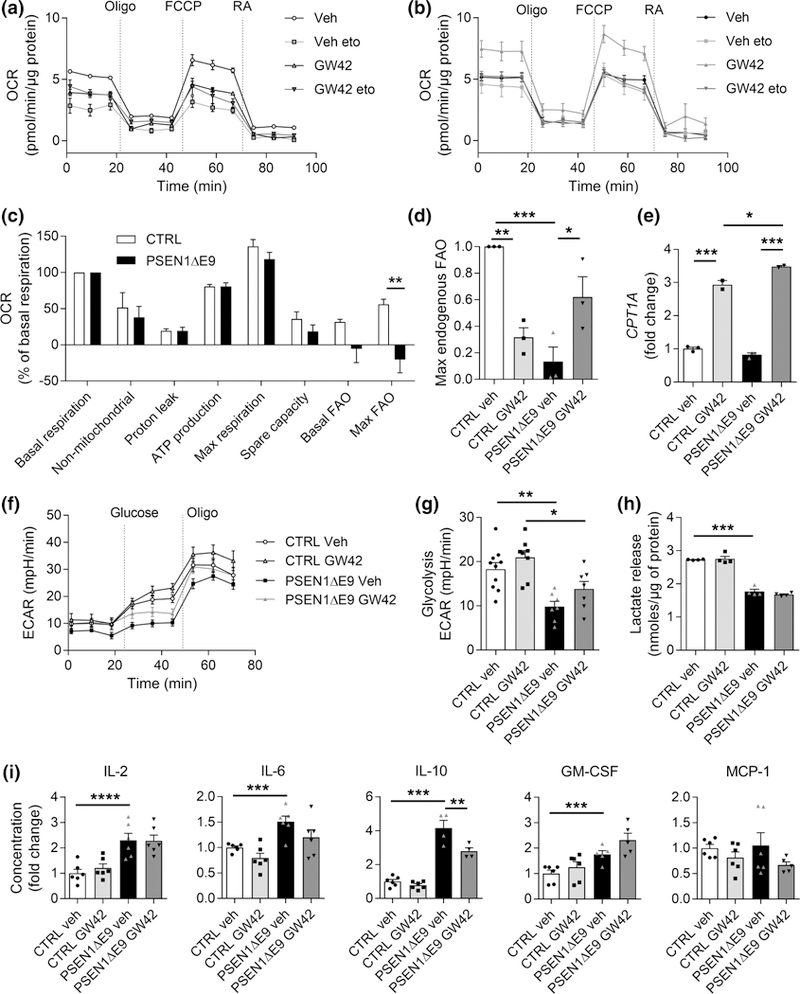
PSEN1ΔE9 iPSC-astrocytes exhibit impaired endogenous fatty acid oxidation (FAO) which is rescued by PPARβ/δ-agonist, GW0742. (a) Representative figure of OCR in control astrocytes after 24 hr culture in limited medium containing 1 mM glucose and 1 hr incubation with 50 μM etomoxir (eto) followed by sequential additions of 1 μM oligomycin (O), FCCP, rotenone, and antimycin A (RA). (b) Corresponding OCR curves for PSEN1ΔE9 astrocytes. (c) Mitochondrial parameters were quantified from OCR values and normalized to basal respiration of each genotype. (d) Maximal respiration due to utilization of endogenous FAs as a fold change to control astrocytes treated with vehicle. (e) Gene expression of CPT1A shown as a fold change to vehicle control group. (a–e) n = 3 biological replicates each with 2–3 technical replicates in three independent experiments. (f ) Representative figure of extracellular acidification rate (ECAR) after 24 hr culture in normal medium followed by sequential additions of 10 mM glucose and 1 μM oligomycin in glucose- and pyruvate-free assay medium. (g) Basal glycolysis was calculated from ECAR values after addition of 10 mM glucose. (f, g) n = 7–9 technical replicates. (h) Lactate release was measured from media with an enzymatic assay and the data were normalized to protein content. n = 4 technical replicates repeated with two biological replicates. (i) Concentrations of IL-2, IL-6, IL-10, GM-CSF, and MCP-1 were quantified from media after stimulation with TNFα (50 ng/ml) and IL-1β (10 ng/ml) for 24 hr using CBA assay shown as fold change to vehicle treated control astrocytes. n = 4–6 technical replicates. All data are presented as mean ± SEM with *p < .05, *p < .01, ***p < .001 as analyzed by unpaired t test or two-way anova followed by Bonferroni post hoc test. FCCP = carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; veh = vehicle containing DMSO the same v/v% as GW0742; GW42 = 1 μM GW0742 for 24 hr;, CTRL = control astrocyte; PSEN1ΔE9 = PSEN1ΔE9 astrocyte; CPT1A = carnitine palmitoyltransferase 1A; GM-CSF = granulocyte-macrophage colony-stimulating factor; MCP-1 = monocyte chemoattractant protein-1
Since FA metabolism appears to be strongly influenced by PPARβ/δ target genes in other cell types (Silva-Veiga et al., 2018), we next determined whether the endogenous FA energy metabolism of iPSC-astrocytes is regulated by the PPARβ/δ-agonist, GW0742. Treatment with 1 μM GW0742 for 24 hr in the presence of limited exogenous energy substrates reversed the impairment in the maximal utilization of endogenous FAs in PSEN1ΔE9 astrocytes and returned the OCR to that of control astrocytes, resulting in genotype × treatment interaction (F(1, 8) = 33.5, p = .0004, two-way anova; Figure 1d). Bonferroni post hoc test further revealed a reduction in endogenous FA utilization upon GW0742 treatment in control (p = .003) astrocytes, whereas in PSEN1ΔE9 astrocytes, GW0742 caused an increase in FA utilization (p = .02; Figure 1d).
3.2. GW0742 enhances the expression of CPT1A, the rate limiting enzyme of FAO
CPT1A is the key enzyme in FA transport across the mitochondrial membrane and controls the rate of mitochondrial β-oxidation of FAs (Jernberg, Bowman, Wolfgang, & Scafidi, 2017). In the brain, CPT1A is dominantly present in astrocytes and neural progenitor cells. Since we observed etomoxir-induced reduction in FAO and since etomoxir directly blocks the function of CPT1A, we measured the mRNA expression level of CPT1A in GW0742 treated iPSC-astrocytes. Although there were no significant differences between PSEN1ΔE9 and control astrocytes in basal conditions, both genotypes showed an increase in expression of CPT1A after 24 hr exposure to 1 μM GW0742 (F(1,6) = 1,136, p < .001, two-way anova main treatment effect, Figure 1e). Further Bonferroni’s post hoc test revealed a significantly higher treatment effect in PSEN1ΔE9 astrocytes compared to control astrocytes (p = .01).
