

Respiratory syncytial virus and human metapneumovirus are leading causes of respiratory illness worldwide, but limited treatment options are available. To better target these viruses, we examined key aspects of the viral life cycle in three-dimensional (3-D) human airway tissues. Both viruses establish efficient infection through the apical surface, but efficient spread and apical release were seen for respiratory syncytial virus (RSV) but not human metapneumovirus (HMPV). Both viruses form inclusion bodies, minimally composed of nucleoprotein (N), phosphoprotein (P), and viral RNA (vRNA), indicating that these structures are critical for replication in this more physiological model. HMPV formed significantly more long, filamentous actin-based extensions in human airway epithelial (HAE) tissues than RSV, suggesting HMPV may promote cell-to-cell spread via these extensions. Lastly, RSV entry and spread were fully inhibited by neutralizing antibodies palivizumab and the novel nirsevimab. In contrast, while HMPV entry was fully inhibited by 54G10, a neutralizing antibody, spread was only modestly reduced, further supporting a cell-to-cell spread mechanism.
KEYWORDS: HAE models, HMPV, RSV, clinical therapeutics, neutralizing antibodies, viral spread
ABSTRACT
Respiratory syncytial virus (RSV) and human metapneumovirus (HMPV) are two of the leading causes of respiratory infections in children and elderly and immunocompromised patients worldwide. There is no approved treatment for HMPV and only one prophylactic treatment against RSV, palivizumab, for high-risk infants. Better understanding of the viral lifecycles in a more relevant model system may help identify novel therapeutic targets. By utilizing three-dimensional (3-D) human airway tissues to examine viral infection in a physiologically relevant model system, we showed that RSV infects and spreads more efficiently than HMPV, with the latter requiring higher multiplicities of infection (MOIs) to yield similar levels of infection. Apical ciliated cells were the target for both viruses, but RSV apical release was significantly more efficient than HMPV. In RSV- or HMPV-infected cells, cytosolic inclusion bodies containing the nucleoprotein, phosphoprotein, and respective viral genomic RNA were clearly observed in human airway epithelial (HAE) culture. In HMPV-infected cells, actin-based filamentous extensions were more common (35.8%) than those found in RSV-infected cells (4.4%). Interestingly, neither RSV nor HMPV formed syncytia in HAE tissues. Palivizumab and nirsevimab effectively inhibited entry and spread of RSV in HAE tissues, with nirsevimab displaying significantly higher potency than palivizumab. In contrast, 54G10 completely inhibited HMPV entry but only modestly reduced viral spread, suggesting HMPV may use alternative mechanisms for spread. These results represent the first comparative analysis of infection by the two pneumoviruses in a physiologically relevant model, demonstrating an interesting dichotomy in the mechanisms of infection, spread, and consequent inhibition of the viral lifecycles by neutralizing monoclonal antibodies.
IMPORTANCE Respiratory syncytial virus and human metapneumovirus are leading causes of respiratory illness worldwide, but limited treatment options are available. To better target these viruses, we examined key aspects of the viral life cycle in three-dimensional (3-D) human airway tissues. Both viruses establish efficient infection through the apical surface, but efficient spread and apical release were seen for respiratory syncytial virus (RSV) but not human metapneumovirus (HMPV). Both viruses form inclusion bodies, minimally composed of nucleoprotein (N), phosphoprotein (P), and viral RNA (vRNA), indicating that these structures are critical for replication in this more physiological model. HMPV formed significantly more long, filamentous actin-based extensions in human airway epithelial (HAE) tissues than RSV, suggesting HMPV may promote cell-to-cell spread via these extensions. Lastly, RSV entry and spread were fully inhibited by neutralizing antibodies palivizumab and the novel nirsevimab. In contrast, while HMPV entry was fully inhibited by 54G10, a neutralizing antibody, spread was only modestly reduced, further supporting a cell-to-cell spread mechanism.
INTRODUCTION
Respiratory syncytial virus (RSV) and human metapneumovirus (HMPV) are single-stranded, negative-sense RNA (nsRNA) enveloped viruses in the Pneumoviridae family (1). They are leading causes of respiratory infections in children; 95% of children by the age of 2 are infected with RSV (2), and nearly all are seropositive for HMPV by the age of 5 (3, 4). Children, immunocompromised, and elderly populations are at significant risk for contracting and developing severe lower respiratory tract infection, with infants at the greatest risk (2–14). While both RSV and HMPV cause severe morbidity and mortality, no vaccines are available and only limited treatment options exist. For RSV, the only FDA-approved therapy is palivizumab, a humanized monoclonal antibody given prophylactically to high-risk infants during the infectious season (15, 16). To better understand how to target these viruses therapeutically, a deeper understanding of viral infection in physiologically relevant model systems is needed.
Pneumoviruses initiate infection by attaching to target cells via their surface glycoproteins, the fusion protein (F) and/or the attachment protein (G), which interact with host receptors and attachment factors. Subsequently, F undergoes a large conformational change to mediate membrane fusion, after which, the viral nucleocapsids are released into the cytoplasm of the infected cell (17, 18). This membrane fusion process is critical, and inhibition of the fusion protein blocks entry and infection. Interestingly, both HMPV and RSV mutants lacking surface glycoproteins G and SH but containing F can mediate entry and infection, albeit attenuated to some degree (19–22), demonstrating that F has an essential role in entry and is involved in attachment. Based on its critical role in infection, targeting the fusion protein is one of the most common strategies for developing therapeutics against HMPV and RSV (23–26).
After entering the target cell, HMPV and RSV nucleocapsids are released and used as templates for synthesis of viral mRNAs and genomic RNA (vRNA) by the viral RNA-dependent RNA polymerase. Research from our lab and others suggests these processes occur in punctate cytosolic structures, termed inclusion bodies (IBs), which are minimally composed of the nucleoprotein (N), the phosphoprotein (P), and vRNA (27, 28). IB-like structures have also been described for other nsRNA viruses, including Ebola virus (29, 30), Marburg virus (31), rabies virus (32, 33), vesicular stomatitis virus (34), parainfluenza virus 3 (35), and parainfluenza virus 5 (36), suggesting a broadly conserved mechanism for viral transcription and genome replication. Once newly synthesized nucleocapsids assemble in IBs, they traffic to assembly sites at the plasma membrane. For HMPV, it has been suggested that the actin cytoskeleton might play a crucial role in nucleocapsid transport and IB coalescence during infection (28), similar to what has been reported for Ebola virus (37). In addition, the actin cytoskeleton has been reported to have a role in movement of ribonucleocapsids to sites of assembly in RSV and measles virus-infected cells, further supporting the importance of the cytoskeleton for viral infection and spread (38–40). After transport, the nucleocapsids coalesce with other viral proteins at the plasma membrane, the proposed assembly site for pneumoviruses, although recent research suggests a cell-to-cell-dependent mechanism may play an important role in pneumovirus spread (41). This mechanism allows for the transfer of infectious particles via en bloc transmission by forming syncytia (9, 10, 42, 43), intercellular extensions (44, 45), or polyploid viruses (3, 46–49) to allow for transmission of multiple genomes from one cell to another, compared with the traditional method of single particle release and reentry.
HMPV has been shown to be primarily cell associated and induce the formation of long, actin-based filamentous extensions, which are important for direct cell-to-cell spread in vitro, even in the presence of neutralizing antibodies (44). RSV has also been shown to form actin-based extensions involving actin related protein-2 (ARP2), suggesting a potential role for these structures during budding and spread in cell culture monolayers (45). RSV additionally induces cell-to-cell fusion in immortalized cell monolayers, generating multinucleated cells termed syncytia, which are also a hallmark of other enveloped virus infections (10). Similarly, HMPV has been shown to generate syncytia, although to a much lesser extent compared to RSV (50, 51). While RSV replication is enhanced through the formation of syncytia in vitro (52), the formation of these structures in animal models and patients is not well understood. A few reports have described the presence of syncytia in postmortem autopsies of patients infected with either RSV or HMPV (9, 43). Currently, the roles of intercellular extensions and syncytia have yet to be fully elucidated in more physiologically relevant model systems in order to further understand how viruses enter and spread within infected tissue.
