

ABSTRACT
Anaplerosis and the associated mitochondrial metabolite transporters generate unique cytosolic metabolic signaling molecules that can regulate insulin release from pancreatic β-cells. It has been shown that mitochondrial metabolites, transported by the citrate carrier (CIC), dicarboxylate carrier (DIC), oxoglutarate carrier (OGC), and mitochondrial pyruvate carrier (MPC) play a vital role in the regulation of glucose-stimulated insulin secretion (GSIS). Metabolomic studies on static and biphasic insulin secretion, suggests that several anaplerotic derived metabolites, including α-ketoglutarate (αKG), are strongly associated with nutrient regulated insulin secretion. Support for a role of αKG in the regulation of insulin secretion comes from studies looking at αKG dependent enzymes, including hypoxia-inducible factor-prolyl hydroxylases (PHDs) in clonal β-cells, and rodent and human islets. This review will focus on the possible link between defective anaplerotic-derived αKG, PHDs, and the development of type 2 diabetes (T2D).
KEYWORDS: Insulin, islets, alpha-ketoglutarate, hypoxia-inducible factor-prolyl hydroxylases
Introduction
Pancreatic β-cell GSIS involves a rise in the ATP:ADP ratio, which leads to closure of ATP-sensitive K+ (KATP) channels, resulting in plasma membrane depolarization, activation of voltage-gated Ca2+ channels (VDCC) and Ca2+-mediated stimulation of insulin granule exocytosis.1–4 Although there are limitations to studying insulin secretion using square wave glucose stimulation assays in β-cells, most studies support the concept that there is a “KATP channel-dependent” mechanism of insulin granule exocytosis. This KATP channel-dependent pathway appears to be particularly important in triggering exocytosis of a small number of granules from a plasma membrane-docked “readily releasable pool” which is responsible for the first, acute phase of insulin release (first 10 minutes of release after stimulation).5,6 In contrast, in the second and sustained phase of insulin secretion (>10 minutes), ATP and Ca2+, along with other glucose-derived second messengers, play an important role.5–7 It is accepted that elevated cytosolic Ca2+ leads to stimulation of exocytosis of insulin granules,8,9 but the Ca2+ signal alone is not the only factor required for sustained secretion since membrane depolarization with high KCl, which leads to a sustained elevation of Ca2+, does not lead to biphasic insulin secretion.10 Under clamped cytosolic Ca2+ concentrations, the addition of glucose can still augment insulin secretion.11 Also, it has been shown that mice lacking KATP channel function retain the ability to secrete insulin and have relatively normal glucose homeostasis.12–17 For example, prior exposure of SUR1 knockout mouse islets to low glucose allows them to exhibit a six-fold increase in insulin secretion in response to 15 mM glucose.13,14 These studies suggest that the KATP-channel pathway cannot fully explain how insulin secretion is regulated. This and other data (reviewed in7,18,19) in rodent and human islets suggest that mitochondrial messengers distinct from ATP and/or Ca2+ are also involved in GSIS.3,4,6,7,11–14,18,20–33 These studies suggest that there are additional regulators of insulin secretion besides ATP and Ca2+. This review will focus on one of them, αKG, and its possible role in the regulation of prolyl hydroxylase domain (PHD) proteins (Figure 1).
Figure 1.
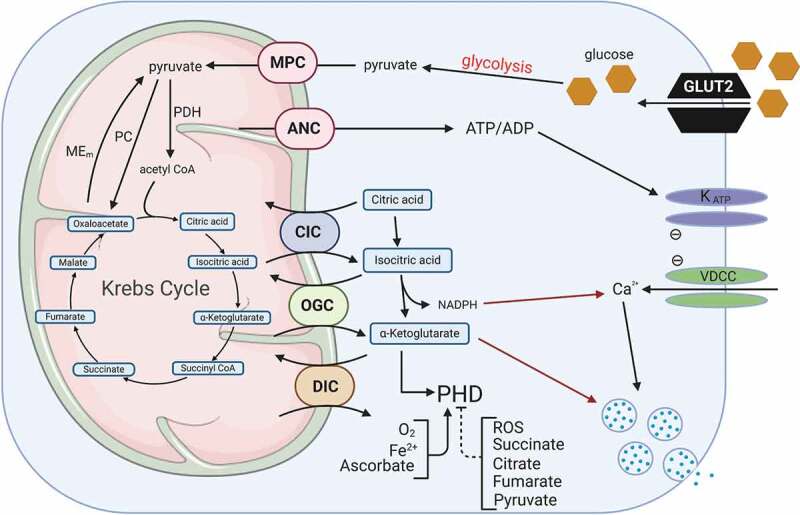
Glucose regulation of insulin secretion and PHDs. In pancreatic β-cells, one important glucose-derived signal is a rise in the ATP:ADP ratio, which stimulates closure of KATP channels, resulting in plasma membrane depolarization, activation of voltage-dependent Ca2+ channels, and Ca2+-mediated stimulation of insulin granule exocytosis. This so-called “KATP channel-dependent” mechanism appears to be particularly important in ‘triggering’ exocytosis of a small number of granules from a plasma membrane-docked “readily releasable pool (RRP)” responsible for the first, acute phase of insulin release. In contrast, in the second and sustained phase of insulin secretion, ATP and Ca2+ may play only a permissive roles, allowing other anaplerotic-derived second messengers, such as NADPH and α-ketoglutarate, to come to the forefront. A number of glucose-derived metabolites can regulate PHDs activity. Glucose transporter 2 (GLUT2), KATP channel (KATP), voltage-dependent Ca2+ channels, mitochondrial pyruvate carrier (MPC), adenine nucleotide carrier (ANC), citrate carrier (CIC), α-ketoglutarate carrier (OGC), dicarboxylate carrier (DIC), malic enzyme (Mem), pyruvate carboxylase (PC), pyruvate dehydrogenase (PDH). Figure was created with BioRender.com.
Prolyl hydroxylases
PHDs belong to the iron and αKG-dependent family of dioxygenase that have several primary substrates, including proteins, methylated nucleotides, lipids, and a wide range of small molecules.34 Members of this family include the lysyl, asparaginyl, and proline hydroxylases (Table 1). There are three procollagen-lysine 2‑oxoglutarate 5‑dioxygenases (PLOD1, PLOD2, and PLOD3) that mediate collagen lysine hydroxylation. The hydroxylated lysine residues of collagen have increased stability, which leads to increased tissue stiffness.37 Bones, cartilage, and tendons have a higher percentage of hydroxylated lysine residues in their collagen fibers compared with soft tissues, such as the skin.37 The factor inhibiting HIF-1α (FIH or FIHAN) is an asparagine hydroxylase that acts on the C-terminal activation domain (C-TAD) of HIF-α proteins.38 Asparagine hydroxylation of HIF-α proteins by FIH inhibits the transcriptional activity of HIFs by preventing its binding to the transcriptional co-activators CBP/p300.38
Table 1.