3.3. GW0742 does not alter glycolytic metabolism in iPSC-derived astrocytes
To confirm that the observed GW0742-induced increase in OCR in PSEN1EΔ9 astrocytes was indeed because of enhanced FAO, and not caused as a secondary effect of glycolytic metabolism, we performed Seahorse MitoStress assay under high glucose levels. As expected, there was a significant reduction in the basal glycolytic metabolism between the genotypes (F(1, 28) = 1,167, p < .001, two-way anova main genotype effect, Figure 1f,g), in line with a previous report (Oksanen et al., 2017), yet GW0742 failed to alter the basal glycolytic metabolism (glycolysis (F(1, 28) = 0.4, p = .55, two-way anova; Figure 1f,g). In addition, GW0742 treatment did not alter the OCR-values of PSEN1ΔE9 astrocytes in the presence of glucose (Supporting Information Figure S1A), although GW0742 increased nonmitochondrial respiration in both genotypes (Supporting Information Figure S1B). Since the glycolytic pathway is tightly intertwined with lactate production, we measured L(+)-lactate release from the cell culture supernatant. As previously reported (Oksanen et al., 2017), lactate secretion was significantly lower in PSEN1ΔE9 astrocytes than in control astrocytes (F(1, 12) = >386.3, p < .001, two-way anova main genotype effect, Figure 1h). GW0742 failed to alter the lactate release (F(1, 12) = >0.4, p < .55, two-way anova) supporting the hypothesis that PPARβ/δ-agonism affects only FAO.
3.4. GW0742 does not alter iPSC-derived astrocyte cytokine secretion
To evaluate the impact of GW0742 treatment on astrocytic pro-inflammatory activation, we stimulated iPSC-astrocytes with two key inflammatory cytokines, 50 ng/ml tumor necrosis factor-α (TNFα) in combination with 10 ng/ml interleukin-1β (IL-1β) in the presence or absence of 1 μM GW0742 for 24 hr and measured the levels of secreted cytokines in the cell culture medium. As demonstrated by Oksanen et al. (2017), PSEN1EΔ9 astrocytes produced higher levels of IL-2, IL-6, IL-10, and GM-CSF (F(1, 16–20) = >13.6, p < .002, two-way anova main genotype effect, Figure 1i) than control astrocytes. Administration of GW0742 failed to significantly alter the production of any of the analyzed cytokines, except for IL-10, that was reduced in GW0742 treated PSEN1ΔE9 astrocytes (F(1, 16) = 144, p < .001, two-way; Figure 1i).
3.5. APP/PS1 astrocytes show impaired FAO
To evaluate whether astrocytes in APP/PS1 TG mice show similar impairment in FAO, we isolated and cultured astrocytes from APP/PS1 mice and their WT controls for seahorse FAO assay. In line with the data obtained from PSEN1ΔE9 iPSC-astrocytes, astrocytes from APP/PS1 mice showed significantly reduced maximal endogenous FAO compared to astrocytes cultured from WT mice (Supporting Information Figure S1C–E). Treatment with GW0742induced a trend toward increase in FAO in TG astrocytes (Supporting Information Figure S1E).
3.6. GW0742 ameliorates memory deficits in APP/PS1 mice and increases Cpt1a expression
To test whether GW0742 is capable of ameliorating AD-related memory impairment, 12-month-old APP/PS1 mice were orally administered with 30 mg/kg GW0742 for 14 days after which the mice underwent a contextual fear conditioning test (Figure 2a). In line with previous studies (Kilgore et al., 2010), vehicle treated APP/PS1 mice exhibited a significant reduction in conditional freezing compared to WT mice (t(38) = 3.3, p = .002, unpaired t test, Figure 2b). Moreover, this short treatment period with GW0742 was capable of reversing the deficits in contextual memory (t(16) = 2.4, p = .03, unpaired t test, Figure 2b).
FIGURE 2.
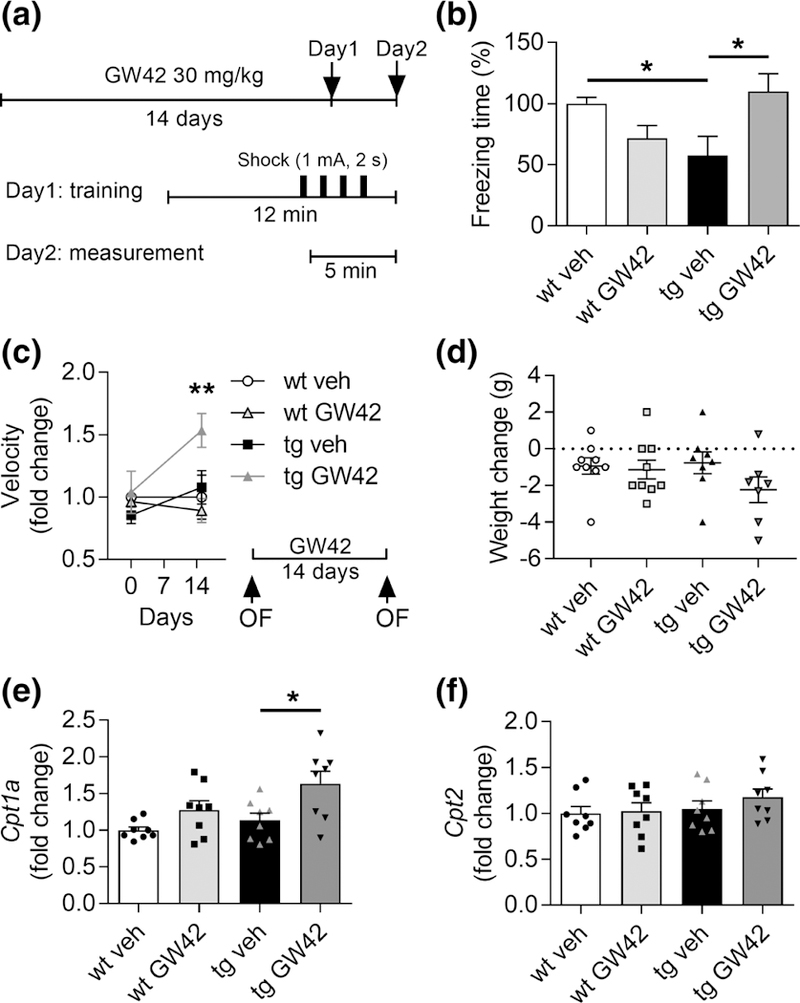
GW0742 ameliorates memory deficits in APP/PS1-mice concomitant with increased Cpt1a expression levels in the brain. (a) Schematic representation of hippocampus-dependent contextual memory test. The mice were daily administered p.o. with vehicle (veh) or 30 mg/kg GW0742 (GW42) for 14 days. The mice were trained in a conditioning chamber for 12 min receiving four foot shocks (1.0 mA, 2 s) in 1-min interval for contextual hippocampal memory formation. After 24 hr, the mice were placed into the same chamber and memory formation was evaluated by scoring freezing behavior for 5 min. (b) Percentage of freezing time to vehicle treated wild type (wt) mice. n = 32 wt veh, n = 7 wt GW42, n = 8–10 APP/PS1 (tg) veh and GW42. (c) Behavioral activity was measured as mean velocity in OF test 1 day before and 14 days after the start of GW0742 administration. Mean velocity is shown as a fold change to wt vehicle at each time point. n = 4–5. (d) Change in the body weight of mice before and after the treatment. n = 7–9. (e) Relative gene expression levels of Cpt1a in cortex, shown as fold change to wt vehicle. (f ) Respective gene expression levels of Cpt2. n = 8. All data are presented as mean ± SEM with *p < .05 and **p < .01 as analyzed by t test or two-way anova followed by Bonferroni post hoc test. cpt1a = carnitine palmitoyltransferase 1a