Human airway epithelial (HAE) tissues have been used previously as model systems to examine respiratory biology and pathogen interactions in cell culture (53–69). This model system utilizes primary human cells differentiated on a trans-well with an air-liquid interface to generate polarized bronchial epithelial tissue composed of multilayered epithelial cells, including basal, apical ciliated, and goblet cells. This composition allows for a more faithful recapitulation of the lung environment, including cellular three-dimensional (3-D) structural organization, functional cilia, and mucus production. This method provides a more accurate model system to study respiratory viruses in vitro, offering significant advantages compared to traditional 2-D cell monolayers (54, 58–61, 66, 70, 71). These tissue models have been used to study both RSV (62, 64, 66–69, 71–74) and HMPV (63, 75–77) infections. For RSV, HAE models have demonstrated that apical ciliated cells are the primary target cell, although occasional infection of nonciliated cells was also observed (62, 64, 72, 74). RSV-induced syncytia, a hallmark in 2-D cell culture, have been observed in several HAE studies, but with varied results. No syncytia were reported in several studies, even up to 36 days postinfection (62, 64), whereas another study observed infrequent syncytia (72). For HMPV, very few studies have been conducted using HAE tissues as a model system (63, 75–77). Consistent with findings from RSV, the primary target for HMPV is also apical ciliated cells (63). However, there are limited studies examining viral and host interactions for both HMPV and RSV in HAE tissues.
In this work, we performed a detailed analysis of RSV and HMPV infection in 3-D HAE cultures, exploring aspects of the viral life cycles that have not been examined previously in HAE tissues. RSV demonstrated significantly higher rates of infection, spread, and apical release than HMPV. Apical ciliated cells infected with either RSV or HMPV generated large cytosolic IBs, consisting of at least N, P, and vRNA, suggesting that these structures are critical replication complexes formed during viral infection in vivo. No syncytium formation was observed for either virus in our HAE studies. Interestingly, HMPV efficiently induced the formation of filamentous extensions in HAE cultures, while RSV formed significantly fewer extensions.
Lastly, we examined monoclonal antibody inhibition of entry and spread in HAE tissues for RSV and HMPV. The only approved FDA treatment against RSV, palivizumab, was able to inhibit RSV entry and spread. We also tested nirsevimab, a novel monoclonal antibody against RSV that has demonstrated potent efficacy against RSV in 2-D cell culture and animal models (78–80). We found that in HAE tissues, nirsevimab is able to block entry and spread of RSV with greater potency than palivizumab, supporting the findings in other model systems and supporting its potential as a novel antiviral therapeutic against RSV. The anti-HMPV antibody 54G10 effectively inhibited the entry of HMPV but only modestly, though significantly, reduced viral spread. Together, our results highlight the conserved and varied aspects of entry, replication, and assembly between two closely related pneumoviruses within HAE tissues and demonstrate an interesting dichotomy between HMPV and RSV and their life cycles.
RESULTS
Both HMPV and RSV have been studied extensively in 2-D nonpolarized monolayers to analyze virus-host interactions. RSV has also been studied in 3-D HAE tissues to assess viral infection and pathology (53, 64, 66–69, 72–74). In contrast, only a few reports examined HMPV in 3-D model systems (63, 75–77). While many important aspects of infection have been identified in 2-D cell studies, additional analysis in more physiologically relevant model systems is needed to further our understanding of the viral life cycle. To address this, we utilized HAE tissues, a 3-D tissue model system which more accurately recapitulates the lung environment, including cellular polarization, mucus production, and functional cilia.
To compare the two pneumoviruses, we first assessed RSV and HMPV infection, spread, and apical release side by side in HAE tissues. Tissues were infected with recombinant green fluorescent protein (GFP) expressing RSV-A2 (rgRSV) or HMPV-A2 CAN97-83 (rgHMPV). At 24 hours postinfection (hpi), GFP expression was analyzed and showed that both HMPV and RSV initiated infection in this model system (Fig. 1A). Initial infection was equivalent between both viruses, but HMPV infection was less efficient than RSV, requiring a multiplicity of infection (MOI) of 3 and 0.3, respectively, to generate a comparable infection at 24 hpi (Fig. 1B). We then examined viral spread within tissues as well as release from the apical surface up to 144 hpi. Using fluorescence threshold analysis (NIS-Elements) to quantify GFP expression, we found that RSV efficiently spreads from 24 hpi up through 144 hpi, with the largest increase in infection seen from 24 to 48 hpi. However, minimal changes in spread were observed past 72 hpi, suggesting spread has plateaued (Fig. 1A, quantified in Fig. 1C). In contrast, HMPV-infected cells increased significantly, but modestly, from 24 hpi to 48 hpi, followed by a decrease from 72 hpi to 144 hpi, with minimal infection remaining at later time points (Fig. 1A, quantified in Fig. 1C). Analysis of apical release of viral particles showed a 3-log difference in the amount of released virus at 24 hpi between HMPV and RSV, even though the numbers of infected cells were similar between the two viruses (Fig. 1B and D). In addition, there was no detectable release of HMPV after 24 hpi. This result demonstrates a rapid spread and sustained release of high titers of RSV particles from the apical surface, whereas HMPV spread and particle release were minimal at all time points, with a decrease in spread after 48 hpi.
FIG 1.

RSV and HMPV infection, spread, and release in HAE tissues. (A) HAE tissues were infected with MOI 0.3 of rgRSV or MOI 3.0 of rgHMPV, and initial infection and spread were examined up to 144 hours postinfection (HPI). (B) RSV and HMPV infection at the 24 hpi time point. (C) Spread analysis of HMPV and RSV were determined using fluorescence threshold analysis. (D) Apical release of virus was determined by washing the apical surface of HAE tissues, determining the titer of the viral wash in 2-D monolayers, and calculating fluorescence-forming units (FFU). (E) Infected cells for HMPV (48 hpi) and RSV (72 hpi) were stained for actin cytoskeleton, as well as keratan sulfate (KS) to stain for ciliated cells. Error bars represent the standard error of the mean (SEM) of 6 different tissues. Scale bar = 10 μm.
Previously, both RSV (62, 64, 72, 74) and HMPV (63) were reported to primarily infect apical ciliated cells in HAE cultures. To verify this in our side-by-side analysis, infected tissues were cryo-sectioned and stained for keratan sulfate (KS), a marker for ciliated airway cells. RSV and HMPV both exclusively infected ciliated cells, confirming this as the primary target cell type for both pneumoviruses (Fig. 1E). In a few instances, infected cells appeared below the apical surface. However, staining with KS demonstrated that these cells were ciliated, though they had not yet reached the apical surface. Based on these findings, the minimal spread and release of HMPV compared to RSV cannot be attributed to a difference in the type of cells infected or the initial rates of infection. Therefore, we examined other aspects of the viral life cycle to better understand how both viruses interact with host cells during infection.
HMPV and RSV induce the formation of replicative inclusion bodies.
The formation of inclusion bodies (IBs) in 2-D cell monolayers has been shown to be critical for viral genome replication and transcription for HMPV and RSV (27, 28). However, to our knowledge, the presence of IBs has not been examined in 3-D HAE tissues or in vivo. To determine if these structures are found in HAE-infected tissues and to assess whether differences in the early stages of infection were observed for RSV and HMPV, we examined the formation of IBs by fluorescently staining RSV or HMPV N and P proteins, the minimal components required for IB-like structure formation (81, 82). Colocalization of both N and P proteins was observed in cytoplasmic structures for RSV (Fig. 2A and B) and HMPV (Fig. 3A and B), suggesting that IBs form in infected HAE tissues.