Expression of αKG-dependent family of dioxygenase hydroxylases in pancreatic β-cells. Procollagen-Lysine, 2-Oxoglutarate 5-Dioxygenase (PLOD), Factor inhibiting HIF (FIH), prolyl-4-hydroxylase domain (PHD), prolyl 4‑hydroxylase (P4H). There are three α-P4Hs (P4HA) and one β-P4H (P4HB). Expression in β-cells was determined from two transcriptomic papers by Segerstolpe35 (http://sandberg.cmb.ki.se/pancreas/) and Mawla36 (http://huisinglab.com/diabetes_2019/index.html). Associations with type 2 diabetes were determined using the type 2 diabetes portal (http://www.type2diabetesgenetics.org/home/portalHome). Amino acid (AA). Not detected (ND).
| Subfamily | Gene | Target AA | Expressed in beta-cells | Association with T2D |
|---|---|---|---|---|
| Procollagen-Lysine,2-Oxoglutarate 5-Dioxygenase | PLOD1 | Lysine | Reference 35, 36 | Weak |
| PLOD2 | Lysine | Reference 35, 36 | Weak | |
| PLOD3 | Lysine | Reference 35, 36 | No | |
| Hypoxia-inducible factor-asparagine dioxygenase | FIH1AN | Asparagine | Reference 35, 36 | Not found |
| Prolyl-4-hydroxylases | PHD1 | Proline | Reference 35, 36 | Weak |
| PHD2 | Proline | Reference 36 | No | |
| PHD3 | Proline | Reference 35, 36 | Weak | |
| PHD-TM | Proline | Reference 35, 36 | Weak | |
| Prolyl-4-Hydroxylase (subunit alpha) | P4HA1 | Proline | Reference 35, 36 | Weak |
| P4HA2 | Proline | Reference 35, 36 | Weak | |
| P4HA3 | Proline | ND Ref 35, low Ref 36 | Weak | |
| Prolyl-4-Hydroxylase (subunit beta) | P4HB | Proline | Reference 35, 36 | Weak |
Prolyl-4-hydroxylases subfamily of proteins consists of the hypoxia-inducible factor (HIF)-prolyl-4-hydroxylases (P4H) (also referred to as prolyl-4-hydroxylase domain (PHD)) and the collagen prolyl hydroxylases (C-P4Hs). PHDs and C-P4Hs are encoded by unique genes with minimal amino acid similarities. The only exception is that PHDs and C-P4Hs have some similarities in their catalytic domains.39 For C-P4Hs, the hydroxylation of proline residues in collagen to 4-hydroxyproline is essential for proper assembly of collagen. The 4-hydroxyproline forms hydrogen bonds between the main chains of neighboring collagen polypeptides.40 There are three known α-subunit isoforms of the collagen prolyl 4‑hydroxylase (P4HA) and include P4HA isoform 1 (P4HA1), P4HA2 and P4HA3, and there is one known β-subunit P4HB. Two α-subunits and two β-subunits form a functional A2B2 tetrameric protein and result in the generation of endoplasmic reticulum localized P4H1 (from P4HA1), P4H2 (from P4HA2) and P4H3 (from P4HA3) holoenzymes.41,42
PHDs, like C-P4H, require oxygen, the tricarboxylic acid cycle intermediate αKG as a substrate, Fe2+, and ascorbate as cofactors (Figure 2).43–46 Unlike the C-P4H, the PHDs have a Km value for oxygen around 100 μM. This Km value is higher than the oxygen concentration found in tissues.44,45 Because of these features, PHDs are considered one of the primary oxygen sensors in cells. There are three isoforms of PHD (PHD1, PHD2, and PHD3) which are also referred to as Egln2, Egln1, and Egln3 or HIF-P4H1, HIF-P4H2, and HIF-P4H3, respectively.39,43–47 In humans, PHD1, PHD2, and PHD3, are composed of 407, 426, and 239 amino acids, respectively, and unlike the tetrameric collagen prolyl hydroxylases, they function as monomeric proteins.39,43–47 A transmembrane PHD (PHD-TM) has also been described that consists of 502 amino acids located in the endoplasmic reticulum with the catalytic domain within the lumen between residues 59 and 82.39,43–45,47 At the amino acid level, PHD1, PHD2, and PHD3 are 42–59% identical to each other.39,43 These isoforms share the hydroxylase domain at the C-terminal end, whereas the N-terminal ends vary within each isoform, with PHD3 having the shortest N-terminal domain.39,43 Each of the PHD isoforms also have several variants that are produced via alternative splicing of exons 4 and 5; however, these may lack sufficient enzymatic activity or stability.43,46 PHDs have unique subcellular localizations with PHD1 exclusively expressed in the nucleus; PHD2 mainly localized in the cytosol, and PHD3 is expressed in almost equal proportions between the cytosol and the nucleus.39,43,45 PHD2 is ubiquitously expressed in most tissues, whereas PHD1 and PHD3 were found at higher levels in the placenta and heart, respectively.43 PHD-TM mRNA levels were highest in the human brain and pancreas, and PHD1 was the sole hydroxylase in the testes.39,43 Whole body knockouts of PHD1 in mice have no overt phenotype, whereas, whole body knockout of PHD2 in mice have placental vascular defect and die prematurely. Mice with whole body knockout of PHD3 have defective development of the sympathetic nervous system and hypotension.48,49
Figure 2.
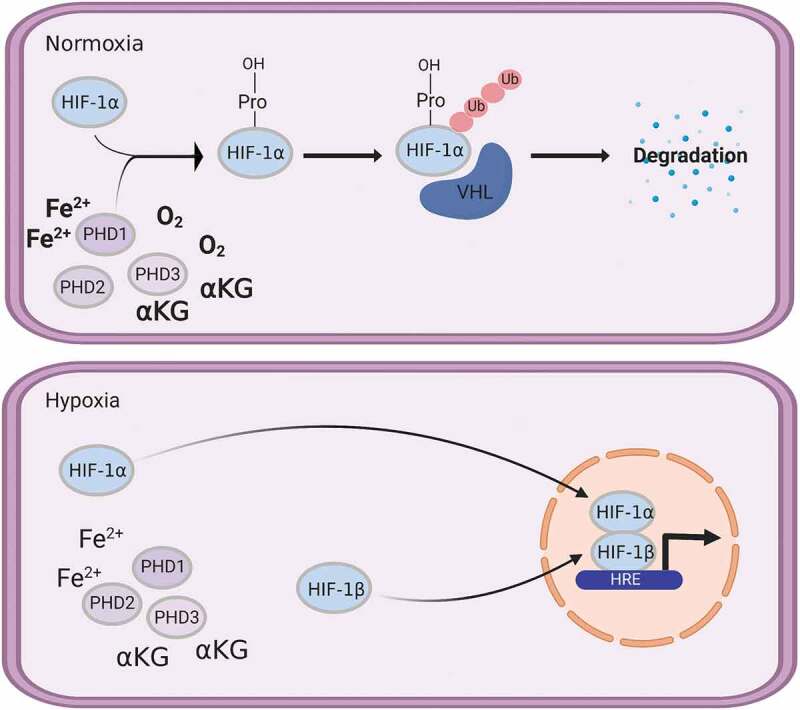
PHD proteins play a key role in regulating hypoxia inducible factor (HIF) stability and activity. Under normoxic conditions, oxygen and α-ketoglutarate are available for PHDs to use to proline hydroxylate HIF proteins which targets them for ubiquitination (Ub) mediated degradation. Under hypoxic conditions, the low oxygen prevents the proline hydroxylation of HIFs, increasing their stability and activity. Von Hippel-Lindau (VHL), hypoxia response element (HRE). Figure was created with BioRender.com.