To further test the effect of GW0742 treatment on locomotor activity of the mice, we performed an OF test before the treatment onset to evaluate basal activity levels, as well as after the treatment period, to determine the treatment effect (Figure 2c). We failed to observe genotype-specific differences on the basal activity level measured as mean velocity of movement (F(1,14) = 0.1, p = .79, two-way ANOVA main genotype effect,), yet the GW0742 treated APP/PS1 mice showed increased activity compared to other groups, resulting in significant genotype × treatment interaction (F(1, 24) = 12.7, p = .003, two-way anova, Bonferroni’s post hoc test p = .002; Figure 2c). Since increased behavioral activity could lead to reduced body weight, we determined the change in body weight before and after the treatment. There were no significant changes in body weight, although there was a trend of weight reduction in APP/PS1 -mice treated with GW0742 compared to TG vehicle group (F(1, 29) = >0.7, p < .42, two-way anova, Figure 2d).
To confirm that the amelioration in memory was indeed related to mechanisms underlying FAO, we measured the relative cortical expression levels of Cpt1a and Cpt2 of the treated APP/PS1. Similar to the human iPSC-astrocytes, we found no genotype difference in the mRNA expression levels of Cpt1a (Figure 2e). However, GW0742 treated mice showed increased levels of Cpt1a in both genotypes, yet the increase reached statistical significance only in APP/PS1 cortices (F(1, 28) = 10.9, p = .003, two-way anova main treatment effect, Bonferroni’s post hoc for APP/PS1 vehicle × APP/PS1 GW0742, p = .03, Figure 2e). The levels of Cpt2 were not altered between the groups (F(1, 28)= > 0.7, p < .39, two-way anova, Figure 2f), suggesting that PPARβ/δ-agonism affects only the rate-limiting Cpt1a.
3.7. PPARβ/δ-agonism by GW0742 fails to alter AD-like pathological hallmarks in vivo
Next we assessed whether the beneficial behavioral outcome of GW0742 treatment was related to effects on AD-related pathological hallmarks. Quantification of the hippocampal ionized calcium binding adaptor molecule 1 (IBA1) and GFAP immunoreactivities revealed that the levels of both markers were unaltered by GW0742 treatment (Supporting Information Figure S3A, B). Further, we evaluated the effect of GW0742 treatment on Ap levels by ELISA (Supporting Information Figure S2), immunohistochemistry using 6E10-antibody (Supporting Information Figure S3C), and by ThT staining (Supporting Information Figure S3D). GW0742 treatment did not alter any of these parameters (Supporting Information Figures S3C, D; S2).
3.8. Inflammation related gene expression is altered by GW0742 treatment
APP/PS1 mice showed a relatively mild phenotype in the expression of cortical inflammatory genes, with approximately twofold up-regulation in the expression levels of II1b, Tnfα, CcI2 (C-C motif chemokine 2, also referred as MCP1), Cxcr2 (C-X-C chemokine receptor type 2), and TIr4 (toll-like receptor 4), while the expression levels of II6, C1qa (complement C1q subcomponent subunit A), and Socs1 (suppressor of cytokine signaling 1) were not different between the genotypes (Supporting Information Figure S3E). GW0742 treated APP/PS1 mice showed reduced levels of CcI2, C1qa, and Socs1. In WT mice, GW0742 similarly reduced CcI2, but additionally also II1b and Tnfα.
3.9. GW0742 attenuates Aβ-induced suppression of LTP ex vivo
To elucidate the impact of Aβ and GW0742 on synaptic plasticity, we investigated whether GW0742 attenuates Aβ-induced impairment in LTP in ex vivo hippocampal slices. LTP was induced in the CA3-CA1 pathway of the hippocampus using TBS (Figure 3a). The field excitatory post-synaptic potential (fEPSP) amplitudes after TBS stimulation were normalized to pre-TBS baseline. There was a significant treatment effect (Figure 3b,d), due to deterioration in LTP in slices incubated with oligomeric Aβ1–42 (F(3, 27) = 7.5, p = .001, RM anova, Tukey’s post hoc adjusted p = .001; Figure 3c,d). GW0742 treatment rescued the Aβ-induced decline in LTP (adjusted p = .002) but did not influence LTP in the control slices (adjusted p = .96) (Figure 3c,d).
FIGURE 3.
