

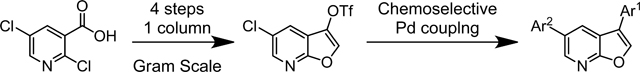
Abstract
A concise 4-step synthesis of furo[2,3-b]pyridines, with handles in the 3- and 5-positions for palladium mediated cross-coupling reactions, is described. The synthetic route has been optimized, with only one step requiring purification by column chromatography. The route is amenable to scale-up, and was successfully executed on a multi-gram scale. Furopyridines are of growing interest in medicinal chemistry, and this route should enable easy access to the core for structure-activity relationship (SAR) studies.
Keywords: Furo[2,3-b]pyridines; Heterocycles; Hinge binders; Kinases; Chemoselectivity
Graphical Abstract
1. Introduction
Protein kinases play an essential role in signal transduction and regulation of cellular activities including metabolism, cell cycle progression, cell differentiation, and apoptosis.1 Deregulation of kinase function has been implicated in cancers, immunological, neurological, metabolic, and infectious diseases.2 Chromosomal mapping of the kinome links 244 kinases to a disease loci or cancer amplicon, emphasizing the potential of therapeutics in this area.3 Kinases are an extremely important group of drug targets, second only to G-protein coupled receptors.4, 5 To date 54 kinase inhibitor have been approved by the FDA and more than 200 are in clinical trials worldwide.6–8
The majority of kinase inhibitors bind to the ATP-binding site, a deep cleft between the N- and C- lobes of the protein’s catalytic domain.4 Most inhibitors bind the active conformation of kinase (type 1), although a significant portion bind to the inactive state (type 2). Other inhibitor types include allosteric inhibitors (type 3), substrate-directed inhibitors (type 4) and covalent inhibitors (type 5).9
There are many heterocyclic hinge binding pharmacophores found in kinase inhibitors, some of which are considered privileged fragments.10, 11 One versatile example is the 7-azaindole core 1 (Fig. 1) which is employed as the hinge binder in vemurafenib (2), an FDA approved serine/threonine-protein kinase B-Raf (B-Raf) inhibitor used in the treatment of melanoma.12 Azaindoles are excellent hinge binders, making two hydrogen bonds with the kinase hinge region. Unfortunately, inhibitors with multiple hinge binding interactions may suffer from poor selectivity across the kinome, leading to off-target toxicity.10, 11, 13
Figure 1:

1 – The azaindole core; 2 – The B-Raf kinase inhibitor vemurafenib; 3 – The furo[2,3-b]pyridine core; 4 – The general structure of the furoquinoline alkaloids isolated from Rutaceae sp.; 5–6: Alkaloids isolated from Sarcomelicope megistophylla - Furomegistine I and II.
One strategy to change a kinase inhibitor’s selectivity profile is to use isosteric replacements, which can elicit changes in binding affinity between closely related kinases.14 There is strong evidence to support that modification of the hydrogen bond interactions between a kinase inhibitor and the enzyme’s hinge region can improve selectivity without sacrificing potency. 11 An isostere of azaindole, the furo[2,3-b]pyridine core (3), has been gaining traction as a hinge-binding template useful for the synthesis of kinase inhibitors. The furopyridine core contains an electron-deficient pyridine ring and an electron-rich furan ring. Furopyridines are relatively uncommon in natural compounds, with the most prominent examples being furochinoline alkaloids (4) isolated from the Rutaceae plant family and furomegistine I and II (5 and 6) alkaloids isolated from the bark of Sarcomelicope megistophylla (Fig. 1).15, 16
Although the furopyridine core is only infrequently found in Nature, it is present in several synthetic drug molecules. One of the earliest reports of the furopyridine core being utilized is in the HIV protease inhibitor L-754,394 (7).17 A recent publication describes a set of substituted furopyridine derivatives based on 8 with activity against multidrug-resistant Mycobacterium tuberculosis (Fig. 2).18 In the kinase field furopyridines have been reported as inhibitors of B-Raf, lymphocyte-specific protein tyrosine kinase (Lck), epidermal growth factor receptor (EGFR) (9), insulin-like growth factor 1 receptor (IGF-1R), and AKT (10).19–23
Figure 2:

7 – The HIV protease inhibitor L-754,394; 8 – A furo[2,3-b]pyridine which has activity against multidrug resistant Mycobacterium tuberculosis; 9 – A furo[2,3-b]pyridine based EGFR inhibitor; 10 – A furo[2,3-b]pyridine based AKT inhibitor.