Both C-P4Hs and PHDs use one oxygen atom in the oxidative decarboxylation of αKG generating succinate and CO2, and the second oxygen atom is used in the hydroxylation of proline residues in target proteins.45 TCA cycle intermediates, succinate, and fumarate have been reported to inhibit PHD activity by competing with αKG for its binding site.45,50 Fumarate and succinate can inhibit all three PHD isoforms, with fumarate having a Ki value between 50–80 μM and succinate between 350 and 460 μM.48 The requirement of Fe2+ for the hypoxic response was shown using iron chelators, which led to the stabilization of HIF1α long before the discovery of PHDs.51,52 Nitric oxide and reactive oxygen species (ROS) have also be recognized to inhibit both C-P4Hs and PHD activity by chelation and oxidization of bound Fe2+ to Fe,3+ respectively.39 Ascorbate plays a role in maintaining maximal C-P4H and PHD catalytic activity and has been reported to reduce Fe3+ from increased ROS production back to Fe2+.39,45
The reactions performed by prolyl hydroxylases are not reversible.44 The primary regulator of the response to low oxygen levels in tissues is the transcription factor hypoxia inducible factors (HIF). HIF is a heterodimer containing an unstable HIFα subunit and a stable HIF1β subunit.44,45 One of the most investigated roles for PHDs is the hydroxylation of proline residues on HIFα. The C-P4Hs are not capable of hydroxylating proline residues in HIFs and are not found in the cytosol. There are three isoforms of human HIFα, HIF1α, HIF2α, and HIF3α. Due to its high expression in many cells, PHD2 has been suggested to be the primary regulator in the hypoxia response pathway. Conditional inactivation of PHD2 in adult mice is enough to lead to excessive angiogenesis, a process that is upregulated with HIFα stabilization.43 A lesser role for PHD1 and PHD3 in the regulation of HIFα is likely due to their lower mRNA levels.44 PHD2 is preferential to HIF1α compared to HIF2α, whereas the opposite is true for PHD3.43,44
PHDs can hydroxylate one or both proline residues (Pro-402 and Pro-564) in the oxygen-dependent degradation domain (ODDD) of HIF1α in normoxic conditions at – Leu-X-X-Leu-Ala-Pro-recognition sites.43 Under normoxic conditions, PHD mediated hydroxylation of proline residues in HIFα are recognized by the von Hippel-Lindau protein (pVHL) E3 ubiquitin ligase complex and targeted for ubiquitination and rapid proteasomal degradation.39,44,53 When hypoxic conditions occur, PHD is inhibited by the lack of oxygen leading to HIFα stabilization. HIFα translocates to the nucleus where it forms a heterodimer with HIF1β and binds to hypoxia-response elements (HREs) located in the regulatory regions of more than 100 genes involved in response to decreasing oxygen levels (Figure 2).39,43,45 These targeted genes include those involved with angiogenesis, such as VEGF, erythropoiesis, energy metabolism, neovascularization, apoptosis, and cell proliferation.46,54
The oxidative state of cells can regulate PHDs activity. PHD activity can be increased with higher levels of antioxidants, such as ascorbate, and their activity can be decreased in response to reactive oxygen species (ROS).39,45,55–58 The lack of cysteine has been shown to lead to oxidative self inactivation of PHD1, which leads to HIF1α accumulation.59 A PHD2 protein complex of 15 proteins might also exist and play a role in regulating PHD2 activity.60 Additionally, there is regulation of PHD expression levels as well. A positive feedback mechanism exists involving HIF-1α-mediated induction of PHD2 and PHD3, but not PHD1, transcript and protein levels via a HRE found in their genes, and this may be important in the removal of HIF1α upon reoxygenation.61–67 FK506 binding protein 38 (FKBP38) has been shown to bind and negatively regulate PHD2 stability by promoting PHD2 entry into an ubiquitin-independent proteasomal pathway.68,69 The effects of FKBP38 on PHD2 stability seems to be independent of FKBP38 peptidyl-prolyl cis-trans isomerase activity.68,69 It has been suggested that FKBP38 may regulate PHD2 by excluding it from the nucleus, whereas, another PHD2 binding protein, ING4, promotes nuclear localization of PHD2 and reoxygenation mediated degradation of HIF.69,70 Although it is not known if FKBP38 can negatively regulate PHD1 and PHD3, hypoxia can increase Siah1a and Siah2 expression, and this can target PHD1 and PHD3 for proteasomal degradation by the E3 ubiquitin ligase pathway.71
Islets, O2, and PHDs
When clonal MIN6 cells and primary mouse islets are stimulated by high glucose, it leads to transient hypoxia (within minutes) that is likely due to increased nutrient stimulated oxygen consumption; this hypoxia is exacerbated when oxygen availability is compromised. This transient hypoxia does not lead to the activation of HIF1α.72 However, exposing β-cells to mild or long-term hypoxia, over a period of a few hours or days, leads to induction in HIF1α, causing a shift to anaerobic metabolism and inhibition of GSIS.72–74 Rat and human islets incubated for 5.5 hours in a hypoxic environment were correlated with a decrease in insulin content and insulin biosynthesis.75 Also, islet oxygen delivery is impaired in both C57BL6/J mice fed a high fat diet (HFD), and in ob/ob mice likely because of poor islet microcirculation and blood supply.72 In adults, T2D is associated with severe hypoxia due to poor islet microcirculation and increased nutrient stimulated oxygen consumption leading to a reduced ability to produce ATP in β-cells.52 The long-term lower oxygen levels in β-cells leads to HIF1α stabilization, which then activates target genes involved in angiogenesis to increase oxygen supply.39,43–47,50,52,54 The upregulation of islet HIF1α during diabetes-induced hypoxia may play a protective/survival role for islets, but upregulated HIF1α comes at the price of inhibition of nutrient regulated insulin secretion.72 Overall, diabetes mediated reduction of PHDs activity due to lower O2 availability leads to elevated HIFα levels which may be beneficial for maintaining insulin sensitivity in peripheral tissues and β-cells. Increased HIFα may promote β-cell survival, but it also blocks nutrient regulated insulin secretion.76 These studies suggest a crucial role for the oxygen sensing PHDs in the development of diabetes; however, we know very little about how these proteins lead to β-cell dysfunction.
Consistent with a negative role for HIF1α in inhibiting β-cell function, the INS-1 832/2 glucose unresponsive cell line have increased HIF1α and anaerobic metabolism.73 Higher HIF1α protein levels were associated with impaired GSIS in rodent and human islets.73,75 These studies suggest that glucose unresponsive β-cells may have lower PHD activity, and this may impair GSIS. HIF1α and HIF2α are required for adaptation of the β-cell to hypoxia seen during embryonic development and β-cell differentiation;77 however, too much HIF1α has a negative effect on β-cell development perhaps by decreasing the ability to produce ATP.78 β-cell specific loss of HIF1α leads to impaired glucose tolerance, reduced islet ATP, and a loss of β-cell function in rodent and human islets.76 The PHD inhibitor/iron chelators deferoxamine (DFO) and deferasirox (DFS) were able to increase HIF1α and significantly improve glucose tolerance, and insulin secretion in wild-type mice fed a HFD.76 HIF1α is also thought to be associated with glucose intolerance and insulin insensitivity in β-cells.79 It appears that β-cells require small amounts of HIF1α to maintain β-cell function; however, too much can lead to β-cell dysfunction and diabetes.76 The primary regulators of HIFα are the PHDs suggesting an important role for this protein in the development of diabetes.
