

Abstract
Osteoclasts are hematopoietic-derived cells that resorb bone. They are required to maintain proper bone homeostasis and skeletal strength. Although osteoclast differentiation depends on receptor activator of NFκB ligand (RANKL) stimulation, additional molecules further contribute to osteoclast maturation. Here, we demonstrate that protocadherin-7 (Pcdh7) regulates formation of multinucleated osteoclasts and contributes to maintenance of bone homeostasis. We found that Pcdh7 expression is induced by RANKL stimulation, and that RNAi-mediated knockdown of Pcdh7 resulted in impaired formation of osteoclasts. We generated Pcdh7-deficient mice and found increased bone mass due to decreased bone resorption but without any defect in bone formation. Using an in vitro culture system, it was revealed that formation of multinucleated osteoclasts is impaired in Pcdh7-deficient cultures, while no apparent defects were observed in differentiation and function of Pcdh7-deficient osteoblasts. Taken together, these results reveal an osteoclast cell-intrinsic role for Pcdh7 in maintaining bone homeostasis.
Keywords: Bone homeostasis, Maturation, Multinucleation, Osteo-clast, Pcdh7
INTRODUCTION
Skeletal homeostasis is maintained via continuous bone formation mediated by osteoblasts and bone destruction mediated by osteoclasts (1). Under pathogenic conditions, excessive osteoclast activity is often observed, which can cause pathogenic bone loss such as is observed in osteoporosis, periodontitis, and rheumatoid arthritis (2-4). Therefore, understanding the cellular and molecular mechanisms that regulate osteoclast differentiation and function will contribute to our knowledge of osteoclast biology, and further provide molecular bases for designing therapeutic strategies for bone diseases that are accompanied by bone destruction.
Osteoclasts are specialized multinucleated giant cells that originate from myeloid progenitors (5). Differentiation of osteoclasts is initiated primarily by the osteoclast differentiation factor RANKL (6), but the process further requires cell surface molecules to mediate cell adhesion and promote osteoclast multinucleation (7), which is itself a hallmark of osteoclast maturation (8). One of the principal factors at sites of cell-cell contact are cadherins. Cadherins are transmembrane, calcium-dependent adhesion molecules that regulate cell-cell contact through homophilic or heterophilic interactions between compatible cadherin extracellular regions (9, 10). The cadherin superfamily consists of classical cadherins, desmosomal cadherins, and protocadherins.
Previously, we reported comparative gene expression profiling designed to identify potential genes that regulate osteoclast maturation through use of multinucleation as a functional readout (8). As a result, the gene Pcdh7, which encodes the protein protocadherin 7 (Pcdh7) was identified (Supplementary Fig. 1). Pcdh7 is a member of non-clustered protocadherin d1 subgroup of the cadherin superfamily (11, 12). It has seven cadherin-like ectodomains, a single transmembrane domain and a C-terminal intracellular domain (13). The expression of Pcdh7 has been identified in osteoclasts, and a potential regulatory role in osteoclast multinucleation has been revealed by RNAi-mediated gene knockdown (14). However, the function of Pcdh7 in regulation of osteoclast differentiation in vivo and its role in bone homeostasis still remain unclear.
Here, we demonstrate that Pcdh7, expression of which is induced by RANKL stimulation, functions as a regulator of bone homeostasis through regulation of osteoclast multinucle-ation. Gene deletion of Pcdh7 resulted in increased bone mass with reduction of osteoclasts but not of osteoblasts in mice. No apparent defects in bone formation were observed. Using in vitro cell culture systems, we showed that Pcdh7 deficiency results in impaired formation of multinucleated osteoclasts, and that the defect could be restored by retroviral transduction of Pcdh7. By contrast, Pcdh7 deficiency did not show significant differences in differentiation or function of osteoblasts. Collectively, these results demonstrate that Pcdh7 controls osteoclast multinucleation/maturation and contributes to maintenance of bone homeostasis.