Despite their interesting chemical and biological activities, there are only a handful of robust synthetic routes to the furopyridine core. Approaches that have been employed in the synthesis of furopyridines are summarized in Scheme 1. Nucleophilic aromatic substitution on 2-halopyridines, followed by subsequent ring closure, has been utilized in several routes to substituted furo[2,3-b]pyridines (Scheme 1 – a), d) and e)).24–27 An intramolecular Diels-Alder reaction between a triazine and alkyne afforded a dihydrofuro[2,3-b]pyridine which was oxidized to the furo[2,3-b]pyridine with DDQ (Scheme 1 – b)).28 There are several examples of palladium catalyzed one-pot syntheses of furopyridines in which Sonogashira couplings were followed by Wacker-type heteroannulations (Scheme 1 – c)).29 The most recent methodology for synthesis of furopyridines was via pyridine N-oxides which yielded 2,3-substituted furo[2,3-b]pyridines (Scheme 1 – f)).30 A comprehensive review on the synthesis of furopyridines has been published by Noravyan and co-workers.31
Scheme 1:

Reported synthetic routes to the furo[2,3-b]pyridine core.
Our interest in the furo[2,3-b]pyridine core was its use as an isosteric hinge binding replacement of a promiscuous azaindole scaffold for the synthesis of a series of kinase inhibitors. We required rapid access on a multigram scale to the furopyridine core that allowed subsequent functionalization at the 3- and 5-positions. Of the synthetic strategies described we decided to utilize the route described by Morita and Shiotani (Scheme 1 – a)).24 Using this methodology and starting with a trisubstituted pyridine such as 12 should give furopyridine product 13 (Scheme 2). We envisioned that a sequence of saponification of the ester in the 2-position followed by decarboxylation would provide 5-chlorofuro[2,3-b]pyridin-3-ol 14. Conversion of the 3-hydroxy functionality into a triflate would provide us with a di-substituted furopyridine core compatible with versatile palladium mediated coupling reactions at the 3- and 5-positions.
Scheme 2:

Initial synthetic route to the furo[2,3-b]pyridine core 15. i) EtOH, H2SO4, 80 °C‚ 16 h, 95%. ii) 1.1 eq. 13, 3.5 eq. NaH, THF, 0–70 °C, 3 h, 88%. iii) 3 eq. KOH, EtOH, 100 °C, 20 min then aq. HCl, 100 °C, 20 min, 0%.
2. Results
2.1. Furo[2,3-b]pyridine core synthesis
Our initial route is described in Scheme 2. 2,5-Dichloronicotinic acid 11 was converted to the ethyl ester 12 under acidic conditions. Ethyl 2-hydroxyacetate 13 was then deprotonated to the nucleophilic alkoxide, which undergoes an SNAr reaction to displace the 2-chloro group of 12 with intramolecular cyclization of the putative intermediate to afford the furo[2,3-b]pyridine 14.
With the furo[2,3-b]pyridine 14 constructed, the hydrolysis and decarboxylation steps were attempted. The conditions described by Morita, aqueous potassium hydroxide in ethanol at reflux, failed to yield any saponified or decarboxylated product with only starting material recovered. Changing the base from potassium to sodium or lithium hydroxide also failed to afford 15 (Table 1 – Entry 1). Increasing the equivalents of base and switching solvent from ethanol to THF, in combination with longer reaction times for both the hydrolysis and acidification steps, yielded the product 15 in moderate yield (46%) (Table 1 – Entry 2). The reaction was attempted under acidic conditions, and also with potassium trimethylsilanolate (Table 1 – Entry 3/4). Although the product was isolated from several of these reaction conditions conversion was not quantitative.
Table 1:
Conditions and yields for the one-pot hydrolysis-decarboxylation reaction.
| Entry | Conditions | Yield |
|---|---|---|
| 1 | i) 3 eq. KOH*, EtOH,100 °C, 20 min, ii) 3 M HCl, 100 °C, 20 min |
0% |
| 2 | i) 10 eq. LiOH, THF, H2O, 60 °C, 16 h ii) 3 M HCl, 100 °C, 1 h |
46% |
| 3 | 6 M HCl, dioxane (1:2), 100 °C, 6 h |
57% |
| 4 | i) KOSiMe3, THF, 60 °C, 16 h, ii) 4 M HCl, 1 h, 100 °C |
63% |
Substituting with NaOH or LiOH afforded the same result.