It is also possible that there may be a link between ROS, PHDs, and diabetes. It is known that islet nutrient overload leads to elevation of ROS,80–82 and the increased ROS may inhibit PHD activity and promote an elevation of HIF and impair islet insulin secretion. It has also been suggested that ROS may be a more relevant physiological regulator of HIF1α stability under aerobic conditions.83 Like HIF1α, neuronal PAS domain protein 4 (NPAS4) is a heterodimerization partner of HIF1β (or aryl hydrocarbon receptor nuclear translocator, ARNT).84 In β-cells, the binding of HIF1α and NPAS4 to HIF1β is competitive, and each binding pair can activate a different subset of genes.85 NPAS4 is rapidly induced by membrane depolarization and calcium influx by the calcineurin, Akt/protein kinase B, and Ca2+/calmodulin-dependent protein kinase signaling pathways in β-cells.86–88 NPAS4 is protective against thapsigargin- and palmitate-induced ER stress, prevents apoptotic cell death and is lower in type 2 diabetic mouse β-cells.88 The Ca2+ induced expression of NPAS4 can prevent ROS induced HIF1α stabilization and maintains maximal mitochondrial OXPHOS function in β-cells.85 Thus like PHDs, NPAS4 can also regulate β-cell HIF1α levels.
In addition to the role of PHDs in the development of β-cell dysfunction, they may also play a role in regulating insulin sensitivity. Hif-p4h-2 gt/gt hypomorphic mice (whole body mutant mice that have lower PHD2 protein levels and increased HIF1α) have less adipose tissue, smaller adipocytes and decreased adipose tissue inflammation resulting in improved glucose tolerance and insulin sensitivity on either normal chow or HFD.89 These mice also had HIF1α stabilization and upregulation of its target genes, involving glucose transporters and enzymes involved in glycolysis.89 These results were replicated with oral administration of the PHD inhibitor FG-4497 in wild type mice.89 These studies suggest a key role for PHD2 in the control of HIF1α. Hepatic knockout of PHD3 leads to improved insulin sensitivity and prevents HFD induced diabetes due to HIF2α stabilization. HIF2α stabilization was associated with increased IRS2 and Akt activity.90 The authors suggest that isoform-specific inhibition of PHD3 may be a viable treatment option for T2D; however, this may not have the intended effect since inhibition of PHD3 blocks GSIS in mouse and human islets.91 Overall, diabetes mediated reduction of PHD activity due to lower oxygen availability leads to elevated HIFα levels which may be beneficial for maintaining insulin sensitivity in peripheral tissues but in β-cells increased HIFα may promote β-cell survival, while also blocking GSIS.
In addition to oxygen, several TCA cycle intermediates are also essential regulators of PHD activity, especially under normoxia.46,50,92 Rodent and human β-cells express high levels of the anaplerotic mitochondrial enzyme pyruvate carboxylase (PC) that plays a crucial role in generating TCA intermediates, including αKG.93,94 Transport of αKG from the mitochondria to the cytosol is facilitated by the αKG carrier (2-oxoglutarate carrier (OGC)).95 Pharmacological or siRNA mediated inhibition of OGC in 832/13 β-cells and primary rat islets significantly reduces GSIS suggesting that αKG needs be transported to the cytosol to affect insulin secretion.95 It has been shown that anaplerotically derived αKG plays a key role in regulating GSIS.33,95–97 αKG may regulate insulin secretion through its metabolism by PHDs leading to proline hydroxylation of crucial proteins involved in nutrient-stimulated insulin release.91,98,99 It has also been proposed that αKG may stimulate insulin secretion via glutamine dehydrogenase, which converts αKG to glutamate, glutamate potentially acts as a direct insulin secretagogue.99 However, this mechanism does not fully explain the role of αKG in regulating GSIS, and we propose that αKG is also oxidized to succinate via PHDs.95,99 It has been shown that succinate and fumarate can regulate PHD activity and HIF stabilization.100,101 A role for succinate has been shown by inhibiting succinate dehydrogenase, which elevated cytosolic succinate levels, and led to a reduction in PHD activity.101 Fumarate, pyruvate, and oxaloacetate can lead to the stabilization of HIF in vitro.100,102,103 The inhibitory effects of some TCA cycle intermediates seem to be specific for different PHD isoforms.50 An essential metabolite in the regulation of GSIS, citrate, has been shown to inhibit PHD3 more effectively than PHD1 or PHD2.50 Certain mutations of isocitrate dehydrogenase isoenzymes 1 and 2 (IDH1, 2) have been shown to decrease αKG in cells and led to the production of the oncometabolite R-2-hydroxyglutarate (also known as D-2-hydroxyglutarate) by the mutated IDH.104,105 Low levels of αKG or the R-2-hydroxyglutarate can negatively affect PHD activity and increase HIF stability.104,105 Whole body PHD1 knockout mice exhibit reduced basal oxygen consumption and were protected from skeletal muscle ischemia.106 The ischemic skeletal muscle protective effects seen in PHD1 knockout mice were partly due to HIF2α-mediated induction of pyruvate dehydrogenase kinase isoforms 1 and 4, which reduced mitochondrial respiration and elevated glycolytic ATP production.106 A role for PHDs in regulating metabolism is also supported by a study showing that PHD1-depleted breast cancer cells had a HIF-independent lowering of mitochondrial O2 consumption.107
PHDs, especially PHD3, have been shown to play a role in regulating GSIS in rodent and human β-cells.91 A recent preprint supports this work by D. Hodson group, suggesting a key role for PHD3 in the regulation of insulin secretion in response to metabolic stress using PHD3 KO mice (https://doi.org/10.1101/2020.04.30.068106). The link between glucose metabolism and PHD regulated insulin release may be anaplerotically derived αKG. Metabolism of αKG by PHDs could lead to proline hydroxylation of critical proteins involved in regulating insulin release. Recent studies have shown that PHDs can regulate several proteins in addition to HIFs, and some of these have been shown to control GSIS. Suggested non-HIFα targets of PHDs include Pax2,108 mTORC1,46 PKM2, ATF4, β2AR, NFkB, p27 (Figure 3).109,110 For example, proline hydroxylation by PHDs is reported to negatively regulate NF-κB.111,112 PHDs have also been shown to interact with p53 tumor suppressor protein, Wnt/β-catenin signaling pathway, and members of the ubiquitin-proteasome system.113–115 PHD1 is positively linked to caspase-3 levels in human inflammatory bowel disease,116 and mice with PHD1 deficiency have reduced apoptosis.117–119 Other novel non-HIFα targets, include MICAL1 and RhoGDI, which are known to be involved in regulating second-phase GSIS.120 Interestingly, PHD3 has the widest range of suggested targets,110 and our published91 and preliminary data, as well as others (https://doi.org/10.1101/2020.04.30.068106) support a strong role for PHD3 in GSIS. Overall, there is support for the concept that long-term changes in PHD activity can elevate HIFα and have a negative impact on β-cell function and lead to T2D. However, in the short-term, it can have a positive effect on β-cell function in an αKG dependent and HIFα independent fashion and promote GSIS.
Figure 3.