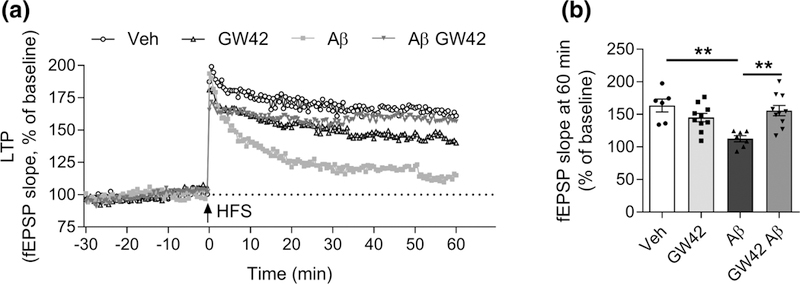
GW0742 attenuates amyloid beta-induced suppression of long-term potentiation (LTP) in ex vivo hippocampal synapses. Hippocampal slices (400 μm) from 3 to 4 months old male C57BL/6J wild type (wt) mice were exposed to 1 μM oligomeric amyloid beta (Aβ) 1–42 to mimic AD-conditions and treated with vehicle or 10 μM GW0742. (a) The electrophysiology recording was started 30 min before high-frequency stimulation (HFS: 1 s burst pulse at 100 Hz) at 0 min, and alterations of LTP were measured for 60 min. LTP presented as percentage of increase in slope of fEPSP normalized to baseline. (b) The quantitative analysis of LTP 60 min after HFS. All data are presented as mean ± SEM with **p < .01. (veh n = 6, GW42 n = 10, Aβ n = 7, Aβ GW42 n = 10)
3.10. GW0742 treatment increases neurogenesis in the hippocampus of APP/PS1 mice
Since neural stem cells have been shown to use FAO to support neurogenic activity (Knobloch et al., 2017), and neurogenesis is linked with amelioration of cognitive deficits, we assessed whether GW0742 treatment enhanced the production of newborn neurons as detected by DCX staining in the hippocampi of the GW0742 treated APP/PS1 mice. Although the number of DCX positive cells was very low in the 12-month-old APP/PS1 mice, it was slightly, yet significantly, increased in the treated mice compared to their vehicle treated controls (t(15) = 2.3, p = .04, unpaired t test, Figure 4a,b).
FIGURE 4.
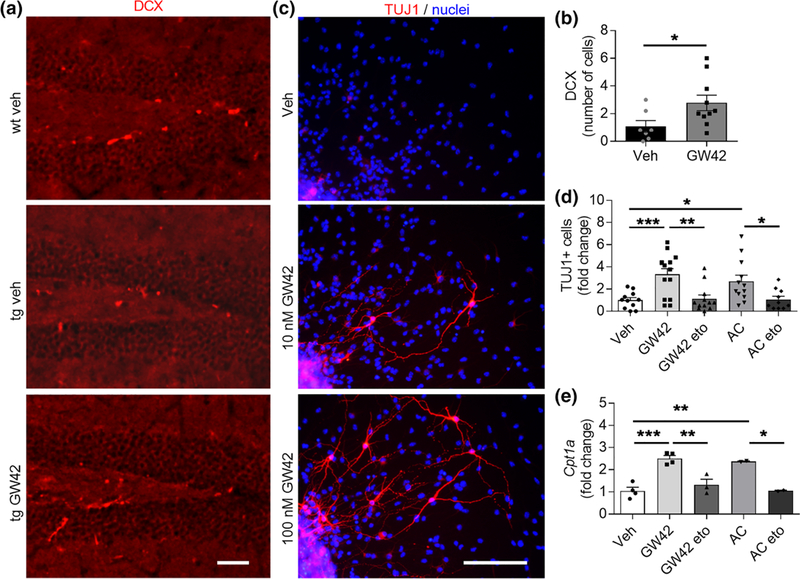
GW0742 treatment increases neurogenesis in vivo and in vitro in conjunction with elevated Cpt1a. (a) Representative immunohistochemistry images of hippocampal slices from wild type (wt) and transgenic (tg) APP/PS1-mice treated with vehicle or 30 mg/kg GW0742 for 14 days and stained for newborn neurons with DCX (red). Scale bars, 100 μm. (b) Corresponding graph for the number of DCX positive cells per hippocampal slice counted from six brain sections at400-μm intervals. n = 7–10. (c) Representative immunocytochemistry images of wt neurospheres differentiated for73 days in vitro and treated with vehicle, 10 or 100 nM GW0742 and stained for mature neurons with TUJ1 (red) and for nuclei (blue). Scale bars, 100 μm. (d) Fold change in the number of TUJ1-positive cells after neurosphere differentiation and treatment for 3 days with 100 nM GW0742, 200 μM etomoxir (eto) to block the activity of CPT1, or 300 μM acylcarnitine (AC) to provide a direct substrate for fatty acid oxidation (FAO). n = 10–14. (e) Corresponding relative gene expression levels of Cpt1a shown as a fold change to vehicle. n = 2–4 biological replicates in independent experiments. All data are presented as mean ± SEM with *p < .05, **p < .01, ***p < .001 as analyzed by t test or two-way anova followed by Bonferroni post hoc test
3.11. GW0742 increases neuronal differentiation of neuroprogenitor cells (NPCs) in vitro via FAO
In order to further understand the role of PPARβ/δ agonism in neurogenesis, WT murine hippocampal embryonic NPC neurospheres were treated for 3 or 7 days with 10 nM or 100 nM GW0742. In the absence of growth factors, NPCs begin to migrate away from the edges of the neurospheres and differentiate toward neurons, astrocytes, and oligodendrocytes. The impact of GW0742 on NPC differentiation was assessed by immunocytochemical staining TUJ1 (a marker of mature neurons). Treatment with 100 nM GW0742 increased TUJ1 immunoreactivity approximately threefold compared to vehicle treated cells (t(23) = 3.8, p = .0009; Figure 4c,d).