To further optimise the hydrolysis-decarboxylation reaction, we decided to switch to an acid liable tert-butyl ester, which we envisioned cleaving efficiently with TFA. The carboxylic acid of 2,5-dichloronicotinic acid was converted to the tert-butyl ester using an acid catalyzed dehydration of concentrated sulfuric acid on magnesium sulfate in the presence of tert-butanol, which mediates the formation of isobutylene in situ.32 The conversion of acid 11 to ester 16 proceeded smoothly in excellent yield (92%). A small excess of tert-butyl 2-hydroxyacetate 17 was deprotonated with 3 equivalents of fresh sodium hydride and utilized in the tandem SNAr-cyclisation reaction to afford the furo[2,3-b]pyridine 18 in excellent yield (86%). Gratifyingly, the TFA mediated tert-butyl ester cleavage and decarboxylation afforded the furo[2,3-b]pyridine 15 in excellent yield (89%). Notably, the three steps from 11 to 15 were conducted on gram scale without the need for column chromatography at any stage. Conversion of the alcohol 15 to triflate 19 proceeded smoothly. Triflate 19 was synthesised in 71% yield, with an overall yield of 50% from 11, with only the final step requiring purification via column chromatography.
2.2. Chemoselectivity testing
Generally, aryl triflates are considered to have greater reactivity than aryl chlorides in palladium catalyzed C-C bond formation. There are only a few reported systems with selectivity for aryl chlorides over triflates. The first was published by Fu and co-workers in 2000.33 Fu used Pd2(dba)3 with the bulky tri-tert-butylphosphine ligand PtBu3 in THF to obtain selectivity for an aryl chloride in the presence of an aryl triflate (Scheme 4). Complementarily, switching to Pd(OAc)2 with the smaller tricyclohexylphosphine (PCy3) ligand reversed selectivity favoring reactivity at the triflate. The selectivity was rationalized by the ligation state of palladium. PtBu3 forms a mono-ligated palladium species which favors C-Cl insertion, but the smaller PCy3 forms a bis-ligated species which favors C-OTf insertion.
Scheme 4:

The chemoselective Suzuki-Miyaura cross-coupling conditions developed by Fu and co-workers.33 Chemoselectivity is achieved with the appropriate palladium and ligand combination.
Proutiere and Schoenebeck later found that solvent polarity plays an important role in the selectivity.34 They demonstrated that, with the Pd2(dba)3/PtBu3 system, changing from THF to DMF switched the selectivity from the chloride to the triflate. The nonpolar toluene retained selectivity for the chloride. Most recently, Neufeldt and co-workers used N-heterocycle carbene ligands to achieve chemoselectivity in the Pd-catalyzed Suzuki-Miyaura cross-coupling of chloroaryl triflates.35
Controlling chemoselectivity in our furopyridine derivative would be a valuable asset in our medicinal chemistry efforts, so we set out to investigate conditions that would allow this. Our primary objective was to first substitute the triflate at the 3-position followed by the chloro in the 5-position. Effective conditions to couple aryl triflates had already been utilized in our group using palladium tetrakis (Pd(PPh3)4) and cesium carbonate. The triflate of 19 selectively reacted in a Suzuki reaction using Pd(PPh3)4 to afford the product 23 in 92% yield. The chlorine was subsequently substituted using a system of Pd2(dba)3/XPhos to efficiently afford 24 in 84% yield.
Our initial attempts to achieve selectivity for the chlorine over the triflate used the reported system of Pd2(dba)3/PtBu3 in THF, either at rt or 70 °C. 4-Acetylphenylboronic acid was used as a coupling partner. We envisioned that the acetyl group could cause a moderate change in polarity and greatly simplify separation of products during chromatography, however after 16 hours only a low yield of unreacted starting material was recovered. We next employed toluene and xylene as solvents to run the reaction at higher temperatures. Frustratingly use of either solvent yielded none of the desired product, although <10% of the triflate substituted product was isolated. Additional aliquots of palladium and ligand with longer reaction times did not improve the result. The NHC carbene pre-catalyst PEPPSI™ SIPr which was reported by Neufeldt as selective and high yielding in coupling of aryl chlorides did not prove effective either, with only starting material recovered.