FIG 2.

RSV inclusion body formation in HAE. (A) HAE tissues were infected with rgRSV and stained for the nucleoprotein, N, and phosphoprotein, P, to assess the formation of inclusion bodies in HAE tissues. (B) Colocalization of N and P was analyzed using colocalization chromatography from NIS-Elements. (C) To confirm inclusion body formation in HAE tissues, FISH analysis was conducted to label vRNA. (D) Both vRNA and RSV N colocalize to cytosolic punctate structures in infected cells. (E) vRNA was also assessed in relation to RSV using fluorescence microscopy. (F) Chromatogram analysis demonstrates that P and vRNA colocalize with one another in infected cells. Scale bar = 10 μm for combined images with differential interference contrast (DIC) and 5 μm for fluorescence insets.
FIG 3.

HMPV inclusion body formation in HAE. (A) HAE tissues were infected with rgHMPV and stained for the nucleoprotein, N, and phosphoprotein, P, to determine the formation of inclusion bodies in HAE tissues. (B) Colocalization of N and P was analyzed using colocalization chromatography from NIS-Elements. (C) To confirm inclusion body formation in HAE tissues, FISH analysis was conducted to label vRNA. (D) Both vRNA and HMPV N colocalize to cytosolic punctate structures in infected cells. (E) vRNA was also assessed in relation to HMPV using fluorescence microscopy. (F) Chromatogram analysis demonstrates that P and vRNA colocalize with one another in infected cells. Scale bar = 10 μm for combined images with DIC and 5 μm for fluorescence insets.
To confirm these structures were IBs, we conducted fluorescence in situ hybridization (FISH) staining for vRNA, known to be localized to IBs during infection of 2-D cells (27, 28). We observed strong colocalization of N and P for both RSV (Fig. 2C to F) and HMPV (Fig. 3C to F) with their respective signal of vRNA, confirming the formation of IBs in infected tissues for both viruses. These findings are, to our knowledge, the first report of the formation of these structures in 3-D HAE tissues for both RSV and HMPV and thus strongly support the hypothesis that these structures are critical for replication and spread of the viruses in 3-D models and likely in vivo, thus providing a viable therapeutic target for pneumoviruses.
N, P, and vRNA had strong colocalization at inclusion bodies and at the plasma membrane, suggesting the coalescence of these viral factors at sites of assembly for both RSV and HMPV. In addition, P also demonstrated localization at the plasma membrane independently of N and vRNA, often associated with cilium-like structures at the apical surface for both RSV- and HMPV-infected cells. However, the presence of P at these sites was much stronger for RSV than HMPV. P has been previously reported to associate with IBs and the plasma membrane to form extensions for HMPV (44) and in proposed assembly complexes for RSV (83), which is supported by our findings. These suggest that differences seen in spread dynamics of the viruses are not a result of differences in the ability to cause initial infection or generate replicative structures.
HMPV and RSV form extensions but no syncytia in HAE tissues.
There are numerous reports of actin cytoskeletal involvement in infectious cycles for both HMPV (44, 84, 85) and RSV (45, 86–90). We have previously shown that HMPV forms actin-based extensions, which were identified as a mechanism for direct cell-to-cell spread in BEAS-2B bronchial epithelial cells (44). HMPV P colocalized with actin, and transient expression of P alone recapitulated some extension formation. Recent reports examining RSV demonstrated that viral infection leads to the formation of actin-based extensions in A549 cells (45). RSV-induced extensions were suggested to be filopodia and could be induced by the expression of F alone. Additionally, disruption of actin architecture in RSV-infected cells decreased viral spread, suggesting that these extensions are critical for infecting new cells.
To compare the formation of actin-based extensions between the two pneumoviruses in different cell types, we infected Vero (monkey kidney), HEp-2 (human laryngeal carcinoma), and BEAS-2B (human bronchial epithelial, nondiseased) cells and analyzed extension formation. For all three cell types, both RSV (Fig. 4A) and HMPV (Fig. 4B) induced the formation of filamentous extensions at 24 hpi. Interestingly, a previous study did not observe extension formation in RSV-infected Vero cells (45). These differences may be due, in part, to other factors such as reagents or cell culture methodology.
FIG 4.

HMPV forms intercellular extensions significantly more than RSV. (A and B) BEAS-2B, Vero, and HEp-2 cells infected with rgRSV (A) or rgHMPV (B) demonstrate the formation of long filamentous extensions. (C) HAE tissues infected with either rgRSV or rgHMPV demonstrate the formation of these filamentous extensions in a 3-D model system. (D) Extension formation is significantly more common in rgHMPV-infected tissues than in those infected with rgRSV. Statistical significance is represented with P < 0.05 (*), P < 0.005 (**), P < 0.0005 (***), and P < 0.0001 (****). Scale bar = 25 μm for 2-D cell culture, 10 μm for HAE tissues, and 5 μm for higher magnification insets.
We next examined extension formation of RSV and HMPV in 3-D HAE tissues. When phenotypically examining cross sections of HAE tissues, HMPV-infected cells demonstrated a high percentage of extensions compared with RSV, where only a small number of infected cells with extensions were observed for RSV (Fig. 4C). RSV- and HMPV-induced extensions had similar morphology but were infrequent in RSV-infected HAE tissues. The presence of extensions in HAE tissues was further confirmed by microtome sectioning two full tissues at the peak of infection with either HMPV (48 hpi) or RSV (72 hpi). Extensions were defined and counted as thin protrusions extending from the cell body which were ≥0.5 of the cell body diameter. For HMPV, 35.8% of infected cells contained extensions (1,541 total infected cells counted), while only 4.4% of RSV-infected cells had extensions (3,859 total infected cells counted) (Fig. 4D). Although both RSV and HMPV have been shown to extensively modify the actin cytoskeleton in nonpolarized cell monolayers, RSV rarely forms extensions in HAE tissues, suggesting these extensions are less important for infection and propagation in vivo. Conversely, HMPV generated intercellular extensions in both 2-D and 3-D model systems, supporting the idea that HMPV is primarily cell associated and may utilize extensions for direct cell-to-cell spread (44).
A hallmark characteristic of RSV infection is the formation of multinucleated cells, termed syncytia, which has been shown to be prominent in 2-D cell monolayers. For HMPV, syncytium formation is less than for RSV but is observed in 2-D culture (50, 91). Studies on RSV-mediated syncytium formation in HAE tissues have been inconclusive (53), while for HMPV, no reports have reported formation in HAE models for HMPV. At the peak of infection for both RSV (72 hpi) and HMPV (48 hpi), we examined syncytium formation in two fully microtome sectioned HAE tissues. Interestingly, no clear syncytia were observed for HMPV or RSV in either tissue examined, nor were syncytia visible from the apical side. To ensure this analysis was correct, any regions with potential multinucleated cells were observed in microtome sections using z-stack confocal microscopy and deconvolution, and none of these were found to correspond to syncytia (data not shown). However, the lack of syncytia did not hinder the infectivity or spread of RSV. Since cell-to-cell fusion for both viruses is mediated by the fusion glycoprotein, we examined the localization of F in cryo-sections using immunofluorescence staining to determine if this finding was due to the localization of the F proteins. RSV F was predominantly localized to the apical surface of infected cells but was also present within extensions and throughout the infected cell, with a similar staining pattern observed for HMPV F (Fig. 4C). Since both viral fusion proteins were present at locations beyond the apical surface, there are likely other variables that may contribute to the lack of syncytium formation for HMPV and RSV. Further investigation into the mechanisms that underlie fusion in tissues is required to better understand the factors that contribute to this phenomenon and whether syncytium formation impacts viral replication and spread in vivo.
Nirsevimab, palivizumab, and 54G10 block viral entry and spread in HAE tissues.