RESULTS
Pcdh7 is involved in in vitro osteoclast differentiation
In order to understand the role of Pcdh7 in osteoclasts, expression dynamics of Pcdh7 during osteoclast differentiation were examined. Mouse bone marrow-derived monocytes (BMMs) were treated with M-CSF + RANKL for up to three days to induce osteoclasts, and then western blotting was performed. Expression of PCDH7 protein was induced, and peaked on day one of culture (Fig. 1A). Next, we investigated the role of Pcdh7 in osteoclasts by RNAi experiments using retrovirus encoding shRNA for Pcdh7. BMMs retrovirally transduced with shRNA for Pcdh7 showed significant reductions in tartrate-resistant acidic phosphatase positive (TRAP+) multinucleated cells (i.e., mature osteoclasts) (Fig. 1B), consis-tent with a previous report (14). These results suggested a potential regulatory function of Pcdh7 in osteoclast maturation.
Fig. 1.
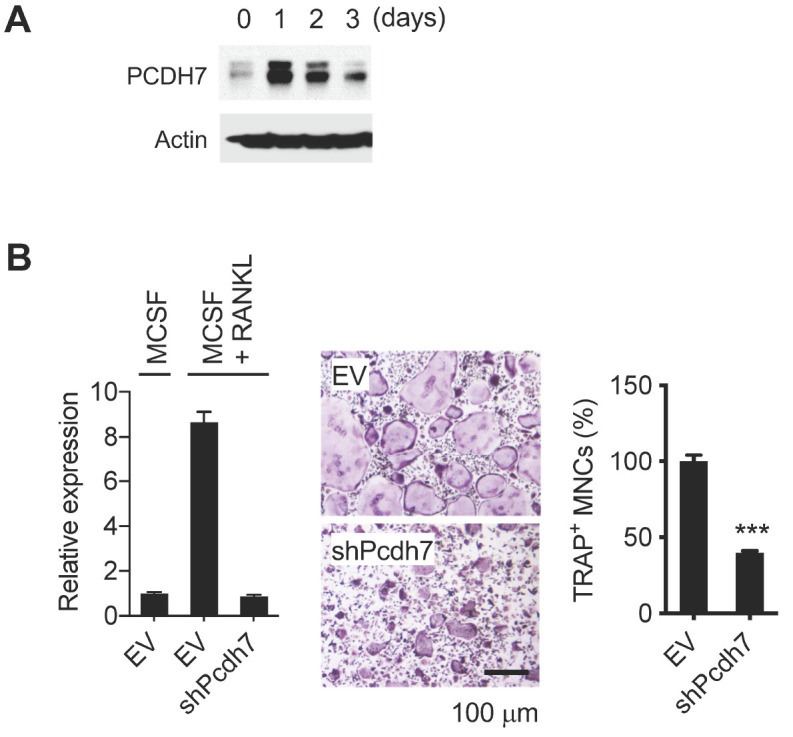
Identification of Pcdh7 as an osteoclast differentiation associated gene. (A) PCDH7 protein expression during osteoclast differentiation. Total cell lysates were prepared from BMMs cultured with M-CSF + RANKL for the indicated days and used for western blotting with the indicated antibodies. (B) Effect of Pcdh7 RNAi on osteoclast differentiation. BMMs retrovirally transduced with the indicated shRNAs were cultured with M-CSF + RANKL for three days. Relative expression of Pcdh7 was determined by Q-PCR (left). Cells were stained for TRAP (middle). Frequency of TRAP+ multinucleated cells is shown (right). Scale bar represents 100 µm.
Pcdh7 regulates bone homeostasis
To better understand the role of Pcdh7 in osteoclasts and its function in bone remodeling, Pcdh7-deficient (Pcdh7−/−) mice were generated using clustered regularly interspersed short palindromic repeats (CRISPR)/Cas9 technology (Supplementary Fig. 2A). Gene knockout mice carrying deletions of 14 base pairs (bps) in exon 1 of Pcdh7 were obtained (Supplementary Fig. 2B). We verified mouse genotypes by genome sequencing, and also confirmed successful gene deletion of Pcdh7 in Pcdh7−/− cultures by western blotting (Supplementary Fig. 2C).