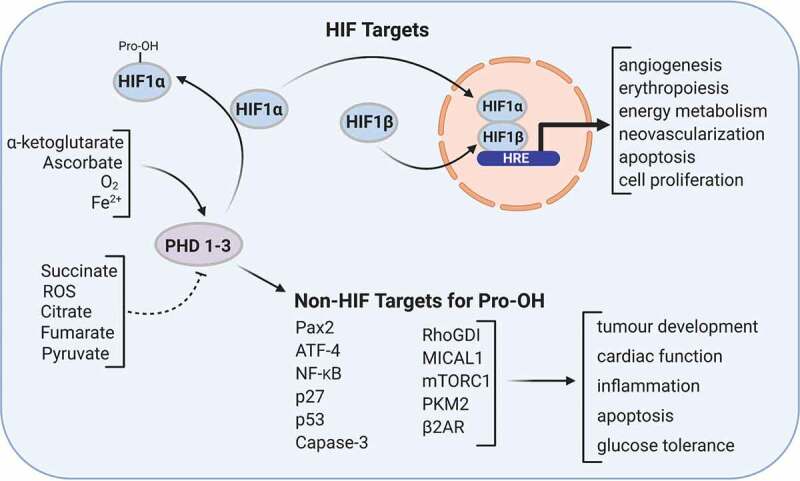
PHD activity can be regulated by a number of metabolic metabolites. In addition to PHD ability to regulate HIF stability and activity through proline hydroxylation, they can proline hydroxylate numerous other non-HIF targets. Figure was created with BioRender.com.
Funding Statement
This work was supported by Canadian Institute of Health Research (CIHR) grants to JWJ (PJT-159552).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Prentki M, Matschinsky FM.. Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev. 1987;67:1185–1248.doi: 10.1152/physrev.1987.67.4.1185. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Prog Biophys Mol Biol. 1989;54(2):87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 3.Newgard CB, Matchinsky FM. Substrate control of insulin release. In: Cryper A, Cherring AD, editors. Handbook of physiology section 7: the endocrine system volume II: the endocrine pancreas and regulation of metabolism. OUP (USA): Oxford University Press; 2001. p. 125–151. [Google Scholar]
- 4.Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic β-cell signal transduction. Annu Rev Biochem. 1995;64(1):689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald PE, Rorsman P. The ins and outs of secretion from pancreatic beta-cells: control of single-vesicle exo- and endocytosis. Physiology (Bethesda). 2007;22:113–121. doi: 10.1152/physiol.00047.2006. [DOI] [PubMed] [Google Scholar]
- 6.Henquin JC, Ravier MA, Nenquin M, Jonas JC, Gilon P. Hierarchy of the beta-cell signals controlling insulin secretion. Eur J Clin Invest. 2003;33(9):742–750. doi: 10.1046/j.1365-2362.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- 7.Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB. Metabolic cycling in control of glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab. 2008;295(6):E1287–97. doi: 10.1152/ajpendo.90604.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariot P, Gilon P, Nenquin M, Henquin JC. Tolbutamide and diazoxide influence insulin secretion by changing the concentration but not the action of cytoplasmic Ca2+ in beta-cells. Diabetes. 1998;47(3):365–373. doi: 10.2337/diabetes.47.3.365. [DOI] [PubMed] [Google Scholar]
- 9.Gilon P, Henquin JC. Influence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin-secreting pancreatic B-cell. J Biol Chem. 1992;267:20713–20720. [PubMed] [Google Scholar]
- 10.Straub SG, Sharp GW. Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab Res Rev. 2002;18:451–463. doi: 10.1002/dmrr.329. [DOI] [PubMed] [Google Scholar]
- 11.Maechler P, Kennedy ED, Pozzan T, Wollheim CB. Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic β-cells. Embo J. 1997;16(13):3833–3841. doi: 10.1093/emboj/16.13.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, et al. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002;277(40):37176–37183. doi: 10.1074/jbc.M206757200. [DOI] [PubMed] [Google Scholar]
- 13.Nenquin M, Szollosi A, guilar-Bryan L, Bryan J, Henquin J-C. Both triggering and amplifying pathways contribute to fuel-induced insulin secretion in the absence of sulfonylurea receptor-1 in pancreatic β-cells. J Biol Chem. 2004;279(31):32316–32324. doi: 10.1074/jbc.M402076200. [DOI] [PubMed] [Google Scholar]
- 14.Szollosi A, Nenquin M, guilar-Bryan L, Bryan J, Henquin J-C. Glucose stimulates Ca2+influx and insulin secretion in 2-week-old β-Cells lacking ATP-sensitive K+ channels. J Biol Chem. 2007;282(3):1747–1756. doi: 10.1074/jbc.M609875200. [DOI] [PubMed] [Google Scholar]
- 15.Remedi MS, Rocheleau JV, Tong A, Patton BL, McDaniel ML, Piston DW, Koster JC, Nichols CG. Hyperinsulinism in mice with heterozygous loss of KATP channels. Diabetologia. 2006;49(10):2368–2378. doi: 10.1007/s00125-006-0367-4. [DOI] [PubMed] [Google Scholar]
- 16.Szollosi A, Nenquin M, Henquin J-C. Overnight culture unmasks glucose-induced insulin secretion in mouse islets lacking ATP-sensitive K+channels by improving the triggering Ca2+ signal. J Biol Chem. 2007;282(20):14768–14776. doi: 10.1074/jbc.M701382200. [DOI] [PubMed] [Google Scholar]
- 17.Ravier MA, Nenquin M, Miki T, Seino S, Henquin J-C. Glucose controls cytosolic Ca2+ and insulin secretion in mouse islets lacking adenosine triphosphate-sensitive K+ channels owing to a knockout of the pore-forming subunit Kir6.2. Endocrinology. 2009;150(1):33–45. doi: 10.1210/en.2008-0617. [DOI] [PubMed] [Google Scholar]
- 18.MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, Kendrick MA. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am J Physiol Endocrinol Metab. 2005;288(1):E1–15. doi: 10.1152/ajpendo.00218.2004. [DOI] [PubMed] [Google Scholar]
- 19.Huypens PR, Huang M, Joseph JW. Overcoming the spatial barriers of the stimulus secretion cascade in pancreatic β-cells. Islets. 2012;4(1):1–116. doi: 10.4161/isl.18338. [DOI] [PubMed] [Google Scholar]
- 20.Maechler P, Wollheim CB. Role of mitochondria in metabolism-secretion coupling of insulin release in the pancreatic β-cell. Biofactors. 1998;8(3–4):255–262. doi: 10.1002/biof.5520080313. [DOI] [PubMed] [Google Scholar]
- 21.Joseph JW, Jensen MV, Ilkayeva O, Palmieri F, Alarcon C, Rhodes CJ, Newgard CB. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J Biol Chem. 2006;281(47):35624–35632. doi: 10.1074/jbc.M602606200. [DOI] [PubMed] [Google Scholar]
- 22.Agascioglu E, Giroix M-H, Malaisse WJ, Sener A. Adenine nucleotide pattern in rat pancreatic islets exposed to nutrient secretagogues. Endocrine. 2006;29(2):325–330. doi: 10.1385/ENDO:29:2:325. [DOI] [PubMed] [Google Scholar]
- 23.Jensen MV, Joseph JW, Ilkayeva O, Burgess S, Lu D, Ronnebaum SM, Odegaard M, Becker TC, Sherry AD, Newgard CB. Compensatory responses to pyruvate carboxylase suppression in islet beta-cells. Preservation of glucose-stimulated insulin secretion. J Biol Chem. 2006;281:22342–22351. [DOI] [PubMed] [Google Scholar]
- 24.Remedi MS, Nichols CG, Koster JC. The mitochondria and insulin release: Nnt just a passing relationship. Cell Metab. 2006;3(1):5–7. doi: 10.1016/j.cmet.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Joseph JW, Odegaard ML, Ronnebaum SM, Burgess SC, Muehlbauer J, Sherry AD, Newgard CB. Normal flux through ATP-citrate lyase or fatty acid synthase is not required for glucose-stimulated insulin secretion.J Biol Chem.2007;282(43):31592–31600.doi: 10.1074/jbc.M706080200. [DOI] [PubMed] [Google Scholar]
- 26.MacDonald PE, Joseph JW, Rorsman P. Glucose-sensing mechanisms in pancreatic β-cells. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2211–2225. doi: 10.1098/rstb.2005.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu D, Mulder H, Zhao P, Burgess SC, Jensen MV, Kamzolova S, Newgard CB, Sherry AD. 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc Natl Acad Sci U S A. 2002;99(5):2708–2713. doi: 10.1073/pnas.052005699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deeney JT, Prentki M, Corkey BE. Metabolic control ofβ-cell function. Semin Cell Dev Biol. 2000;11(4):267–275. doi: 10.1006/scdb.2000.0175. [DOI] [PubMed] [Google Scholar]