Given that PPARβ/δ agonism increased neuronal differentiation, we next explored whether this was connected with FAO, and hence exposed the NPC neurospheres either to 200 μM etomoxir to block the activity of CPT1, or to 300 μM acylcarnitine, a direct substrate for FAO. Similar to GW0742, acylcarnitine significantly increased the proportion of TUJ1-positive neurons by approximately threefold (t(21) = 2.7, p = .01, unpaired t test) compared to vehicle treatment after 7 days of differentiation (Figure 4e). The observed increase in TUJ1-immunoreactivity was prevented when the GW0742 (t(24) = 3.0, p = .002) or acylcarnitine (t(20) = 2.5, p = .02) treated cells were co-exposed to etomoxir. Corresponding relative gene expression levels of Cpt1a were measured from NPCs and the levels were concomitant with TUJ1-immunoreactivity (Figure 4f). Both GW0742 (t(6)= 6.9, p = .0005, unpaired t test) and acylcarnitine (t(4) = 5.3, p = .006) increased the expression of Cpt1a compared to vehicle treated cells, and etomoxir decreased the expression of Cpt1a back to the level of vehicle when co-treated with GW0742 (t(5) = 4.5, p = .006) or acylcarnitine (t(2) = 39.7, p = .0006).
4. DISCUSSION
Neurons depend on astrocytes for energy production in the form of lactate and glutamine. Although the main astrocytic energy consumption occurs through glycolysis, 20% of the energy is produced by FAO. Here we show for the first time that AD patient iPSC-astrocytes carrying PSEN1EA9 mutation show impaired FAO. This deficit is ameliorated by GW0742 treatment. To demonstrate the functional relevance of FAO enhancement, we also show that GW0742 treatment is able to ameliorate the AD-related behavioral deficits in APP/PS1 mice. Although impairment in FAO is unlikely to be the cause for AD-related pathology, our study suggests that FAO may be therapeutically targeted.
AD pathology is known to impair brain energy metabolism. Glucose hypometabolism has been suggested to be an early sign of AD even prior to the appearance of Aβ deposition (Chen & Zhong, 2013; Mosconi, Pupi, & De Leon, 2008; Nashold Jr. & Goldner, 1975). Exposure of astrocytes to Aβ and proinflammatory cytokines has been shown to increase astrocytic glycolysis without enhancing lactate production (Allaman et al., 2010; Gavillet, Allaman, & Magistretti, 2008), suggesting impairment in astrocytic metabolic activity. Given the dependency of neurons on astrocytic energy supply, it is not surprising that this influences neuronal functions (Allaman et al., 2010). Indeed, astrocytic production of lactate is required for the hippocampal memory formation and LTP (Suzuki et al., 2011). Importantly, lactate production is impaired in the PSEN1EΔ9 iPSC-astrocytes used in this study (Oksanen et al., 2017), implying that an AD-causing genotype directly impairs astrocytic energy metabolism. Although FAs are oxidized in healthy astrocytes, they become an even more important source of energy when brain health and normal glycolysis are compromised. Glucose deprivation induces FAO in astrocytes in hypoxic/ischemic conditions and may serve as an attempt to cope with the injury (Cabodevilla et al., 2013). For example, ischemia has been reported to induce fatty acid binding protein 3 expression in astrocytes in adult monkeys subjected to ischemic injury 15 days post injury (Boneva et al., 2011). Impairment in FAO is also evident in AD, indicated as reduced levels of the FAO metabolites, acylcarnitines, and ketones in the plasma of AD patients (Ciavardelli et al., 2016). Moreover, octanoate, a substrate for FAO has been shown to counteract the Aβ-induced memory deficits in chicks (Gibbs et al., 2009). It has been proposed that once astrocytes cannot compensate for comprised neuronal mitochondrial functions by producing lactate and ketones, the onset of neurodegenerative disease, such as AD is initiated (Iglesias, Morales, & Barreto, 2017).
Interestingly, GW0742 increased FAO only in PSEN1ΔE9 carrying astrocytes in the current study. Although this study did not assess the reason for this specificity, it is likely to be related to increased Aβ production of the cells. Aβ1–42 has been shown to activate nSMase, which participates in the production of ceramides (Mou, Yang, Qu, Chen, & Zhang, 2016). In contrast, ceramides have been shown to further increase the production of Aβ1–42 (Grimm et al., 2005). Ceramides are converted to sphingosines, which feed as substrates for FA oxidation. Although PPARβ/δ has not been shown to drive the function of ceramidase, it does participate into catabolism of ceramides by activating other enzymes taking part in this process (Aleshin & Reiser, 2014). Thus, this may be one possible pathway explaining why GW0742 drives FAO specifically in PSEN1AE9 astrocytes and not in control astrocytes, although proving this mechanism was beyond the scope of this study.
PPARs are ligand activated transcription factors capable of regulating a wide variety of cellular functions, such as glucose metabolism, energy balance, differentiation, inflammation, and importantly, to drive FAO. There are three main isoforms of PPARs, of which PPARβ/δ is ubiquitously expressed in the brain. PPARβ/δ is activated by unsaturated and saturated FAs, prostacyclin, 4-hydroxy-2-nonenal (4-HNE), and synthetic ligands, such as GW0742 used in this study, to drive FA uptake and catabolism (Wang et al., 2004), as well as differentiation (D’Angelo et al., 2011; Ji et al., 2015). The impact of PPARβ/δ in memory improvement and the enhancement of neurogenesis have not been previously reported in the context of AD; however, earlier studies have demonstrated the impact of PPARβ/δ activation on enhancing neurogenesis (D’Angelo et al., 2011; Ji et al., 2015) and improving memory impairments in a rodent model of Parkinson’s disease (Das, Gangwal, Damre, Sangamwar, & Sharma, 2014). In accordance, inhibition of PPARβ/δ impairs hippocampal dependent cognitive functions (Benedetti et al., 2016).
These studies are well in line with our study showing that administration of GW0742 to APP/PS1 mice reversed the contextual fear conditioning deficit and increased hippocampal neurogenesis, the latter of which we previously demonstrated to be reduced in the hippocampi of APP/PS1 mice (Puli et al., 2012; Taniuchi et al., 2007). To prove further the ability of PPARβ/δ to enhance neurogenesis, we used a mouse NPC model of neurogenesis and showed that GW0742 induces the production of Tuj-1 positive neurons. Since there was no genotype difference in differentiation of embryonic NPCs between WT and APP/PS1 as previously published (Karkkainen, Magga, Koistinaho, & Malm, 2012), the effect of GW0742 was only studied in WT NPCs. Benedetti et al. reported a similar finding in cortical neurons in which maturation was induced by administration of GW0742 (Benedetti et al., 2016). In addition, it has been shown that PPARβ/δ promotes the differentiation of other brain cell types, such as oligodendrocytes (D’Angelo et al., 2011). Neurogenesis and neuronal differentiation are driven by multiple factors. Yet neuronal stem cells in the subventricular zone of adult animals have been shown to rely on FAO for energy production and FAs, in particular, support their neurogenic activity (Stoll et al., 2015). This is in line with our study showing that GW0742 induced neurogenesis is FAO dependent.