In order to examine the impact of Pcdh7 deficiency on bone homeostasis, gender- and age-matched Pcdh7+/+ and Pcdh7−/− mice were prepared and representative 3D trabecular bone reconstruction analyses were performed. Bone microstructure imaging by high-resolution microcomputed tomography (µCT) of Pcdh7−/− mice revealed increases in bone mass, characterized by augmented bone indices including trabecular bone volume per tissue volume (BV/TV) and trabecular thickness (Tb.Th), with concomitant decreases in trabecular spacing (Tb.Sp) (Fig. 2A). Trabecular number (Tb.N) was comparable between Pcdh7+/+ and Pcdh7−/− mice (Fig. 2A). There were no significant differences in cortical bone thickness (Ct.Th) between Pcdh7+/+ and Pcdh7−/− mice (Fig. 2B). Dynamic histomorphometry by sequential injection of calcein and xylenol orange revealed that bone formation rates is normal in Pcdh7−/− mice (Fig. 2C). Osteoclasts and osteoblasts in Pcdh7+/+ and Pcdh7−/− mice were enumerated using bone sections. TRAP-stained bone sections exhibited a reduction in osteoclast numbers on the bone surface (N.Oc/BS) in Pcdh7−/− bone compared to numbers in Pcdh7+/+ (Fig. 2D). By contrast, quantitation of osteoblast numbers per bone surface (N.Ob/BS) revealed comparable numbers of osteoblasts in bone sections between Pcdh7+/+ and Pcdh7−/− mice (Fig. 2D). These results suggest that Pcdh7 deficiency results in increased bone mass with impaired osteoclast differentiation but apparently normal osteoblast development and function in male mice.
Fig. 2.

Pcdh7 deficiency results in increased bone mass in mice. (A) Microcomputed tomography (µCT) images of femurs from Pcdh7+/+ and Pcdh7−/− mice. The femurs of 12-week-old male mice were analyzed. Bone volume per tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular spacing (Tb.Sp) are shown. (B) Microcomputed tomography (µCT) images of femurs cortical thickness (Ct.Th) from 12-week-old male Pcdh7+/+ and Pcdh7−/− mice is shown. (C) Dynamic histomorphometry of tibias from 12-week-old Pcdh7+/+ and Pcdh7−/− mice. Mineral apposition rate (MAR) and bone formation (BFR) are shown. Scale bar represents 50 µm. (D) Histological analysis of tibias from 12-week-old Pcdh7+/+ and Pcdh7−/− mice. Tibial sections were stained with TRAP or H&E. Osteoclast number per bone surface (N.Oc/BS) and osteoblast number per bone surface (N.Ob/BS) are shown. Data are shown as mean ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
Cell-intrinsic role of Pcdh7 in osteoclast differentiation
To further characterize the specific cell-intrinsic role of Pcdh7, osteoclasts and osteoblasts were generated in vitro, and differentiation and cellular functions were examined. First, expression levels of Pcdh7 message in osteoclasts and osteoblasts were examined. Pcdh7+/+ BMMs or bone marrow-derived stromal cells (BMSCs) were cultured, respectively, with M-CSF + RANKL to generate osteoclasts, or with glycerophosphate + ascorbic acid to generate osteoblasts. Pcdh7 message was significantly increased in osteoclasts cultures compared to levels in BMMs, while Pcdh7 message levels were lower in BMSCs or BMSC-derived osteoblasts than in BMMs (Fig. 3A). Next, osteoclasts were generated from Pcdh7+/+ and Pcdh7−/− BMMs with M-CSF + RANKL. Significant reductions in the frequency of multinucleated (>3 nuclei per cell) TRAP+ cells in Pcdh7−/− cultures were observed compared to Pcdh7+/+ cultures (Fig. 3B). Message levels of osteoclast differentiation markers, Acp5 and Ctsk, were comparable between Pcdh7+/+ and Pcdh7−/− cultures (Fig. 3C). Formation of pit area per mature osteoclasts was slightly reduced in Pcdh7−/− cultures (Fig. 3D). These results suggest that Pcdh7 is required for osteoclast multinucleation rather than for differentiation or bone resorbing functions. Retroviral transduction of Pcdh7−/− BMMs with full-length Pcdh7 completely rescued TRAP+ multinucleated cell formation in Pcdh7−/− cultures (Fig. 3E), suggesting that the phenotypes observed in Pcdh7−/− osteoclast cultures can be solely attributed to deletion of the Pcdh7 gene.