- 29.Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis.Nature.1999;402(6762):685–689. doi: 10.1038/45280. [DOI] [PubMed] [Google Scholar]
- 30.Ivarsson R, Quintens R, Dejonghe S, Tsukamoto K, in’t Veld P, Renstrom E, Schuit FC. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes. 2005;54:2132–2142. [DOI] [PubMed] [Google Scholar]
- 31.Corkey BE, Glennon MC, Chen KS, Deeney JT, Matschinsky FM, Prentki M. A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic beta-cells. J Biol Chem. 1989;264:21608–21612. [PubMed] [Google Scholar]
- 32.Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem. 1992;267:5802–5810. [PubMed] [Google Scholar]
- 33.Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, Stevens RD, Becker TC, Sherry AD, Newgard CB, Jensen MV. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem. 2006;281(41):30593–30602. doi: 10.1074/jbc.M511908200. [DOI] [PubMed] [Google Scholar]
- 34.Hausinger RP. fe(II)/α-Ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol.2004;39(1):21–68. Doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 35.Segerstolpe Å, Palasantza A, Eliasson P, Andersson E-M, Andreasson A-C, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 2016;24(4):593–607. doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mawla AM, Huising MO. Navigating the depths and avoiding the shallows of pancreatic islet cell transcriptomes.Diabetes.2019;68(7):1380–1393. doi: 10.2337/dbi18-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Slot AJ, Zuurmond A-M, van den Bogaerdt AJ, Ulrich MMW, Middelkoop E, Boers W, Karel Ronday H, DeGroot J, Huizinga TWJ, Bank RA. Increased formation of pyridinoline cross-links due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. Matrix Biol. 2004;23(4):251–257. doi: 10.1016/j.matbio.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Lando D. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fong G-H, Takeda K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008;15(4):635–641. doi: 10.1038/cdd.2008.10. [DOI] [PubMed] [Google Scholar]
- 40.Bhattacharjee A, Bansal M. Collagen structure: the Madras triple helix and the current scenario. IUBMB Life.2005;57(3):161–172.doi: 10.1080/15216540500090710. [DOI] [PubMed] [Google Scholar]
- 41.Annunen P, Helaakoski T, Myllyharju J, Veijola J, Pihlajaniemi T, Kivirikko KI. Cloning of the human prolyl 4-hydroxylase alpha subunit isoform alpha(II) and characterization of the type II enzyme tetramer. The alpha(I) and alpha(II) subunits do not form a mixed alpha(I)alpha(II)beta2 tetramer. J Biol Chem. 1997;272:17342–17348. [DOI] [PubMed] [Google Scholar]
- 42.Kukkola L, Hieta R, Kivirikko KI, Myllyharju J. Identification and characterization of a third human, rat, and mouse collagen prolyl 4-hydroxylase isoenzyme. J Biol Chem. 2003;278(48):47685–47693. doi: 10.1074/jbc.M306806200. [DOI] [PubMed] [Google Scholar]
- 43.Myllyharju J, Koivunen P. Hypoxia-inducible factor prolyl 4-hydroxylases: common and specific roles. Biol Chem.2013;394(4):435–448. doi: 10.1515/hsz-2012-0328. [DOI] [PubMed] [Google Scholar]
- 44.Appelhoff RJ, Tian Y-M, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 45.Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. MolCell.2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 46.Boulahbel H, Duran RV, Gottlieb E. Prolyl hydroxylases as regulators of cell metabolism. Biochem Soc Trans. 2009;37(1):291–294. doi: 10.1042/BST0370291. [DOI] [PubMed] [Google Scholar]
- 47.Rishi MT, Selvaraju V, Thirunavukkarasu M, Shaikh IA, Takeda K, Fong G-H, Palesty JA, Sanchez JA, Maulik N. Deletion of prolyl hydroxylase domain proteins (PHD1, PHD3) stabilizes hypoxia inducible factor-1 alpha, promotes neovascularization, and improves perfusion in a murine model of hind-limb ischemia. Microvasc Res. 2015;97:181–188. doi: 10.1016/j.mvr.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 48.Takeda K, Ho VC, Takeda H, Duan L-J, Nagy A, Fong G-H. Placental but not heart defects are associated with elevated hypoxia-inducible factor α levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26(22):8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, Ortega-Saenz P, Oster H, Wijeyekoon B, Sutherland AI, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol Cell Biol. 2008;28(10):3386–3400. doi: 10.1128/MCB.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282(7):4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- 51.Bruick RK. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 52.Bento CF, Pereira P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia. 2011;54(8):1946–1956. doi: 10.1007/s00125-011-2191-8. [DOI] [PubMed] [Google Scholar]
- 53.Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr.. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111(6):3236–3244. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puri S, Cano DA, Hebrok M. A role for von hippel-lindau protein in pancreatic -cell function. Diabetes. 2009;58(2):433–441. doi: 10.2337/db08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III Stabilize hypoxia-inducible factor-1α during hypoxia. J Biol Chem. 2000;275(33):25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 56.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95(20):11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flashman E, Davies SL, Yeoh KK, Schofield CJ. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem J. 2010;427(1):135–142. doi: 10.1042/BJ20091609. [DOI] [PubMed] [Google Scholar]
- 58.Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003;63:1764–1768. [PubMed] [Google Scholar]
- 59.Briggs KJ, Koivunen P, Cao S, Backus KM, Olenchock BA, Patel H, Zhang Q, Signoretti S, Gerfen GJ, Richardson AL, et al. Paracrine induction of HIF by glutamate in breast cancer: eglN1 senses cysteine. Cell. 2016;166(1):126–139. doi: 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, Yang H, Sorokina I, Conaway RC, Conaway JW, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2002;99(21):13459–13464. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Metzen E, Stiehl DP, Doege K, Marxsen JH, Hellwig-Burgel T, Jelkmann W. Regulation of the prolyl hydroxylase domain protein 2 (phd2/egln-1) gene: identification of a functional hypoxia-responsive element. BiochemJ.2005;387(3):711–717. doi: 10.1042/BJ20041736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO, del Peso L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene.Biochem J.2005;390(1):189–197. doi: 10.1042/BJ20042121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-α-prolyl-4-hydroxylases. Biochem J. 2004;381(3):761–767. doi: 10.1042/BJ20040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.D’Angelo G, Duplan E, Boyer N, Vigne P, Frelin C. Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J Biol Chem. 2003;278(40):38183–38187. doi: 10.1074/jbc.M302244200. [DOI] [PubMed] [Google Scholar]