Astrocytes and NPCs are the main cell types in the brain that utilize FAO for their energy expenditure (Edmond, Robbins, Bergstrom, Cole, & de Vellis, 1987; Knobloch et al., 2017) and FAs have been shown to be produced endogenously in neuronal stem cells (Knobloch et al., 2013). This prompted us to focus this study on astrocytes and NPCs. The produced FAs can be utilized as a source of energy, which is vital especially when the brain encounters neurodegeneration and compromised neuronal functions. Upon induction of FAO, FAs are esterified to acyl-CoA by acyl-CoA synthase and enter the mitochondria. In the intermembrane space of mitochondria, the fatty acyl-CoA is converted to a fatty acylcarnitine by CPT1 and subsequently transported across the mitochondrial inner membrane space. An increase in the expression level of CPT1a, the main isoform of CPT1 expressed in astrocytes, is indicative of increased FAO since it is the rate-limiting transport enzyme of FAs (Blazquez et al., 1999; Jernberg et al., 2017). Etomoxir, which blocks the activity of CPT1, is widely used to inhibit β-oxidation. Indeed, in the current study etomoxir blocked the astrocytic FAO and resulted in loss of GW0742-induced differentiation of NPCs.
Although we failed to observe an anti-inflammatory effect of GW0742 in iPSC-astrocytes, others have reported that PPARβ/δ activation alleviates astrocytic pro-inflammatory activation (Chistyakov, Aleshin, Astakhova, Sergeeva, & Reiser, 2015). In contrast, the impact of GW0742 on suppression of inflammation may be model dependent, as Schnegg et al. reported that GW0742 inhibited neuroinflammation, yet failed to restore neurogenesis or prevent hippocampal dependent cognitive deficits (Schnegg et al., 2013). Similarly, we have previously shown that GW0742 administration to 5×FAD mice suppresses microglial activation and prompts the clearance of Aβ deposits (Malm, Mariani, Donovan, Neilson, & Landreth, 2015), whereas in the current study, when administered to APP/PS1 mice, we failed to observe changes in microgliosis. The pathology progression of 5xFAD mice is aggressive leading to severe brain amyloidosis already at the age of 4 months whereas at the same age the APP/PS1 mice are only starting to build up plaques (Jankowsky et al., 2004; Oakley et al., 2006). The same applies for the brain inflammatory profile: the levels of pro-inflammatory cytokines are higher in 5×FAD mice compared to APP/PS1. Similarly, mouse model dependent alterations are also possible in astrocytic FA oxidation.
Taken together, this study shows for the first time that human iPSC-derived astrocytes carrying AD mutation PSEN1ΔE9 exhibit impaired FAO. This deficit is rescued by activation of PPARβ/δ with a synthetic agonist, GW0742. A similar deficit in FAO is also observed in astrocytes isolated from adult APP/PS1 mice. Treatment of APP/PS1 mice with GW0742 improves cognitive deficits of the mice. This study is the first to address the AD related defect in FA oxidation specifically in astrocytes and suggests that astrocytic lipid metabolism provides a potential target for AD therapy.
Supplementary Material
ACKNOWLEDGEMENTS
We thank K. Puttonen and S. Lehtonen for help in generation and characterization of iPSCs and iPSC-derived astrocytes and S.-C. Zhang and A. J. Petersen for help in generation of isogenic iPSC lines and setting up astrocyte differentiation protocol. M. Viitanen provided the skin samples for PSEN1ΔE9 iPSCs. We thank L. Kaskela, E. Korhonen, and M. Tikkanen, for technical assistance. This study was supported by University of Eastern Finland, Academy of Finland (298071), EU Joint Programme - Neurodegenerative Disease Research (JPND) project (301234), and European Union’s Horizon 2020 Research and Innovation Program (643417).
Funding information
2020 European Institute of Innovation and Technology, Grant/Award Number: 643417; Academy of Finland, Terveyden Tutkimuksen Toimikunta, Grant/Award Number: 298071301234; Horizon 2020; Academy of Finland; University of Eastern Finland
Footnotes
CONFLICT OF INTEREST
The authors declare they have no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- Acosta C, Anderson HD, & Anderson CM (2017). Astrocyte dysfunction in Alzheimer disease. Journal of Neuroscience Research, 95(12), 2430–2447. 10.1002/jnr.24075 [DOI] [PubMed] [Google Scholar]
- Aleshin S, & Reiser G (2014). Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) protects against ceramide-induced cellular toxicity in rat brain astrocytes and neurons by activation of ceramide kinase. Molecular and Cellular Neurosciences, 59, 127–134. 10.1016/j.mcn.2014.01.008 [DOI] [PubMed] [Google Scholar]
- Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, & Magistretti PJ (2010). Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: Impact on neuronal viability. The Journal of Neuroscience, 30(9), 3326–3338. 10.1523/JNEUROSCI.5098-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti E, Di Loreto S, D’Angelo B, Cristiano L, d’Angelo M, Antonosante A … Cimini A (2016). The PPARbeta/delta agonist GW0742 induces early neuronal maturation of cortical post-mitotic neurons: Role of PPARbeta/delta in neuronal maturation. Journal of Cellular Physiology, 231(3), 597–606. 10.1002/jcp.25103 [DOI] [PubMed] [Google Scholar]
- Bigl M, Apelt J, Eschrich K, & Schliebs R (2003). Cortical glucose metabolism is altered in aged transgenic Tg2576 mice that demonstrate Alzheimer plaque pathology. Journal of Neural Transmission (Vienna, Austria : 1996), 110(1), 77–94. 10.1007/s00702-002-0772-x [DOI] [PubMed] [Google Scholar]
- Blazquez C, Sanchez C, Daza A, Galve-Roperh I, & Guzman M (1999). The stimulation of ketogenesis by cannabinoids in cultured astrocytes defines carnitine palmitoyltransferase I as a new ceramide-activated enzyme. Journal of Neurochemistry, 72(4), 1759–1768. [DOI] [PubMed] [Google Scholar]
- Boneva NB, Kaplamadzhiev DB, Sahara S, Kikuchi H, Pyko IV, Kikuchi M … Yamashima T (2011). Expression of fatty acid-binding proteins in adult hippocampal neurogenic niche of postischemic monkeys. Hippocampus, 21(2), 162–171. 10.1002/hipo.20732 [DOI] [PubMed] [Google Scholar]