Fig. 3.

Pcdh7 deficiency results in impaired osteoclast differentiation but not osteoblast differentiation or function. (A) Relative expression of Pcdh7 in BMMs, osteoclasts (three days culture with M-CSF + RANKL), BMSCs, and osteoblasts (14 days culture with glycerophosphate + ascorbic acid). Total RNAs were collected and expression of Pcdh7 was measured by Q-PCR. (B) Osteoclast differentiation of Pcdh7+/+ and Pcdh7−/− cells. BMMs were cultured with M-CSF + RANKL for three days. Frequency of TRAP+ multinucleated cells (3 nuclei or more per cell) and hyper-multinucleated cells (>100 µm) are shown. (C) Gene expression during osteoclast differentiation. Total RNAs were collected from Pcdh7+/+ and Pcdh7−/− cultures and indicated genes were measured by Q-PCR. (D) Bone resorption activity of Pcdh7+/+ and Pcdh7−/− osteoclasts. Resorption area per cell is shown. (E) Osteoclast differentiation rescued by retroviral transduction of Pcdh7 in Pcdh7−/− BMMs. BMMs were retrovirally transduced with empty vector (EV) or Flag-tagged Pcdh7 expression vector followed by culture with M-CSF + RANKL for three days. Frequency of TRAP+ multinucleated cells (3 nuclei or more per cell) is shown. Expression of exogeneous Pcdh7 was confirmed by western blotting with anti-Flag antibody. (F) Pcdh7+/+ and Pcdh7−/− BMSCs were cultured with osteogenic medium for the indicated days, and then stained with alizarin red. (G) Gene expression during osteoblast differentiation. Total RNAs were collected from Pcdh7+/+ and Pcdh7−/− cultures and indicated genes were measured by Q-PCR. (H) Pcdh7+/+ and Pcdh7−/− BMMs and BMSC-derived osteoblasts were cocultured in the presence of 1,25-dihydroxyvitamin D3 and prostaglandin E2, and the formation of multinucleated osteoclasts was determined by TRAP staining. Frequency of TRAP+ multinucleated cells (3 nuclei or more per cell) are shown. Data are shown as mean ± S.D. *P < 0.05, ***P < 0.001.
Next, osteoblasts were generated from Pcdh7+/+ and Pcdh7−/− BMSCs with glycerophosphate + ascorbic acid. In vitro osteo-genic differentiation assays with alizarin red staining revealed no differences in terms of mineralized nodule formation and calcium deposits in Pcdh7+/+ and Pcdh7−/− osteoblasts (Fig. 3F). Message levels of osteoblast differentiation markers such as Alpl, osteocalcin (also known as Bglap), and Bmp2 were also comparable between Pcdh7+/+ and Pcdh7−/− cells (Fig. 3G). Additionally, when BMSC-derived osteoblasts were co-cultured with BMMs in the presence of 1,25-dihydroxyvitamin D3 + prostaglandin E2, Pcdh7−/− osteoblasts promoted the formation of Pcdh7+/+ osteoclasts to the same extent as Pcdh7+/+ osteoblasts (Fig. 3H). These results suggest that Pcdh7 is not required for osteoblast differentiation or function. Taken together, these results clarify the role of Pcdh7 in bone homeostasis as a regulator of osteoclast multinucleation/maturation, but not of osteoblast differentiation or function.