Recently, a novel monoclonal anti-F antibody, nirsevimab, showed more potent neutralizing capacity in 2-D cultures and animal models with an extended serum half-life than palivizumab (78, 79, 92). Conversely, no FDA-approved anti-F antibodies are available against HMPV, but a neutralizing monoclonal antibody, 54G10, has been described that has subnanomolar efficacy in vitro against all clades of HMPV and also has cross-reactivity to RSV, although efficacy against RSV is 6× less potent than palivizumab (93). To better understand how HMPV and RSV infection might be targeted in HAE tissues, we compared nirsevimab and palivizumab for RSV, and 54G10 for HMPV, for their ability to inhibit viral entry and spread in 3-D HAE tissues.
We first conducted a microneutralization assay to assess the neutralizing capacity of palivizumab and nirsevimab on RSV in HAE tissues. We preincubated the virus in the presence or absence of neutralizing antibody for 1 hour and then inoculated tissues at the apical surface. As expected, both nirsevimab and palivizumab were able to completely neutralize RSV entry at a concentration of 0.5 μg/ml and 10 μg/ml, respectively (Fig. 5A and B). Congruent with previous findings, nirsevimab demonstrated significantly higher neutralizing potency than palivizumab (approximately 20-fold) (Fig. 5C). Similarly, 54G10 demonstrated a complete block of infection at 10 μg/ml (Fig. 6A and B, quantified in Fig. 6C), indicating therapeutic potential for 54G10 against HMPV.
FIG 5.
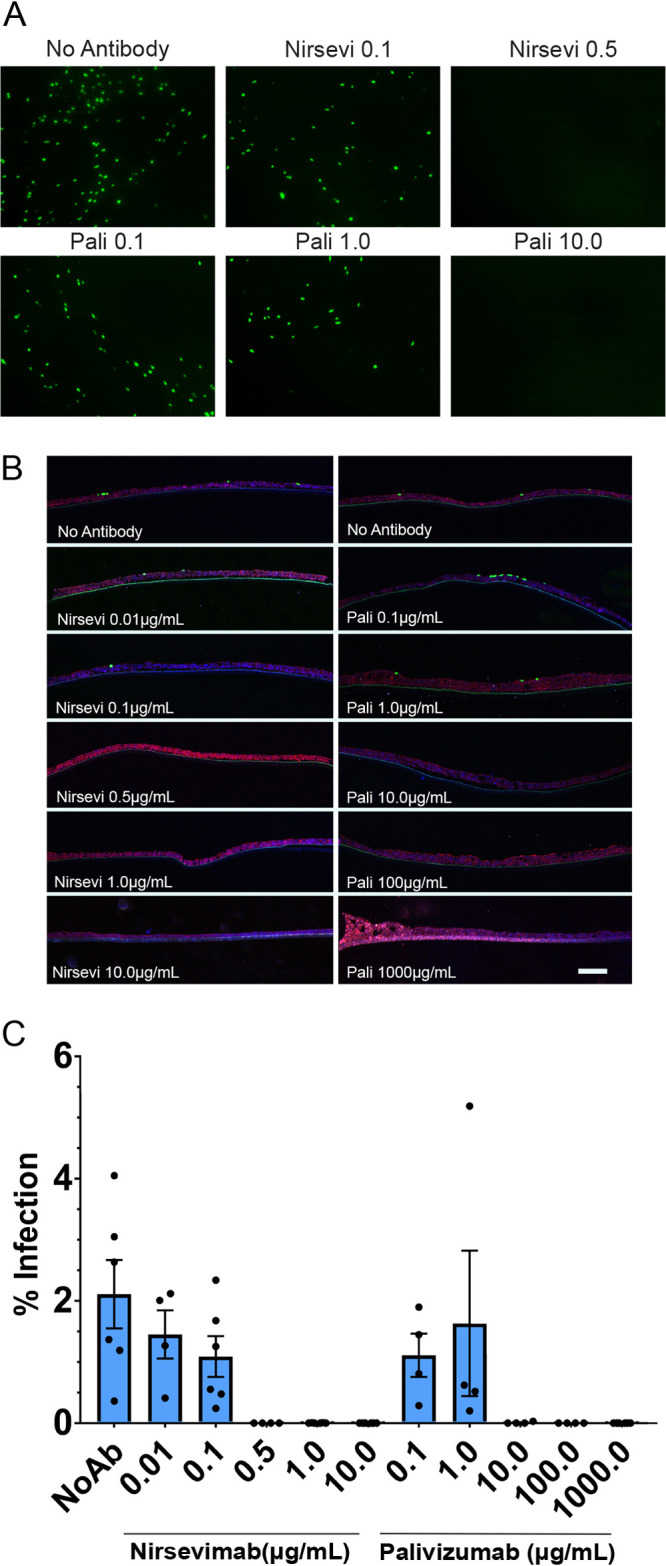
Nirsevimab and palivizumab inhibition of RSV entry. (A) HAE tissues were infected with rgRSV at MOI 1.0 preincubated with or without palivizumab (pali) or nirsevimab (nirsevi) for 1 h at 37°C. Then, 48 hours postinfection, fluorescence was examined by inverted or confocal microscopy to determine inhibition of infection as well as microtome cross sections (B) of infected tissues to examine infection inhibition. (C) Threshold analyses of inverted fluorescence microscopy images were quantified using ImageJ adaptive threshold analysis. Scale bar = 10 μm. Error bars represent SEM of 4 to 6 tissues for each treatment group.
FIG 6.
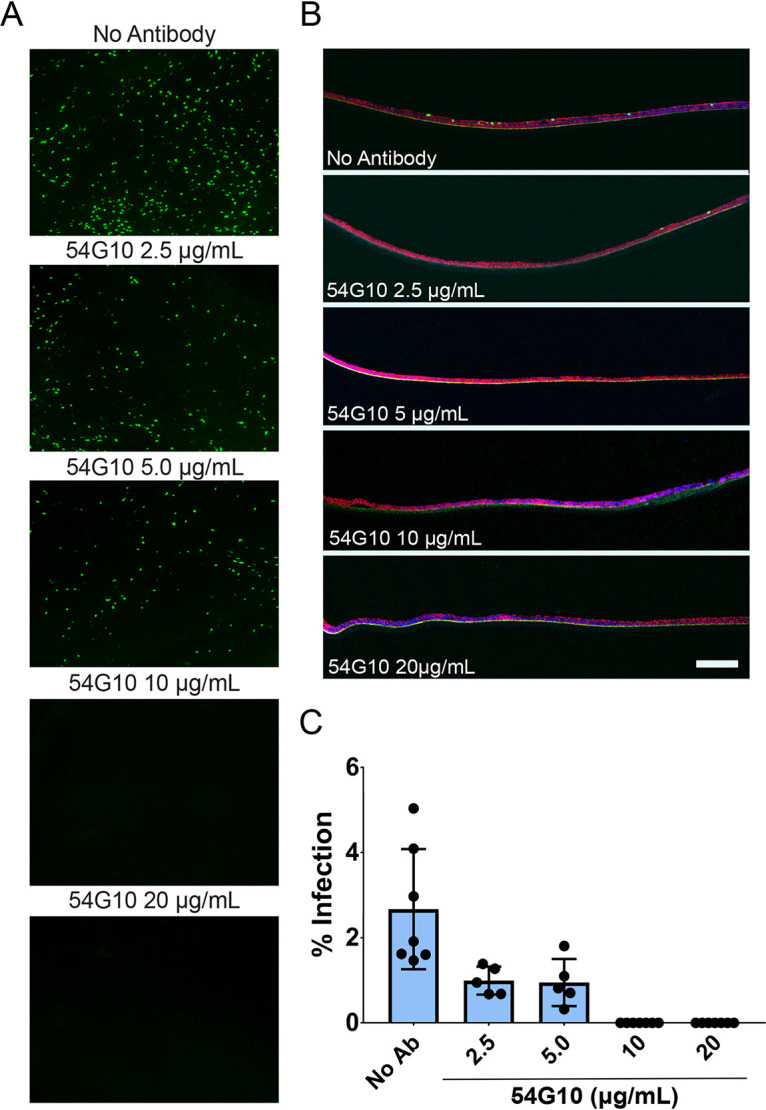
54G10 inhibition of HMPV entry. (A) HAE tissues were infected with rgHMPV at MOI 3.0 preincubated with or without 54G10 for 1 hour at 25°C. Then, 48 hours postinfection, fluorescence was examined by inverted or confocal microscopy to determine inhibition of infection (B) as well as microtome cross sections of infected tissues to examine infection inhibition. (C) Threshold analyses of inverted fluorescence microscopy images were quantified using ImageJ adaptive threshold analysis. Scale bar = 10 μm. Error bars represent SEM of 4 to 6 tissues for each treatment group.