As we could not find conditions that led to reaction at the chlorine instead of the triflate, we opted to make the bromo analogue of compound 19. Aryl bromides are often considered to have similar reactivity to triflates, but we wanted to investigate if selectivity for the bromide could be achieved over the triflate. Following the same route as shown in Scheme 3, the bromo-triflate 26 was synthesized in 62% overall yield.
Scheme 3:

Revised route to the furo[2,3-b]pyridine 19. i) H2SO4, Mg2SO4, tBuOH, CH2Cl2, rt, 16 h, 92%. ii) 1.1 eq. 17, 3.5 eq.NaH, THF, 0–50 °C, 3 h, 86%. iii) TFA, CH2Cl2, rt, 16 h. 89%. iv) Tf2O, DIPEA, CH2Cl2, −10–25 °C, 3 h, 71%.
Initial attempts to couple the bromide utilized Fu’s conditions of Pd2(dba)3 and PtBu3 in THF at room temperature (Scheme 7 and Table 3). The reaction failed to afford product, and only limited starting material was isolated after purification. Increasing the reaction temperature to 70 °C did afford the product 28, but in poor yield (16%) along with small amounts of 27 and 29. Multiple conditions were subsequently explored, affording various mixtures of mono- and di-substituted products. Several conditions reported to selectively couple the triflate over the bromide were also attempted33–35 (Table 3), but all failed to give clean reactions.
Scheme 7:

Couplings of the furo[2,3-b]pyridine 26 afforded mixtures mono-substituted 27 and 28, and di-substituted product 29. The conditions and isolated yields are shown in Table 3.
Table 3:
Conditions attempted to selectively couple the triflate or bromide of 26. Yields were calculated after isolation.
| Entry | System§ | Conditions | Selϕ | 26:27:28:29 |
|---|---|---|---|---|
| 1 | Pd2(dba)3, PtBu3 | THF, KF, rt, 16 h | Br | SM recovered† |
| 2 | Pd2(dba)3, PtBu3 | THF, KF, 70 °C, 16 h | Br | 37%:<5%:16%:<5% |
| 3 | Pd2(dba)3, PtBu3 | PhMe, KF, 110 °C, 16 h | Br | 61%:<5%:14%:<5% |
| 4 | Pd2(dba)3, PtBu3 | Dioxane, KF, 50 °C, 16 h | Br | 63%:<5%:17%:9% |
| 5 | Pd2(dba)3, PtBu3 | Dioxane, KF, 100 °C, 16 h | Br | 12%:9%:29%:15% |
| 6 | Pd2(dba)3, PtBu3 | Xylene, KF, 170 °C, 16 h | Br | 37%:<5%:36%:6% |
| 7 | Pd2(dba)3, PtBu3 | Xylene, KF, 200 °C, μW, 30 min | Br | * |
| 8 | PEPPSI-SiPr | THF, KF, rt, 16 h | Br | SM recovered† |
| 9 | PEPPSI-SiPr | PhMe, KF, 110 °C, 16 h | Br | SM recovered† |
| 10 | Pd(OAc)2/PCy3 | MeCN, KF, rt, 16 h | OTf | SM recovered† |
| 11 | Pd(OAc)2/PCy3 | MeCN, KF, 70 °C, 16 h | OTf | 51%:44%:<5%:<5% |
| 12 | Pd(OAc)2/PCy3 | Dioxane/H2O, KF, 100 °C, 16 h | OTf | 12%:19%:<5%:13% |
| 13 | Pd(PPh3)4 | Dioxane/H2O, KF, rt, 16 h | OTf | 17%:36%:<5%:<5% |
| 14 | Pd(PPh3)4 | Dioxane/H2O KF, 50 °C, 16 h | OTf | 13%:42%:11%:<5% |
| 15 | Pd(PPh3)4 | MeCN, KF, 70 °C, 16 h | OTf | 15%:19%:<5%:<5% |
All reaction were carried out with 5 mol% of catalyst and 10 mol% of ligand.
Predicted selectivity is based on the results in the relevant literature.
These conditions resulted in the precipitation of palladium metal in the microwave vial, causing intense localised heating and shattering of the microwave vial. This happened on two attempts after which safety concerns led us to stop using the microwave reactor for this reaction.
The amount of starting material recovered varied from 0–71%.