DISCUSSION
Cell-cell interaction through surface receptors is required for proper osteoclast differentiation and multinucleation (7, 15-17). In this study, we demonstrated that Pcdh7, a transmembrane protein belonging to the protocadherins within the cadherin superfamily, is required for proper bone homeostasis through regulation of osteoclast multinucleation/maturation (Fig. 4). We generated Pcdh7 gene deletion mice and revealed that Pcdh7 deficiency resulted in increased bone mass without affecting bone formation in male mice. Determining whether the requirement for Pcdh7 for proper bone homeostasis is influenced by sexual difference will need further investigation. Employing an in vitro culture system using Pcdh7-deficient cells further revealed a requirement for Pcdh7 in osteoclast multinucleation, while Pcdh7 deficiency resulted in no significant impact on osteoblast differentiation or function. A previous report identified Pcdh7 as a gene that is epigenetically regulated during osteoclastogenesis, and implied a role for Pcdh7 in osteoclast multinucleation (14). Our findings reported here are consistent with those previously reported findings with respect to the role of Pcdh7 in osteoclast multinucleation, and provide the first in vivo evidence of involvement of Pcdh7 in bone homeostasis.
Fig. 4.

A summarizing diagram. Deficiency of Pcdh7 results in in-creased bone mass with reduction of osteoclasts but not of osteo-blasts in mice.
We have identified an osteoclast cell-intrinsic role of Pcdh7. However, the principal function and molecular mechanism of Pcdh7 in the context of osteoclast differentiation/multinucleation still remain to be determined. Cadherins function by mediating cell-cell adhesion through homophilic interactions, whereas protocadherins appear to have more varied physiological functions as mediators of both cell-cell adhesion and as regulators of signaling molecules (12, 18, 19). Indeed, in addition to exhibiting binding capacity via homophilic interactions (20), Pcdh7 has been shown to control signal transduction pathways via binding cytoplasmic signaling molecules such as SET (also known as TAF1) and PP2a (21). Involvement of PP2a in osteoclasts has been shown using a chemical inhibitor (22), and the findings suggest that Pcdh7 regulates PP2a during osteoclast differentiation. There is another possibility that the Pcdh7 intracellular region recruits additional signaling molecules. Pcdh7 has been shown to express four unique isoforms with different cytoplasmic tails that are generated by alternative splicing (23). It is unclear which isoform(s) is expressed in osteoclasts, and whether the different isoforms have functional differences. Further studies will be required to understand the molecular mechanisms of Pcdh7-mediated regulation of osteoclast maturation.
Bone remodeling is tightly controlled by coupling between bone resorbing osteoclasts and bone forming osteoblasts (24, 25). Coupling is critical for preserving bone architecture and strength. Current treatments for bone loss such as bisphosphonate and anti-RANKL antibody (Denosmab), which primarily target early osteoclast commitment and/or viability (26), often fail to uncouple bone degradation and formation. Consequently, bone strength is compromised due to unintended inhibition of coupled bone formation (27). In this study, we report having identified Pcdh7 in the process of searching for genes related to the maturation/late stage of osteoclast development, which we believe will lead to better treatment targets (i.e., inhibition of osteoclastic bone resorption without preventing osteoblastic new bone formation), and we have shown that Pcdh7 uncouples bone degradation and formation. Our results suggest that Pcdh7 might be a good candidate target for selectively inhibiting bone loss.
Taken together, although a full understanding of how Pcdh7 contributes to osteoclast differentiation remains to be addressed, we provide evidence of a non-redundant function for Pcdh7 in osteoclast multinucleation/maturation and bone homeostasis. Pcdh7 may be a promising candidate for a therapeutic target for bone diseases.
MATERIALS AND METHODS
Detailed information is provided in the Supplementary Information.