The effect of neutralizing antibodies on RSV and HMPV spread at the apical surface of tissues after infection was examined. We inoculated tissues at the apical surface with RSV or HMPV and then monitored fluorescence after infection. RSV infection alone had a significant increase in spread from 24 hpi through 72 hpi (Fig. 1A and B, 7A to C). In the presence of palivizumab and nirsevimab, RSV spread was almost completely inhibited compared with the 24 hpi time point, demonstrating that RSV spread occurs mostly through apical release and reentry, which can be blocked by antibody present at the apical surface. Our results also confirm previous observations for palivizumab and its ability to prevent spread in HAE tissues (64). Nirsevimab again demonstrated a significantly higher potency (approximately 10-fold) than palivizumab, showing similar inhibition of spread at lower concentrations (Fig. 7A to C).
FIG 7.
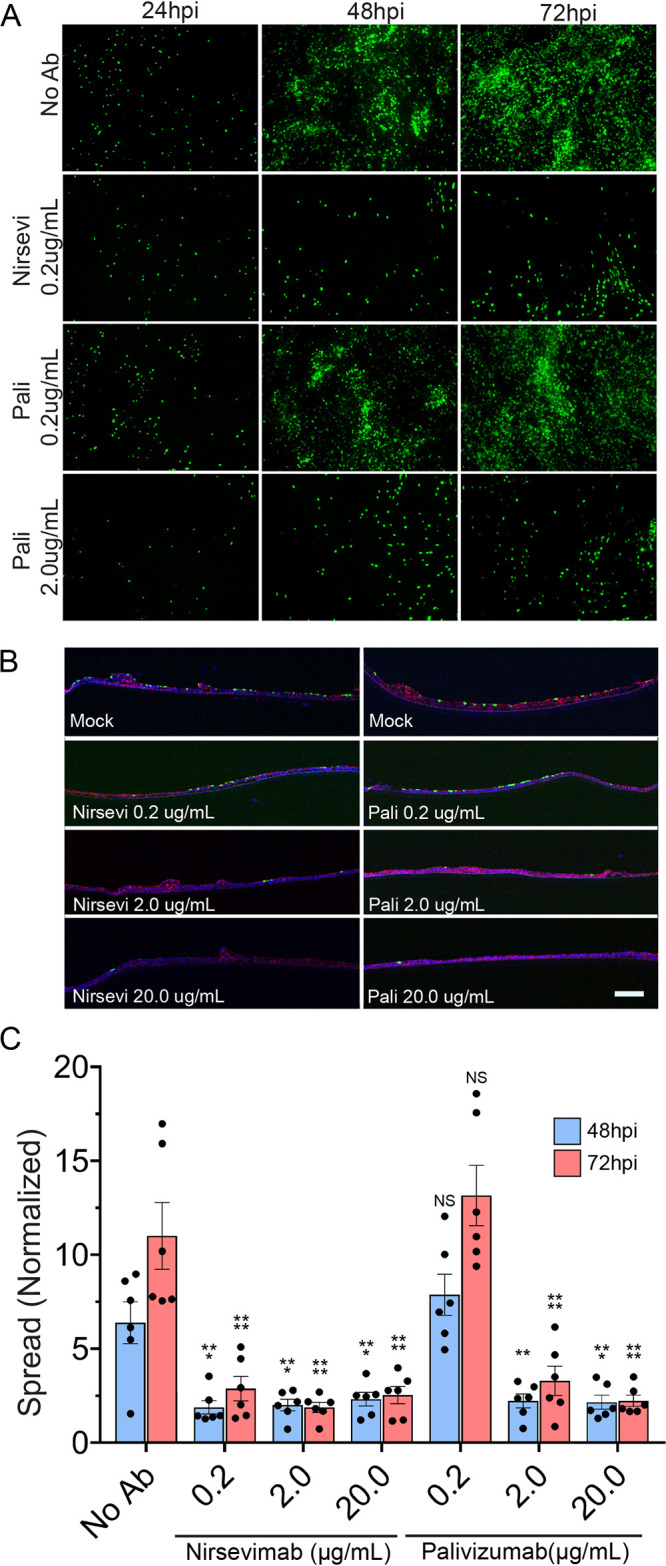
Nirsevimab inhibits spread significantly more potently than palivizumab. (A) HAE tissues were infected with rgRSV at MOI of 0.3. Tissues were treated apically with either palivizumab or nirsevimab 6 hours postinoculation in 50 μl of TEER buffer. Fluorescence microscopy images were taken up to 72 hours postinfection. (B) Tissues were fixed at 72 hpi and microtome-sectioned for each treatment group and examined for viral spread. (C) Fluorescence threshold analyses of inverted fluorescence microscopy images demonstrate that both pali and nirsevi are able to prevent the spread of rgRSV in HAE-infected tissues, with nirsevi able to inhibit spread at lower concentrations than pali. Scale bar = 10 μm. Error bars represent SEM of 6 tissues per treatment group. Statistical significance is represented with P < 0.05 (*), P < 0.005 (**), P < 0.0005 (***), and P < 0.0001 (****).
We also examined 54G10 inhibition of HMPV spread from 24 hpi through 48 hpi. There was a modest but statistically significant inhibition in HMPV spread with increasing amounts of 54G10 (Fig. 8A to C). Spread in the presence of 2.5 μg/ml and 25 μg/ml of neutralizing antibody was only reduced by 25%, suggesting that a large fraction of HMPV spread likely still occurs via a direct cell-to-cell transmission mechanism likely utilizing extensions as shown in 2-D cell monolayers (44). Further investigation into the mechanisms for HMPV cell-to-cell spread in 3-D tissues will help to elucidate how HMPV spread can be targeted therapeutically. Altogether, our results indicate the mechanisms by which neutralizing antibodies act to prevent RSV infections, although a different strategy may be needed to fully inhibit HMPV spread.
FIG 8.