3. Discussion
Our initial synthetic route to the furopyridine core 12 was successful, although the hydrolysis-decarboxylation to 14 was not as efficient as anticipated. Under basic conditions, the stability of the ethyl ester 13 could be due to the ability of the deprotonated starting material to chelate with the positively charged metal counter ion, forming a pseudo 6-membered ring (Fig. 3 – 30a/b). This intermediate is analogous to the acetylacetonate anion (31a/b), which is commonly used as bidentate ligand in metal complexes.36 The chelation could hinder nucleophilic attack of hydroxide into the ester carbonyl group and suppress the saponification reaction.
Figure 3:
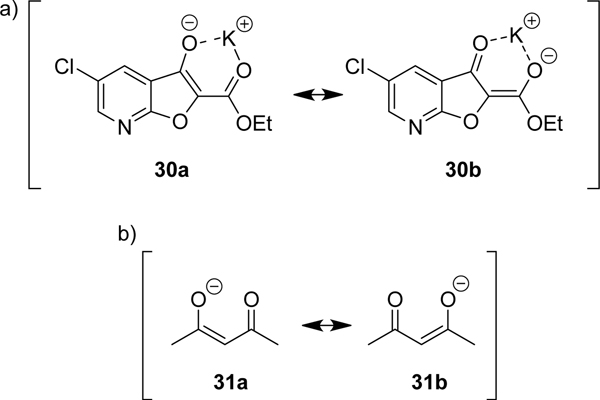
a) The deprotonated species 30 can coordinate a metal counter-ion forming a pseudo 6-membered ring stabilized by resonance forms. This may hinder the nucleophilic attack of hydroxide anions, preventing the saponification of the ester. b) The acetylacetonate (acac−) anion 31 which is often used as a bidentate ligand in inorganic metal complexes.
Switching to acidic hydrolysis conditions did afford product, but longer reaction times led to decomposition of the product and reduction in yield. Potassium trimethylsilanolate proved effective but capricious, with the yield on repeated reactions proving variable. Switching to the acid labile tert-butyl ester proved to be an effective strategy to facilitate the hydrolysis-decarboxylation step, proceeding cleanly in high yield and on multigram scale with TFA. The conversion of the aryl-alcohol to the triflate proceeded smoothly to afford the furo[2,3-b]pyridine with handles in the 3- and 5-positions. The triflate and chloro of 19 could be subjected to Pd-catalyzed coupling reaction sequentially and chemoselectively, which accomplished our primary objective.
Unfortunately, all attempts to switch the chemoselectivity by coupling the chloride of 19 before the triflate proved futile. The chlorine group is positioned at the least reactive carbon of the pyridine ring, hindering the oxidative insertion of the active palladium species. This observation is in line the with predictive model of regioselectivity developed by Handy and Zhang based on the 1H NMR chemical shift values.37 Review of published NMR data and predicted NMR spectrums indicate the 5-position has the smallest 1H chemical shift and therefore is often the least reactive.
Switching intermediates to the more reactive bromine did afford some success, but the yields of bromo-substituted product were not high enough for this to be an effective method to support analog synthesis. This approach also led to problems in the chemoselective coupling of the triflate of 26 over the bromide. Having a 5-chloro instead of a 5-bromo increased our ability to achieve selectivity for the triflate in the 3-position. Conditions which couple the triflate in 19 are indiscriminate when used on the bromo-triflate analogue 26, resulting in non-selective addition to the bromine and triflate. Reducing the temperature from 100 °C to rt or 50 °C, did enhance selectivity of the triflate, but the yields were greatly reduced, leading us back to chloro triflate 19 as our key intermediate.
Literature conditions reported to chemoselectively couple aryl halides and triflates performed poorly, and is likely reflective of these challenging furopyridine substrates, which push the current methodologies for Pd-catalyzed reactions to their limits. Initial attempts to couple the bromine afforded only starting material. Changing to higher boiling non-polar solvents, with accompanying increases in temperature did produce low yields of product. The most successful condition employed xylene at 170 °C, but only afforded the isolated product in 36% yield. Longer reaction times may have improved the yield, but running reactions at 170 °C for over 24 hours did not meet our goal of a practical synthesis.
In conclusion, we successfully developed a 4-step high yielding and scalable synthesis of chloro-triflate furopyridine 19, requiring only one chromatographic purification. The route is, to the best of our knowledge, the most efficient way to access a furo[2,3-b]pyridine core containing synthetic handles for further derivatization. This intermediate has allowed us to undertake a large kinase medicinal chemistry program, the results of which will be published soon.