Supplemental Materials
ACKNOWLEDGEMENTS
This work was supported in part by the Penn Center for Musculoskeletal Disorders Histology Core (NIH P30-AR069619).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- 2.Moutsopoulos NM, Konkel J, Sarmadi M, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 2014;6:229ra40. doi: 10.1126/scitranslmed.3007696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Souza PP, Lerner UH. The role of cytokines in inflammatory bone loss. Immunol Invest. 2013;42:555–622. doi: 10.3109/08820139.2013.822766. [DOI] [PubMed] [Google Scholar]
- 4.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 5.Walsh MC, Kim N, Kadono Y, et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol. 2006;24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646. [DOI] [PubMed] [Google Scholar]
- 6.Boyle JW, Simonet SW, Lacey LD. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 7.Aguilar PS, Baylies MK, Fleissner A, et al. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013;29:427–437. doi: 10.1016/j.tig.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H, Walsh MC, Takegahara N, et al. The purinergic receptor P2X5 regulates inflammasome activity and hyper-multinucleation of murine osteoclasts. Sci Rep. 2017;7:196. doi: 10.1038/s41598-017-00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeichi M. Dynamic contacts: rearranging adherens junctions to drive epithelial remodelling. Nat Rev Mol Cell Biol. 2014;15:397–410. doi: 10.1038/nrm3802. [DOI] [PubMed] [Google Scholar]
- 10.Leckband DE, de Rooij J. Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol. 2014;30:291–315. doi: 10.1146/annurev-cellbio-100913-013212. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi S, Takeichi M. Emerging roles of protocadherins: from self-avoidance to enhancement of motility. J Cell Sci. 2015;128:1455–1464. doi: 10.1242/jcs.166306. [DOI] [PubMed] [Google Scholar]
- 12.Kim SY, Yasuda S, Tanaka H, Yamagata K, Kim H. Non-clustered protocadherin. Cell Adh Migr. 2011;5:97–105. doi: 10.4161/cam.5.2.14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peek SL, Mah KM, Weiner JA. Regulation of neural circuit formation by protocadherins. Cell Mol Life Sci. 2017;74:4133–4157. doi: 10.1007/s00018-017-2572-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura H, Nakashima T, Hayashi M, et al. Global epigenomic analysis indicates protocadherin-7 activates osteoclastogenesis by promoting cell-cell fusion. Biochem Biophys Res Commun. 2014;455:305–311. doi: 10.1016/j.bbrc.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Helming L, Gordon S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009;19:514–522. doi: 10.1016/j.tcb.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Oren-Suissa M, Podbilewicz B. Cell fusion during development. Trends Cell Biol. 2007;17:537–546. doi: 10.1016/j.tcb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Kim H, Takegahara N, Walsh MC, et al. IgSF11 regulates osteoclast differentiation through association with the scaffold protein PSD-95. Bone Res. 2020;8:5. doi: 10.1038/s41413-019-0080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahr I, Vandepoele K, van Roy F. Delta-proto-cadherins in health and disease. Prog Mol Biol Transl Sci. 2013;116:169–192. doi: 10.1016/B978-0-12-394311-8.00008-X. [DOI] [PubMed] [Google Scholar]
- 19.Heggem M, Bradley R. The cytoplasmic domain of Xenopus NF-protocadherin interacts with TAF1/set. Dev Cell. 2003;4:419–429. doi: 10.1016/S1534-5807(03)00036-4. [DOI] [PubMed] [Google Scholar]
- 20.Bradley R, Espeseth A, Kintner C. NF-protocadherin, a novel member of the cadherin superfamily, is required for Xenopus ectodermal differentiation. Curr Biol. 1998;8:325–334. doi: 10.1016/S0960-9822(98)70132-0. [DOI] [PubMed] [Google Scholar]
- 21.Zhou X, Updegraff BL, Guo Y, et al. PROTOCA DHERIN 7 Acts through SET and PP2A to Potentiate MAPK Signaling by EGFR and KRAS during Lung Tumorigenesis. Cancer Res. 2017;77:187–197. doi: 10.1158/0008-5472.CAN-16-1267-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Guo X, Zhou W, et al. Protein phosphatase 2A as a new target for downregulating osteoclastogenesis and alleviating titanium particle-induced bone resorption. Acta Biomater. 2018;73:488–499. doi: 10.1016/j.actbio.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Vanhalst K, Kools P, Staes K, van Roy F, Redies C. delta-Protocadherins: a gene family expressed differentially in the mouse brain. Cell Mol Life Sci. 2005;62:1247–1259. doi: 10.1007/s00018-005-5021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- 25.Matsuo K, Irie N. Osteoclast-osteoblast communication. Arch Biochem Biophys. 2008;473:201–209. doi: 10.1016/j.abb.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 26.Miyazaki T, Tokimura F, Tanaka S. A review of denosumab for the treatment of osteoporosis. Patient Prefer Adherence. 2014;8:463–471. doi: 10.2147/PPA.S46192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone. 2011;48:677–692. doi: 10.1016/j.bone.2010.11.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.