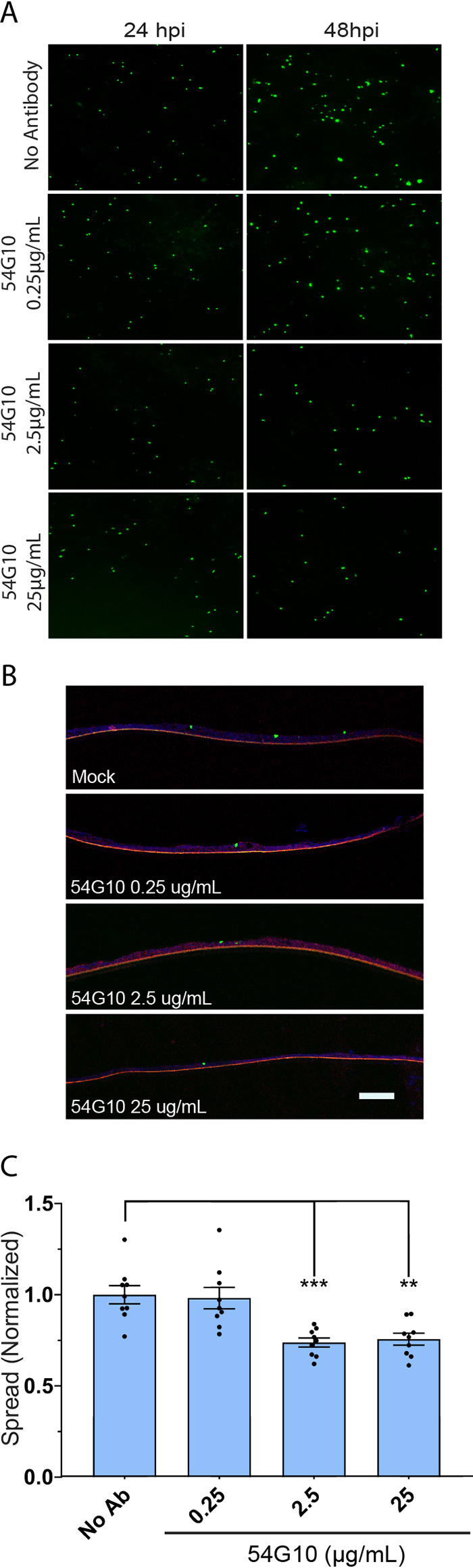
54G10 significantly inhibits spread of HMPV. (A) HAE tissues were infected with rgHMPV at MOI of 3.0. Tissues were treated apically with 54G10 6 hours postinoculation in 50 μl of TEER buffer. Fluorescence microscopy images were taken up to 72 hours postinfection. (B) Tissues were fixed at 48 hpi and microtome-sectioned for each treatment group and examined for viral spread. (C) Fluorescence threshold analyses of inverted fluorescence microscopy images demonstrate that 54G10 modestly but significantly prevents the spread of rgHMPV in HAE infected tissues. Scale bar = 10 μm. Error bars represent SEM of 6 tissues per treatment group. Statistical significance is represented with P < 0.05 (*), P < 0.005 (**), P < 0.0005 (***), and P < 0.0001 (****).
DISCUSSION
In this study, we conducted a comparative analysis of HMPV and RSV infection in HAE tissues. Altogether, our results demonstrate that two closely related human respiratory pathogens may utilize significantly different mechanisms of spread in a 3-D model system. RSV can infect, replicate, and release large amounts of virus apically, resulting in very efficient spread. In striking contrast, HMPV is also able to infect ciliated cells in the HAE tissue, and productive establishment of replication centers is seen, as judged by production of viral RNA, but little apical release of virus was observed, leading to poor spread in this system.
Initial infection mediated by HMPV required higher MOIs to achieve similar infection rates, suggesting that RSV infects HAE tissues more efficiently than HMPV. RSV spread in HAE tissues increased significantly from 24 hpi up to 72 hpi, compared with HMPV which reached a peak of infection at 48 hpi and significantly decreased thereafter. One potential reason for this observation is the antiviral response from infected tissues. RSV encodes two additional nonstructural (NS) proteins, NS1 and NS2, which show significant inhibition of this response (94). Some proteins from HMPV, including G, M2-2, and SH, are also able to inhibit the interferon response (95), but potentially to a lesser extent than RSV, which could decrease spread in comparison. Alternatively, HMPV has been shown to establish persistent infections (96–101). Thus, it is possible that the low replication rates of HMPV in HAE tissues may drive the virus toward persistence rather than an acute infection. In support of this, HMPV peaked at 48 hpi, but a low, residual infection was present up to 144 hpi. When infected tissues were examined, both HMPV and RSV primarily infected apical ciliated cells, as previously demonstrated (53, 63, 64, 71). Therefore, the differences observed in entry and replication between HMPV and RSV do not appear to be due to the cell types infected. A recent publication utilized HMPV CAN97-82 (B strain) infection in human airway epithelium obtained from nasal biopsy specimens and showed limited apical release up to 5 days postinfection (76). These studies and ours suggest strain differences may affect how HAE tissues release and spread virus to some degree. The presence of both the HMPV SH and G proteins, as well as the strain used, was recently reported to impact spread in HAE tissues (77), with the deletion of HMPV G being especially deleterious. Interestingly, HMPV infection occurred at a higher level in these studies, and further investigation is needed to understand differences involved in efficient HMPV entry in 3-D tissues (63, 75–77).
Once cells have been infected, both HMPV and RSV form IBs. The formation and characterization of these replication organelles in 2-D model systems have been previously reported (27, 28), but IB formation has not been previously examined in 3-D tissues. We showed that these IB structures form within HAE-infected cells and contain markers for IBs, including P, N, and viral RNA. This is the first report of the formation of these structures in 3-D HAE tissues for both RSV and HMPV and suggests these structures are broadly important for pneumovirus infection. As these structures appear in 2-D models for a number of negative-sense, single-stranded RNA viruses (28–36), identifying and understanding critical host and viral components of these organelles may provide a unique antiviral approach against a wide range of human viral pathogens.
Recent reports have shown both RSV and HMPV form actin-based filamentous structures, important for viral spread within 2-D monolayers (44, 45). Viral titer for RSV was significantly reduced when these structures were inhibited (45). For HMPV, these filamentous structures were shown to be involved in cell-to-cell spread of the virus, independent of neutralizing antibodies in BEAS-2B cells. Similar to RSV, when these cytoskeletal structures were inhibited, viral spread was significantly reduced (44).
To assess the importance of cellular polarization on extension formation in 2-D monolayers, we examined extension formation in 3-D HAE tissues. Interestingly, extensions similar to those observed in 2-D cells were also detected in approximately a third of HMPV-infected cells within HAE tissues, whereas RSV-infected cells had few extensions. Thus, we hypothesize that infection by RSV relies primarily on abundant release of particles from the apical side of tissues and reinfection of new target cells. However, the low apical release of virus and high percentage of cells with extensions suggests that HMPV infection of new target cells is likely dependent primarily on direct cell-to-cell contacts, similar to previous findings in 2-D models. Furthermore, HMPV was also demonstrated to be primarily associated with minimal release, and therefore extensions may be the primary mechanism of spread (44). However, even with the formation of extensions, spread of HMPV was minimal, suggesting that additional factors may be needed for efficient cell-to-cell spread in HAE tissues.
RSV forms large, multinucleated cells termed syncytia, which are a hallmark of infection in 2-D monolayers. In addition, while not as prominent, HMPV also mediates cell-to-cell fusion during monolayer infection (50, 51). Some studies have indicated that RSV can form minimal and infrequent syncytia in HAE tissues; however, there are conflicting results (53, 73). Analysis of lung autopsy specimens for both viruses suggested that syncytium formation can occur in vivo (9, 43). We were unable to identify any syncytia in either the fully sectioned tissues or apical images for RSV or HMPV. Studies in polarized cell monolayers or in HAE tissues demonstrated that RSV F was primarily localized to the apical surface (53, 102). Here, we also found that RSV and HMPV F were primarily localized to the apical surface. In addition, we also observed the presence of staining at the basolateral portion of infected cells. Other factors for cell-to-cell fusion may be required in order to mediate the formation of syncytia in addition to the cellular distribution of F. It is also unknown if syncytium formation is beneficial for viral replication. An RSV strain containing a hyperfusogenic fusion protein led to larger syncytia in 2-D monolayers and higher pathogenesis in vivo, suggesting that higher fusion is beneficial for viral replication and spread (52).
One caveat in HAE tissues that is different from in vivo studies is the lack of immune cells, which play a major role during infection. Their absence may contribute to our observations related to initial infection rate, apical release, spread kinetics, and lack of cytopathic effects. The inflammatory response has been suggested to be important for RSV infection (103) and may also play roles in HMPV infection as well. The inflammatory response yields damage to the lung epithelium layer, which may expose proteins and other factors present in tight junctions, not normally accessible to the virus (104, 105), and this could aid in HMPV spread. Further experiments are needed to understand how immune cells and the inflammatory response could modulate HMPV infection of airways.