- Cabodevilla AG, Sanchez-Caballero L, Nintou E, Boiadjieva VG, Picatoste F, Gubern A, & Claro E (2013). Cell survival during complete nutrient deprivation depends on lipid droplet-fueled beta-oxidation of fatty acids. The Journal of Biological Chemistry, 288(39), 27777–27788. 10.1074/jbc.M113.466656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, & Zhong C (2013). Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Progress in Neurobiology, 108, 21–43. 10.1016/j.pneurobio.2013.06.004 [DOI] [PubMed] [Google Scholar]
- Chistyakov DV, Aleshin SE, Astakhova AA, Sergeeva MG, & Reiser G (2015). Regulation of peroxisome proliferator-activated receptors (PPAR) alpha and -gamma of rat brain astrocytes in the course of activation by toll-like receptor agonists. Journal of Neurochemistry, 134(1), 113–124. 10.1111/jnc.13101 [DOI] [PubMed] [Google Scholar]
- Ciavardelli D, Piras F, Consalvo A, Rossi C, Zucchelli M, Di Ilio C, … Sensi SL (2016). Medium-chain plasma acylcarnitines, ketone levels, cognition, and gray matter volumes in healthy elderly, mildly cognitively impaired, or Alzheimer’s disease subjects. Neurobiology of Aging, 43, 1–12. 10.1016/j.neurobiolaging.2016.03.005 [DOI] [PubMed] [Google Scholar]
- Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H,… Haltia M (1998). A variant of alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nature Medicine, 4(4), 452–455. [DOI] [PubMed] [Google Scholar]
- D’Angelo B, Benedetti E, Di Loreto S, Cristiano L, Laurenti G, Ceru MP, & Cimini A (2011). Signal transduction pathways involved in PPARbeta/delta-induced neuronal differentiation. Journal of Cellular Physiology, 226(8), 2170–2180. 10.1002/jcp.22552 [DOI] [PubMed] [Google Scholar]
- Das NR, Gangwal RP, Damre MV, Sangamwar AT, & Sharma SS (2014). A PPAR-beta/delta agonist is neuroprotective and decreases cognitive impairment in a rodent model of Parkinson’s disease. Current Neurovascular Research, 11(2), 114–124. [DOI] [PubMed] [Google Scholar]
- Ebert D, Haller RG, & Walton ME (2003). Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. The Journal of neuroscience, 23(13), 5928–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmond J (1992). Energy metabolism in developing brain cells. Canadian Journal of Physiology and Pharmacology, 70(Suppl), S118–S129. [DOI] [PubMed] [Google Scholar]
- Edmond J, Higa TA, Korsak RA, Bergner EA, & Lee WN (1998). Fatty acid transport and utilization for the developing brain. Journal of Neurochemistry, 70(3), 1227–1234. [DOI] [PubMed] [Google Scholar]
- Edmond J, Robbins RA, Bergstrom JD, Cole RA, & de Vellis J (1987). Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. Journal of Neuroscience Research, 18(4), 551–561. 10.1002/jnr.490180407 [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, … Frosch MP (2006). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiology of Disease, 24(3), 516–524. [DOI] [PubMed] [Google Scholar]
- Gavillet M, Allaman I, & Magistretti PJ (2008). Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia, 56(9), 975–989. 10.1002/glia.20671 [DOI] [PubMed] [Google Scholar]
- Gibbs ME, Gibbs Z, & Hertz L (2009). Rescue of abeta(1–42)-induced memory impairment in day-old chick by facilitation of astrocytic oxidative metabolism: Implications for Alzheimer’s disease. Journal of Neuro-chemistry, 109(Suppl 1), 230–236. 10.1111/j.1471-4159.2009.05800.x [DOI] [PubMed] [Google Scholar]
- Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, … Hartmann T (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nature Cell Biology, 7(11), 1118–1123. [DOI] [PubMed] [Google Scholar]
- Grimm MO, Mett J, Grimm HS, & Hartmann T (2017). APP function and lipids: A bidirectional link. Frontiers in Molecular Neuroscience, 10, 63 10.3389/fnmol.2017.00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm MO, Rothhaar TL, & Hartmann T (2012). The role of APP proteolytic processing in lipid metabolism. Experimental Brain Research, 217(3–4), 365–375. 10.1007/s00221-011-2975-6 [DOI] [PubMed] [Google Scholar]
- Gritti A, Parati EA, Cova L, Frolichsthal P, Galli R, Wanke E, … Vescovi AL (1996). Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. The Journal of neuroscience, 16(3), 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist S, Lehtonen S, Chumarina M, Puttonen KA, Azevedo C, Lebedeva O, … Roybon L (2016). Creation of a library of induced pluripotent stem cells from parkinsonian patients. NPJ Parkinson’s Disease, 2, 16009 10.1038/npjparkd.2016.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias J, Morales L, & Barreto GE (2017). Metabolic and inflammatory adaptation of reactive astrocytes: Role of PPARs. Molecular Neurobiology, 54(4), 2518–2538. 10.1007/s12035-016-9833-2 [DOI] [PubMed] [Google Scholar]
- Iram T, Trudler D, Kain D, Kanner S, Galron R, Vassar R, … Frenkel D (2016). Astrocytes from old Alzheimer’s disease mice are impaired in abeta uptake and in neuroprotection. Neurobiology of Disease, 96, 84–94. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Rudinskiy N, Herrmann AG, Croft S, Kim JM, Petrova V, … Spires-Jones TL (2016). Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer’s disease. The European Journal of Neuroscience, 44(12), 3056–3066. 10.1111/ejn.13442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, … Borchelt DR (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Human Molecular Genetics, 13(2), 159–170. 10.1093/hmg/ddh019 [DOI] [PubMed] [Google Scholar]
- Jernberg JN, Bowman CE, Wolfgang MJ, & Scafidi S (2017). Developmental regulation and localization of carnitine palmitoyltransferases (CPTs) in rat brain. Journal of Neurochemistry, 142(3), 407–419. 10.1111/jnc.14072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji MJ, Yu XB, Mei ZL, An YQ, Tang SS, Hu M, … Hong H (2015). Hippocampal PPARdelta overexpression or activation represses stress-induced depressive behaviors and enhances neurogenesis. The International Journal of Neuropsychopharmacology, 19(1). 10.1093/ijnp/pyv083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, … Hol EM (2012). GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One, 7(8), e42823. 10.1371/journal.pone.0042823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen V, Magga J, Koistinaho J, & Malm T (2012). Brain environment and Alzheimer’s disease mutations affect the survival, migration and differentiation of neural progenitor cells. Current Alzheimer Research, 9(9), 1030–1042. [DOI] [PubMed] [Google Scholar]