Supplementary Material
Scheme 5:

Coupling reaction of 19. i) Pd(PPh3)4 (5 mol%), Cs2CO3, dioxane/water (3:1), 100 °C, 16 h, 92%. ii) 4-methylphenylboronic acid, Pd2(dba)3 (5 mol%), XPhos (10 mol%), Cs2CO3, dioxane/water (3:1), 100 °C, 16 h, 84%.
Scheme 6:

Attempted chemoselective Suzuki coupling of the chloride of 19. Reaction conditions are listed in Table 2.
Table 2:
Conditions utilized towards the chemoselective coupling of furo[2,3-b]pyridine 19.
| Conditions | Outcome |
|---|---|
| Pd2(dba)3 (5 mol%), PtBu3 (10 mol%), KF, THF, 70 °C, 16 h | SM recovered† |
| Pd2(dba)3 (5 mol%), PtBu3 (10 mol%), KF, PhMe, 110 °C, 16 h | SM recovered† |
| Pd2(dba)3 (5 mol%), PtBu3 (10 mol%), KF, Xylene, 170 °C, 16 h | SM recovered† |
| PEPPSI-SiPr (5 mol%), KF, PhMe, 110 °C, 16 h | SM recovered† |
Amounts of starting material recovered varied from 0–48%.
Acknowledgements
We thank Brandie M Ehrmann and the UNC Department of Chemistry Mass Spectrometry Core Laboratory for collecting HR-MS data. We thank Christopher R.M Asquith for valuable input and reviewing of the manuscript.
This research was funded in part by the National Cancer Institute of the National Institutes of Health (D.H.D.) grant number R01CA218442 and also by the NIH Illuminating the Druggable Genome program (TMW and DHD) grant number 1U24DK116204-01. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome [106169/ZZ14/Z].,
Footnotes
Supplementary data
Supplementary data associated with this article can be found in the online version and contains experimental procedures and characterization data for all new compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Zhang J; Yang PL; Gray NS Nat. Rev. Cancer, 2009, 9, 28. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson FM; Gray NS Nat. Rev. Drug Discov, 2018, 17, 353. [DOI] [PubMed] [Google Scholar]
- 3.Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S Science 2002, 298, 1912. [DOI] [PubMed] [Google Scholar]
- 4.Cohen P Nat. Rev. Drug Discov, 2002, 1, 309. [DOI] [PubMed] [Google Scholar]
- 5.Sriram K; Insel PA Molecular Pharmacology, 2018, 93, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roskoski R Pharmacological Research, 2019, 144, 19. [DOI] [PubMed] [Google Scholar]
- 7.Compiled by Roskoski, R. J.; available online from http://www.brimr.org/PKI/PKIs.htm
- 8.Carles F; Bourg S; Meyer C; Bonnet P Molecules, 2018, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhullar KS; Lagarón NO; McGowan EM; Parmar I; Jha A; Hubbard BP; Rupasinghe HP V. Mol. Cancer, 2018, 17, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghose AK; Herbertz T; Pippin DA; Salvino JM; Mallamo JP J. Med. Chem, 2008, 51, 5149. [DOI] [PubMed] [Google Scholar]
- 11.Xing L; Klug-Mcleod J; Rai B; Lunney EA Bioorg. Med. Chem, 2015, 23, 6520. [DOI] [PubMed] [Google Scholar]
- 12.Bollag G; Tsai J; Zhang J; Zhang C; Ibrahim P; Nolop K; Hirth P Nat. Rev. Drug Discov, 2012, 11, 873. [DOI] [PubMed] [Google Scholar]
- 13.Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP Nat. Biotech, 2011, 29, 1046. [DOI] [PubMed] [Google Scholar]
- 14.Meanwell NA J. Med. Chem, 2011, 54, 2529. [DOI] [PubMed] [Google Scholar]
- 15.Aldona A-S; Kazimierz G; Tomasz B Current Issues in Pharmacy and Medical Sciences 2016, 29, 33. [Google Scholar]