Lastly, therapeutic monoclonal antibodies against RSV and HMPV were evaluated for their inhibition of viral infection and spread in this study. Preincubation of RSV with palivizumab or nirsevimab showed neutralization and spread inhibition for RSV in HAE tissues. Nirsevimab demonstrated a significantly higher neutralizing capacity than palivizumab (79). These results confirm the potential for nirsevimab, which is currently in late-stage clinical study, for immunoprophylaxis against RSV. Studies in 2-D in vitro cultures and in vivo studies have demonstrated increased efficacy and potency against RSV compared with the only available therapeutic, palivizumab (78, 79, 92), which are further supported by the results of this study.
We also examined the effect of 54G10, a potent neutralizing monoclonal antibody against HMPV F (93) for inhibition HMPV infection and spread in HAE. Preincubation of HMPV with 54G10 at a concentration of 5 μg/ml completely inhibited viral entry. However, spread was only modestly, but significantly, reduced at 2.5 μg/ml, and increasing the concentration of 54G10 to 25 μg/ml did not result in further spread reduction. These results suggest that at least a portion of HMPV spread in HAE tissues may occur by a neutralizing antibody-independent cell-to-cell mechanism. Thus, targeting the F protein of HMPV may not be the most effective antiviral therapy. The RSV and HMPV antibodies have different potencies in 2-D cell culture and bind the F protein at different sites (palivizumab: site II, nirsevimab: site 0, and 54G10: site IV). It is unclear if the modest inhibition of spread by 54G10 is related to a certain F binding mechanism of action. Future studies evaluating a large panel of antibodies could help to determine if this phenomenon is specific for HMPV or related to F protein binding. It is important that future studies assess other cross-neutralizing antibodies that bind alternative epitopes, including MPE8 and 25P13 (106, 107), to better assess their effects on viral infection and spread. This information would provide better understanding of which epitopes are most potent for viral inhibition and potentially lead to generating a highly potent and cross-reactive antibody for both RSV and HMPV.
While this study furthers our understanding of two important respiratory pathogens in a more physiologically relevant model, there is further research needed to characterize pneumovirus infection. Here, we utilize A2 subtypes of both HMPV and RSV to model viral replication in HAE tissues. However, it is possible there is strain-to-strain variation which can only be determined with additional studies in this model system. We also exploited monoclonal antibodies to understand entry and spread of RSV and HMPV in HAE tissues and examined the efficacy of these antibodies as potential antiviral therapeutics. However, additional antibodies against both RSV and HMPV should be analyzed to determine which viral protein sites are more important to entry and spread and use this information to identify optimal sites for antiviral targets. HAE tissues offer a unique model system to better understand viral infection in 3-D human tissue using an in vitro culture method, with caveats. An important role of the respiratory tract is a protective barrier for invading pathogens. In response, the lung signals the immune system to fight infection. This results in the inhibition and clearance of infection but also generates tissue damage as a result of inflammation and infection. This critical aspect is absent in the HAE system, and therefore, this level of complexity is inaccessible using this method.
Altogether, these results demonstrate a significant dichotomy between two very closely related pneumoviruses and illustrate important aspects of their viral life cycles. RSV spreads primarily through release and reentry of viral particles. Current approaches to combat RSV entry and spread by targeting the F protein are a promising avenue for RSV antiviral therapeutics. However, HMPV spread is poorly affected via this approach, suggesting other aspects of the viral life cycle may be more effective. Both RSV and HMPV, in addition to other nsRNA viruses, form punctate replication organelles in the cytoplasm of infected cells. It is possible that targeting components involved in the formation and maintenance of these viral organelles may prove to be a potent, broad-spectrum antiviral for many significant viral pathogens. As there are limited studies analyzing these structures, more research is needed to better understand their formation, function, and key role in viral replication.
MATERIALS AND METHODS
Cell lines, viruses, and antibodies.
HEp-2 and BEAS-2B cells (ATCC) were grown in Opti-MEM (Gibco) supplemented with 2% fetal bovine serum (FBS; Sigma). Vero cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FBS. All cells were maintained at 37°C and 5% CO2. Recombinant respiratory syncytial virus A2 expressing green fluorescent protein (GFP, rgRSV-A2) was propagated in HEp-2 cells, and recombinant human metapneumovirus strain CAN97-83 expressing GFP (rgHMPV) (a kind gift from Peter Collins and Ursula Buchholz, NIAID) was propagated in Vero cells. Monoclonal antibody 54G10 against HMPV F was a kind gift from John Williams (University of Pittsburgh Children’s Hospital).
Infection of human airway epithelial (HAE) tissues.
Human tracheal bronchial differentiated airway (Epiairway) tissues were purchased from MatTek and maintained in 3 ml of Air-100 medium at 37°C for 1 week prior to infection, with the medium changed and the apical surface washed with 0.9% NaCl (in dH2O) every other day to remove mucus. Prior to infection, the basal surface was washed with HEPES buffered saline (HBS) for 30 minutes and the apical surface was washed three times with 0.9% NaCl every 10 minutes with incubation. For HMPV, the apical surface washes were performed using alpha-lysophosphatidylcholine (Sigma) in HBS (75 μg/ml) as previously described by our lab and others (63, 75). Tissues were infected with either rgRSV or rgHMPV on the apical surface and incubated for 3 hours at 37°C. The apical side of tissues was then washed once with HBS and incubated at 37°C. Medium containing 0.3 μg/ml TPCK-trypsin was added to the basal side of rgHMPV-infected tissues and replenished daily due to the requirement of exogenous protease for F protein cleavage. Images were obtained using a Zeiss Axiovert 100 or a Nikon Ti-2. After the final time point, both apical and basal sides were washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 20 minutes at room temperature.
Viral microneutralization in HAE tissues.
The rgRSV (MOI 1) was preincubated with nirsevimab or palivizumab for 1 hour at 37°C. rgHMPV (MOI 3) was preincubated with 54G10 monoclonal antibody (MAb) for 1 hour at room temperature. Both rgRSV and rgHMPV incubations were completed in a total volume of 150 μl TEER buffer (MatTek). The total microneutralization volume was added to the apical surface of tissues and incubated for 3 hours at 37°C. After incubation, the inoculation mixture was aspirated and the apical surface was washed 1× with HBS and incubated at 37°C for up to 48 hours.
Viral spread in HAE tissues.
HAE tissues were infected with rgRSV (MOI 0.3) or rgHMPV (MOI 3.0) for 3 hours at 37°C. Inoculation mixture was aspirated and washed 1× with HBS. Antibody dilutions were completed in 50 μl of TEER buffer and added to the apical surface. The initial antibody at indicated concentrations was added at 6 hpi and replenished every 24 hours postinoculation until experiments were completed.
Analysis of apical release of virus.
To collect viruses released from the 3-D HAE culture, 100 μl of Opti-MEM was added to the apical surface of infected HAE tissues and incubated for 10 minutes at room temperature. After incubation, apical supernatant was removed and flash frozen in dry ice/MeOH and stored at –80°C. The following day, serial dilutions of each virus were completed starting at 10−1 and going up to 10−8. Viral dilutions were added to fully confluent Vero cells and incubated overnight. Fluorescence-forming units (FFU) were counted, and viral titers were determined as Log10 FFU/ml.
Confocal microscopy.
HAE tissues were removed from trans-wells and frozen in O.C.T compounding embedding medium (EM Sciences). Tissue sections at 10 to 50 μm were cut using a Microm HM 525 cryostat and collected on Superfrost Plus slides and heat fixed at 55°C for 30 minutes. All sections were stored at –20°C for further use and brought to room temperature prior to processing. Sections were rinsed 1× with PBS and permeabilized in 1.0% Triton X-100 for 15 minutes at 4°C followed by blocking in 1% normal goat serum. For RSV, sections were incubated with primary antibodies for RSV F (1:600), N (1:200), P (1:600) (Abcam), or keratan sulfate (1:1,200) (EMD Millipore), and for HMPV, the primary antibodies were HMPV F (1:500), N (1:100), P (1:500) (Abcam), or keratan sulfate. All primary antibodies were diluted in 1% normal goat serum and incubated at 4°C overnight.