- 65.Stiehl DP, Wirthner R, Koditz J, Spielmann P, Camenisch G, Wenger RH. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensingsystem.JBiolChem. 2006;281:23482–23491. [DOI] [PubMed] [Google Scholar]
- 66.To KK, Huang LE. Suppression of hypoxia-inducible factor 1α (HIF-1α) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J Biol Chem. 2005;280(45):38102–38107.doi: 10.1074/jbc.M504342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. doi: 10.1016/S0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 68.Barth S, Edlich F, Berchner-Pfannschmidt U, Gneuss S, Jahreis G, Hasgall PA, Fandrey J, Wenger RH, Camenisch G. Hypoxia-inducible factor prolyl-4-hydroxylase PHD2 protein abundance depends on integral membrane anchoring of FKBP38. J Biol Chem. 2009;284(34):23046–23058. doi: 10.1074/jbc.M109.032631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barth S, Nesper J, Hasgall PA, Wirthner R, Nytko KJ, Edlich F, Katschinski DM, Stiehl DP, Wenger RH, Camenisch G.The peptidyl prolyl cis/trans isomeraseFKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol. 2007;27(10):3758–3768. doi: 10.1128/MCB.01324-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soilleux EJ, Turley H, Tian YM, Pugh CW, Gatter KC, Harris AL. Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology. 2005;47:602–610. [DOI] [PubMed] [Google Scholar]
- 71.Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell. 2004;117(7):941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 72.Sato Y, Endo H, Okuyama H, Takeda T, Iwahashi H, Imagawa A, Yamagata K, Shimomura I, Inoue M. Cellular hypoxia of pancreatic β-cells due to high levels of oxygen consumption for insulin secretion in vitro. J Biol Chem. 2011;286(14):12524–12532. doi: 10.1074/jbc.M110.194738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spegel P, Malmgren S, Sharoyko VV, Newsholme P, Koeck T, Mulder H. Metabolomic analyses reveal profound differences in glycolytic and tricarboxylic acid cycle metabolism in glucose-responsive and -unresponsive clonal β-cell lines. Biochem J. 2011;435(1):277–284. doi: 10.1042/BJ20100655. [DOI] [PubMed] [Google Scholar]
- 74.Ma Z, Moruzzi N, Catrina S-B, Grill V, Bjorklund A. Hyperoxia inhibits glucose-induced insulin secretion and mitochondrial metabolism in rat pancreatic islets. Biochem Biophys Res Commun. 2014;443(1):223–228. doi: 10.1016/j.bbrc.2013.11.088. [DOI] [PubMed] [Google Scholar]
- 75.Ma Z, Moruzzi N, Catrina S-B, Hals I, Oberholzer J, Grill V, Bjorklund A, Nadal A. Preconditioning with associated blocking of Ca2+ inflow alleviates hypoxia-induced damage to pancreatic β-cells. PLoS ONE. 2013;8(7):e67498. doi: 10.1371/journal.pone.0067498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ, O’Connell PJ, Loudovaris T, Kay TW, Kulkarni RN, et al. Hypoxia-inducible factor-1α regulates β cell function in mouse and human islets. J Clin Invest. 2010;120(6):2171–2183. doi: 10.1172/JCI35846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cantley J, Grey ST, Maxwell PH, Withers DJ. The hypoxia response pathway and β-cell function. Diabetes Obes Metab. 2010;12(Suppl 2):159–167. doi: 10.1111/j.1463-1326.2010.01276.x. [DOI] [PubMed] [Google Scholar]
- 78.Heinis M, Soggia A, Bechetoille C, Simon M-T, Peyssonnaux C, Rustin P, Scharfmann R, Duvillie B. HIF1α and pancreatic β-cell development. Faseb J. 2012;26(7):2734–2742. doi: 10.1096/fj.11-199224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Catrina S-B, Okamoto K, Pereira T, Brismar K, Poellinger L.Hyperglycemiaregulates hypoxia-inducible factor-1 protein stability and function.Diabetes.2004;53(12):3226–3232. doi: 10.2337/diabetes.53.12.3226. [DOI] [PubMed] [Google Scholar]
- 80.Li N, Brun T, Cnop M, Cunha DA, Eizirik DL, Maechler P. Transient oxidative stress damages mitochondrial machinery inducing persistent β-cell dysfunction. J Biol Chem. 2009;284(35):23602–23612. doi: 10.1074/jbc.M109.024323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Robertson RP, Harmon JS. Pancreatic islet β-cell and oxidative stress: the importance of glutathione peroxidase. FEBS Lett. 2007;581(19):3743–3748. doi: 10.1016/j.febslet.2007.03.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roma LP, Pascal SM, Duprez J, Jonas J-C. Mitochondrial oxidative stress contributes differently to rat pancreatic islet cell apoptosis and insulin secretory defects after prolonged culture in a low non-stimulating glucose concentration. Diabetologia. 2012;55(8):2226–2237. doi: 10.1007/s00125-012-2581-6. [DOI] [PubMed] [Google Scholar]
- 83.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8(6):425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Greb-Markiewicz B, Kolonko M. Subcellular localization signals of bHLH-PAS proteins: their significance, current state of knowledge and future perspectives. Int J Mol Sci. 2019;20(19):4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sabatini PV, Speckmann T, Nian C, Glavas MM, Wong CK, Yoon JS, Kin T, Shapiro AMJ, Gibson WT, Verchere CB, et al. Neuronal PAS domain protein 4 suppression of oxygen sensing optimizes metabolism during excitation of neuroendocrine cells. Cell Rep. 2018;22(1):163–174. doi: 10.1016/j.celrep.2017.12.033. [DOI] [PubMed] [Google Scholar]
- 86.Speckmann T, Sabatini PV, Nian C, Smith RG, Lynn FC. Npas4 transcription factor expression is regulated by calcium signaling pathways and prevents tacrolimus-induced cytotoxicity in pancreatic beta cells. J Biol Chem. 2016;291(6):2682–2695. doi: 10.1074/jbc.M115.704098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sabatini PV, Lynn FC. All-encomPASsing regulation of β-cells: PAS domain proteins in β-cell dysfunction and diabetes. Trends Endocrinol Metab. 2015;26(1):49–57. doi: 10.1016/j.tem.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 88.Sabatini PV, Krentz NA, Zarrouki B, Westwell-Roper CY, Nian C, Uy RA, Shapiro AM, Poitout V, Lynn FC. Npas4 is a novel activity–regulated cytoprotective factor in pancreatic β-cells. Diabetes. 2013;62(8):2808–2820. doi: 10.2337/db12-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rahtu-Korpela L, Karsikas S, Horkko S, Blanco SR, Lammentausta E, Makela KA, Herzig K-H, Walkinshaw G, Kivirikko KI, Myllyharju J, et al. HIF prolyl 4-hydroxylase-2 inhibition improves glucose and lipid metabolism and protects against obesity and metabolic dysfunction. Diabetes. 2014;63(10):3324–3333. doi: 10.2337/db14-0472. [DOI] [PubMed] [Google Scholar]
- 90.Taniguchi CM, Finger EC, Krieg AJ, Wu C, Diep AN, LaGory EL, Wei K, McGinnis LM, Yuan J, Kuo CJ, et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med. 2013;19(10):1325–1330. doi: 10.1038/nm.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang M, Paglialunga S, Wong JM, Hoang M, Pillai R, Joseph JW. Role of prolyl hydroxylase domain proteins in the regulation of insulin secretion. Physiol Rep. 2016;4(5):e12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tennant DA, Gottlieb E. HIF prolyl hydroxylase-3 mediates alpha-ketoglutarate-induced apoptosis and tumor suppression. J Mol Med (Berl). 2010;88(8):839–849. doi: 10.1007/s00109-010-0627-0. [DOI] [PubMed] [Google Scholar]