- 16.Fokialakis N; Magiatis P; Aligiannis N; Mitaku S; Tillequin F; Sévenet T Phytochemistry 2001, 57, 593. [DOI] [PubMed] [Google Scholar]
- 17.Vacca JP; Dorsey BD; Schleif WA; Levin RB; McDaniel SL; Darke PL; Zugay J; Quintero JC; Blahy OM; Roth EP Nat. A Sci, 1994, 91, 4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fumagalli F; de Melo SMG; Ribeiro CM; Solcia MC; Pavan FR; da Silva Emery F Bioorg. Med. Chem. Lett, 2019, 29, 974. [DOI] [PubMed] [Google Scholar]
- 19.Buckmelter AJ; Ren L; Laird ER; Rast B; Miknis G; Wenglowsky S; Schlachter S; Welch M; Tarlton E; Grina J; Lyssikatos J; Brandhuber BJ; Morales T; Randolph N; Vigers G; Martinson M; Callejo M Bioorg. Med. Chem. Lett, 2011, 21, 1248. [DOI] [PubMed] [Google Scholar]
- 20.Ren L; Wenglowsky S; Miknis G; Rast B; Buckmelter AJ; Ely RJ; Schlachter S; Laird ER; Randolph N; Callejo M; Martinson M; Galbraith S; Brandhuber BJ; Vigers G; Morales T; Voegtli WC; Lyssikatos J Bioorg. Med. Chem. Lett, 2011, 21, 1243. [DOI] [PubMed] [Google Scholar]
- 21.Wu Z; Robinson RG; Fu S; Barnett SF; Defeo-Jones D; Jones RE; Kral AM; Huber HE; Kohl NE; Hartman GD; Bilodeau MT Bioorg. Med. Chem. Lett, 2008, 18, 2211. [DOI] [PubMed] [Google Scholar]
- 22.Martin MW; Newcomb J; Nunes JJ; Bemis JE; McGowan DC; White RD; Buchanan JL; DiMauro EF; Boucher C; Faust T; Hsieh F; Huang X; Lee JH; Schneider S; Turci SM; Zhu X Bioorg. Med. Chem. Lett, 2007, 17, 2299. [DOI] [PubMed] [Google Scholar]
- 23.Hempel C; Najjar A; Totzke F; Schächtele C; Sippl W; Ritter C; Hilgeroth A Med. Chem. Comm, 2016, 7, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morita H; Shiotani SJ Het. Chem, 1986, 23, 1465. [Google Scholar]
- 25.Cailly T; Lemaître S; Fabis F; Rault S Synthesis 2007, 2007, 3247. [Google Scholar]
- 26.Cartwright MW; Parks EL; Pattison G; Slater R; Sandford G; Wilson I; Yufit DS; Howard JAK; Christopher JA; Miller DD Tetrahedron 2010, 66, 3222. [Google Scholar]
- 27.Jasselin-Hinschberger A; Comoy C; Chartoire A; Fort Y Eur. J. Org.Chem, 2015, 2015, 2321. [Google Scholar]
- 28.Taylor EC; Macor JE Tetrahedron Lett, 1986, 27, 431. [Google Scholar]
- 29.Eastman KJ; Parcella K; Yeung K-S; Grant-Young KA; Zhu J; Wang T; Zhang Z; Yin Z; Beno BR; Sheriff S; Kish K; Tredup J; Jardel AG; Halan V; Ghosh K; Parker D; Mosure K; Fang H; Wang Y-K; Lemm J; Zhuo X; Hanumegowda U; Rigat K; Donoso M; Tuttle M; Zvyaga T; Haarhoff Z; Meanwell NA; Soars MG; Roberts SB; Kadow JF Med. Chem. Comm 2017, 8, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fumagalli F; da Silva Emery FJ Org. Chem, 2016, 81, 10339. [DOI] [PubMed] [Google Scholar]
- 31.Sirakanyan SN; Hovakimyan AA; Noravyan AS Russ. Chem. Rev, 2015, 84, 441. [Google Scholar]
- 32.Wright SW; Hageman DL; Wright AS; McClure LD Tetrahedron Lett. 1997, 38, 7345. [Google Scholar]
- 33.Littke AF; Dai C; Fu GC J. Am. Chem. Soc, 2000, 122, 4020. [Google Scholar]
- 34.Proutiere F; Schoenebeck F Angew. Chem. Int. Ed, 2011, 50, 8192. [DOI] [PubMed] [Google Scholar]
- 35.Reeves EK; Humke JN; Neufeldt SR J. Org.Chem, 2019, 84, 11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manbeck KA; Boaz NC; Bair NC; Sanders AMS; Marsh AL J. Chem. Ed, 2011, 88, 1444. [Google Scholar]
- 37.Handy ST; Zhang Y Chem. Comm, 2006, 299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.