- Karkkainen V, Pomeshchik Y, Savchenko E, Dhungana H, Kurronen A, Lehtonen S, … Koistinaho J (2014). Nrf2 regulates neurogenesis and protects neural progenitor cell against abeta toxicity. Stem Cells (Dayton, Ohio), 32(7), 1904–1916. 10.1002/stem.1666 [DOI] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, & Rumbaugh G (2010). Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology, 35(4), 870–880. 10.1038/npp.2009.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, & Ohno M (2009). Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiology of Disease, 33(2), 229–235. 10.1016/j.nbd.2008.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Arauzo-Bravo MJ, … Jessberger S (2013). Metabolic control of adult neural stem cell activity by fasn-dependent lipogenesis. Nature, 493(7431), 226–230. 10.1038/nature11689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Pilz GA, Ghesquiere B, Kovacs WJ, Wegleiter T, Moore DL, … Jessberger S (2017). A fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Reports, 20(9), 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Kim HD, Maxwell JA, & Fukuchi K (2005). Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neuroscience Letters, 390(2), 87–92. [DOI] [PubMed] [Google Scholar]
- Malm T, Mariani M, Donovan LJ, Neilson L, & Landreth GE (2015). Activation of the nuclear receptor PPARdelta is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. Journal of Neuroinflammation, 12, 7 10.1186/s12974-014-0229-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Iivonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Kanninen K, … Koistinaho J (2007). Pyrrolidine dithio-carbamate activates akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. The Journal of Neuroscience, 27(14), 3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani MM, Malm T, Lamb R, Jay TR, Neilson L, Casali B, … Landreth GE (2017). Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Scientific Reports, 7, 42270 10.1038/srep42270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Ihalainen J, Malm T, Matilainen O, Keksa-Goldsteine V, Goldsteins G, … Tanila H (2008). Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. Journal of Neurochemistry, 105(3), 584–594. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Pupi A, & De Leon MJ (2008). Brain glucose hypometabolism and oxidative stress in preclinical alzheimer’s disease. Annals of the New York Academy of Sciences, 1147, 180–195. 10.1196/annals.1427.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou D, Yang H, Qu C, Chen J, & Zhang C (2016). Pharmacological activation of peroxisome proliferator-activated receptor {delta} increases sphingomyelin synthase activity in THP-1 macrophage-derived foam cell. Inflammation, 39(4), 1538–1546. 10.1007/s10753-016-0389-0 [DOI] [PubMed] [Google Scholar]
- Nashold BS Jr., & Goldner JL (1975). Electrical stimulation of peripheral nerves for relief of intractable chronic pain. Medical Instrumentation, 9(5), 224–225. [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, … Vassar R (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. The Journal of neuroscience, 26(40), 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen M, Petersen AJ, Naumenko N, Puttonen K, Lehtonen S, Gubert Olive M, … Koistinaho J (2017). PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Reports, 9(6), 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puli L, Pomeshchik Y, Olas K, Malm T, Koistinaho J, & Tanila H (2012). Effects of human intravenous immunoglobulin on amyloid pathology and neuroinflammation in a mouse model of Alzheimer’s disease. Journal of Neuroinflammation, 9,105 10.1186/1742-2094-9-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, & Weiss S (1992). A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. The Journal of Neuroscience, 12(11), 4565–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnegg CI, Greene-Schloesser D, Kooshki M, Payne VS, Hsu FC, & Robbins ME (2013). The PPARdelta agonist GW0742 inhibits neuroinflammation, but does not restore neurogenesis or prevent early delayed hippocampal-dependent cognitive impairment after whole-brain irradiation. Free Radical Biology & Medicine, 61, 1–9. 10.1016/j.freeradbiomed.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sun X, Sun Y, Hou L, Yao M, Lian K, … Jiang L (2016). Elevation of cortical C26:0 due to the decline of peroxisomal beta-oxidation potentiates amyloid beta generation and spatial memory deficits via oxidative stress in diabetic rats. Neuroscience, 315, 125–135. 10.1016/j.neuroscience.2015.11.067 [DOI] [PubMed] [Google Scholar]
- Silva-Veiga FM, Rachid TL, de Oliveira L, Graus-Nunes F, Mandarim-de-Lacerda CA, & Souza-Mello V (2018). GW0742 (PPAR-beta agonist) attenuates hepatic endoplasmic reticulum stress by improving hepatic energy metabolism in high-fat diet fed mice. Molecular and Cellular Endocrinology, 474, 227–237. [DOI] [PubMed] [Google Scholar]
- Stoll EA, Makin R, Sweet IR, Trevelyan AJ, Miwa S, Horner PJ, & Turnbull DM (2015). Neural stem cells in the adult subventricular zone oxidize fatty acids to produce energy and support neurogenic activity. Stem Cells (Dayton, Ohio), 33(7), 2306–2319. 10.1002/stem.2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, & Alberini CM (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell, 144(5), 810–823. 10.1016/j.cell.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi N, Niidome T, Goto Y, Akaike A, Kihara T, & Sugimoto H (2007). Decreased proliferation of hippocampal progenitor cell in APPswe/PS1dE9 transgenic mice. Neuroreport, 18(17), 1801–1805. 10.1097/WNR.0b013e3282f1c9e9 [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, & Jones JM (1998). Embryonic stem cell lines derived from human blastocysts. Science, 282(5391), 1145–1147. [DOI] [PubMed] [Google Scholar]
- Volianskis A, Kostner R, Molgaard M, Hass S, & Jensen MS (2010). Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1deltaE9-deleted transgenic mice model of ss-amyloidosis. Neurobiology of Aging, 31(7), 1173–1187. 10.1016/j.neurobiolaging.2008.08.005 [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, … Evans RM (2004). Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biology, 2(10), e294. 10.1371/journal.pbio.0020294 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.