- 93.Muoio DM, Newgard CB. Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(3):193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 94.Maechler P, Carobbio S, Rubi B. In beta-cells, mitochondria integrate and generate metabolic signals controlling insulin secretion. Int J Biochem Cell Biol. 2006;38(5–6):696–709. doi: 10.1016/j.biocel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 95.Odegaard ML, Joseph JW, Jensen MV, Lu D, Ilkayeva O, Ronnebaum SM, Becker TC, Newgard CB. The mitochondrial 2-oxoglutarate carrier is part of a metabolic pathway that mediates glucose- and glutamine-stimulated insulin secretion. J Biol Chem. 2010;285(22):16530–16537.doi: 10.1074/jbc.M109.092593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang M, Joseph JW. Metabolomic analysis of pancreatic β-cell insulin release in response to glucose. Islets. 2012;4(3):210–222. doi: 10.4161/isl.20141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huypens P, Pillai R, Sheinin T, Schaefer S, Huang M, Odegaard ML, Ronnebaum SM, Wettig SD, Joseph JW. The dicarboxylate carrier plays a role in mitochondrial malate transport and in the regulation of glucose-stimulated insulin secretion from rat pancreatic beta cells. Diabetologia. 2011;54(1):135–145. doi: 10.1007/s00125-010-1923-5. [DOI] [PubMed] [Google Scholar]
- 98.Lorenz MA, El Azzouny MA, Kennedy RT, Burant CF. Metabolome response to glucose in the β-cell line INS-1 832/13. J Biol Chem. 2013;288(15):10923–10935. doi: 10.1074/jbc.M112.414961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rabaglia ME, Gray-Keller MP, Frey BL, Shortreed MR, Smith LM, Attie AD. α-Ketoisocaproate-induced hypersecretion of insulin by islets from diabetes-susceptible mice. Am J Physiol Endocrinol Metab. 2005;289(2):E218–E24.doi: 10.1152/ajpendo.00573.2004. [DOI] [PubMed] [Google Scholar]
- 100.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung Y-L, Merino M, Trepel J, Zbar B, Toro J, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8(2):143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 101.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 102.Dalgard CL, Lu H, Mohyeldin A, Verma A. Endogenous 2-oxoacids differentially regulate expression of oxygen sensors. Biochem J. 2004;380:419–424. doi: 10.1042/bj20031647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem. 2005;280:41928–41939. doi: 10.1074/jbc.M508718200. [DOI] [PubMed] [Google Scholar]
- 104.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, Ito S, Yang C, Wang P, Xiao M-T, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-Ketoglutarate-Dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Aragones J, Schneider M, Van GK, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40(2):170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 107.Zhang J, Wang C, Chen X, Takada M, Fan C, Zheng X, Wen H, Liu Y, Wang C, Pestell RG, et al. EglN2 associates with the NRF1-PGC1α complex and controls mitochondrial function in breast cancer. Embo J. 2015;34(23):2953–2970. doi: 10.15252/embj.201591437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yan B, Jiao S, Zhang H-S, Lv -D-D, Xue J, Fan L, Wu G-H, Fang J. Prolyl hydroxylase domain protein 3 targets Pax2 for destruction. Biochem Biophys Res Commun. 2011;409(2):315–320. doi: 10.1016/j.bbrc.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 109.Zeng H, Chen J-X, Fan G-C. Conditional knockout of prolyl hydroxylase domain protein 2 attenuates high fat-diet-induced cardiac dysfunction in mice. PLoS One. 2014;9(12):e115974. doi: 10.1371/journal.pone.0115974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jaakkola PM, Rantanen K. The regulation, localization, and functions of oxygen-sensing prolyl hydroxylase PHD3. Biol Chem. 2013;394(4):449–457. doi: 10.1515/hsz-2012-0330. [DOI] [PubMed] [Google Scholar]
- 111.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, et al. Prolyl hydroxylase-1 negatively regulates I B kinase-beta, giving insight into hypoxia-induced NF B activity. Proc Natl Acad Sci U S A. 2006;103(48):18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKβ independent of hydroxylase activity. Gastroenterology. 2010;138(2):606–615. doi: 10.1053/j.gastro.2009.09.049. [DOI] [PubMed] [Google Scholar]
- 113.Mazzone M, Dettori D, de Oliveira RL, Loges S, Schmidt T, Jonckx B, Tian Y-M, Lanahan AA, Pollard P, de Almodovar CR, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136(5):839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rodriguez J, Pilkington R, Garcia Munoz A, Nguyen LK, Rauch N, Kennedy S, Monsefi N, Herrero A, Taylor CT, von Kriegsheim A. Substrate-trapped interactors of PHD3 and FIH cluster in distinct signaling pathways. Cell Rep. 2016;14(11):2745–2760. doi: 10.1016/j.celrep.2016.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scholz CC, Rodriguez J, Pickel C, Burr S, Fabrizio J-A, Nolan KA, Spielmann P, Cavadas MA, Crifo B, Halligan DN, et al. FIH regulates cellular metabolism through hydroxylation of the deubiquitinase OTUB1. PLoS Biol. 2016;14(1):e1002347. doi: 10.1371/journal.pbio.1002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Van Welden S, Laukens D, Ferdinande L, De Vos M, Hindryckx P. Differential expression of prolyl hydroxylase 1 in patients with ulcerative colitis versus patients with Crohn’s disease/infectious colitis and healthy controls. J Inflamm (Lond). 2013;10(1):36. doi: 10.1186/1476-9255-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schneider M, Van Geyte K, Fraisl P, Kiss J, Aragones J, Mazzone M, Mairbaurl H, De Bock K, Jeoung NH, Mollenhauer M, et al. Loss or silencing of the PHD1 prolyl hydroxylase protects livers of mice against ischemia/reperfusion injury. Gastroenterology. 2010;138(3):e1–2. doi: 10.1053/j.gastro.2009.09.057. [DOI] [PubMed] [Google Scholar]
- 118.Adluri RS, Thirunavukkarasu M, Dunna NR, Zhan L, Oriowo B, Takeda K, Sanchez JA, Otani H, Maulik G, Fong GH, et al. Disruption of hypoxia-inducible transcription factor-prolyl hydroxylase domain-1 (PHD-1-/-) attenuates ex vivo myocardial ischemia/reperfusion injury through hypoxia-inducible factor-1alpha transcription factor and its target genes in mice. Antioxid Redox Signal. 2011;15:1789–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fitzpatrick SF, Fabian Z, Schaible B, Lenihan CR, Schwarzl T, Rodriguez J, Zheng X, Li Z, Tambuwala MM, Higgins DG, et al. Prolyl hydroxylase-1 regulates hepatocyte apoptosis in an NF-κB-dependent manner. Biochem Biophys Res Commun. 2016;474(3):579–586. doi: 10.1016/j.bbrc.2016.04.085. [DOI] [PubMed] [Google Scholar]
- 120.Wang Z, Thurmond DC. Differential phosphorylation of RhoGDI mediates the distinct cycling of Cdc42 and Rac1 to regulate second-phase insulin secretion. J Biol Chem. 2010;285(9):6186–6197. doi: 10.1074/jbc.M109.072421. [DOI] [PMC free article] [PubMed] [Google Scholar]