The following day, tissues were washed with 0.05% Tween-PBS, and Alexa647 goat anti-mouse IgG (1:300) (Jackson ImmunoResearch) and TRITC-Phalloidin (1:1,000) (Invitrogen) were added at room temperature for 1 hour. Tissues were then washed and mounted using SloFade plus DAPI (4′,6-diamidino-2-phenylindole; Invitrogen). Immunolabelling of P and N within infected cells utilized isotype-specific antibodies (Jackson ImmunoResearch). Imaging was performed on a Nikon A1R confocal microscope and analyzed with NIS-Elements software.
Stellaris fluorescent in situ hybridization (FISH) for viral RNA detection.
All FISH probes were obtained from and generated using software from BioSearch Technologies (Novato, CA). For HMPV, 48 DNA probes targeting the HMPV vRNA genome between nucleotides (nt) 1 and 5467 were generated. Each probe is 20 nt long and linked at the 3′ end to Quasar 570 fluorophore. For RSV, 48 DNA probes targeting the RSV vRNA genome between nt 1 and 7324 were generated. Each probe is 20 nt long and linked at the 3′ end to Quasar 670 fluorophore. HAE cultures were fixed, permeabilized, blocked as described above, and immunostained with antibodies against P or N for the respective virus. After washing in PBS-Tween, sections were washed twice with 2× SSC-10% formamide buffer and then incubated overnight at 30°C in hybridization buffer (4× SSC, [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate] 1× Denhardt’s solution, 150 μg/ml single-stranded DNA [ssDNA], 2 mM EDTA, 50% formamide in diethyl pyrocarbonate [DEPC]-treated water) containing the probes at a concentration of 2.5 μM for 18 hours. The sections were washed two times for 20 min with 2× SSC-10% formamide buffer, and slides were mounted using SloFade plus DAPI (Invitrogen). Imaging was performed using a Nikon A1R confocal microscope and analyzed with NIS-Elements software.
Fluorescence threshold analysis.
GFP cells present within infected tissues were quantified with either NIS-Elements “object count” or ImageJ (FIJI) using the “adaptive threshold” analysis plugin. Briefly, for ImageJ quantification of fluorescence microscopy images of HAE tissues, background was subtracted for each image and converted to black and white (B&W). Using several test images, the adaptive threshold was set to ensure that all data points with minimal background were obtained, and this macro was used for all images within that replicate. Finally, particle density was quantified using “analyze particles” and total density was quantified as percent area. GFP expression was also analyzed with NIS-Elements software using threshold analysis based on area coverage using the “object count” plug-in and a selected fluorescence threshold. All counts were taken as percent area coverage. A total of 4 to 5 fields were taken for each tissue, and the counts averaged.
Analysis of extension formation.
BEAS-2B, HEp-2, and VERO cells were infected with either rgRSV or rgHMPV. At 24 hpi, cells were fixed and processed for immunofluorescence (IF) as described above. Cells were stained for the presence of RSV or HMPV F and counterstained for actin using TRITC-phalloidin. For HAE tissues, two full tissues were cut in 50-μm sections, and all infected cells and those exhibiting extensions were manually counted. Due to lower infection rates for HMPV, all sections were used. For RSV, 4 random slides were counted to match the number of cells for both viruses.
Statistical analysis.
Statistical analysis was preformed using Prism 7 for Windows (GraphPad). A P value of <0.05 was considered statistically significant. Multiple-comparison tests were generated using a one-way analysis of variance (ANOVA) with a Bonferroni multiple comparison correction.
ACKNOWLEDGMENTS
We thank John Williams (University of Pittsburgh Children’s Hospital) for providing the 54G10 antibody used in these studies and Peter Collins and Ursula Buchholz (NIAID) for providing rgHMPV-GFP.
Financial support was provided by AstraZeneca and NIH R01AI140758 to R.E.D.
REFERENCES
- 1.Afonso CL, Amarasinghe GK, Bányai K, Bào Y, Basler CF, Bavari S, Bejerman N, Blasdell KR, Briand F-X, Briese T, Bukreyev A, Calisher CH, Chandran K, Chéng J, Clawson AN, Collins PL, Dietzgen RG, Dolnik O, Domier LL, Dürrwald R, Dye JM, Easton AJ, Ebihara H, Farkas SL, Freitas-Astúa J, Formenty P, Fouchier RAM, Fù Y, Ghedin E, Goodin MM, Hewson R, Horie M, Hyndman TH, Jiāng D, Kitajima EW, Kobinger GP, Kondo H, Kurath G, Lamb RA, Lenardon S, Leroy EM, Li C-X, Lin X-D, Liú L, Longdon B, Marton S, Maisner A, Mühlberger E, Netesov SV, Nowotny N, et al. . 2016. Taxonomy of the order Mononegavirales: update 2016. Arch Virol 161:2351–2360. doi: 10.1007/s00705-016-2880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. 2009. The burden of respiratory syncytial virus infection in young children. N Engl J Med 360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7:719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leung J, Esper F, Weibel C, Kahn JS. 2005. Seroepidemiology of human metapneumovirus (hMPV) on the basis of a novel enzyme-linked immunosorbent assay utilizing hMPV fusion protein expressed in recombinant vesicular stomatitis virus. J Clin Microbiol 43:1213–1219. doi: 10.1128/JCM.43.3.1213-1219.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain S, Williams DJ, Arnold SR, Ampofo K, Bramley AM, Reed C, Stockmann C, Anderson EJ, Grijalva CG, Self WH, Zhu Y, Patel A, Hymas W, Chappell JD, Kaufman RA, Kan JH, Dansie D, Lenny N, Hillyard DR, Haynes LM, Levine M, Lindstrom S, Winchell JM, Katz JM, Erdman D, Schneider E, Hicks LA, Wunderink RG, Edwards KM, Pavia AT, McCullers JA, Finelli L. 2015. Community-acquired pneumonia requiring hospitalization among U.S. children. N Engl J Med 372:835–845. doi: 10.1056/NEJMoa1405870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahn JS. 2006. Epidemiology of human metapneumovirus. Clin Microbiol Rev 19:546–557. doi: 10.1128/CMR.00014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebihara T, Endo R, Kikuta H, Ishiguro N, Yoshioka M, Ma X, Kobayashi K. 2003. Seroprevalence of human metapneumovirus in Japan. J Med Virol 70:281–283. doi: 10.1002/jmv.10391. [DOI] [PubMed] [Google Scholar]
- 8.Wolf DG, Zakay-Rones Z, Fadeela A, Greenberg D, Dagan R. 2003. High seroprevalence of human metapneumovirus among young children in Israel. J Infect Dis 188:1865–1867. doi: 10.1086/380100. [DOI] [PubMed] [Google Scholar]
- 9.Vargas SO, Kozakewich HP, Perez-Atayde AR, McAdam AJ. 2004. Pathology of human metapneumovirus infection: insights into the pathogenesis of a newly identified respiratory virus. Pediatr Dev Pathol 7:478–486. Discussion 421. doi: 10.1007/s10024-004-1011-2. [DOI] [PubMed] [Google Scholar]
- 10.Hamelin M-È, Abed Y, Boivin G. 2004. Human metapneumovirus: a new player among respiratory viruses. Clin Infect Dis 38:983–990. doi: 10.1086/382536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraha HY, Lanctot KL, Paes B. 2015. Risk of respiratory syncytial virus infection in preterm infants: reviewing the need for prevention. Expert Rev Respir Med 9:779–799. doi: 10.1586/17476348.2015.1098536. [DOI] [PubMed] [Google Scholar]
- 12.Matias G, Taylor R, Haguinet F, Schuck-Paim C, Lustig R, Shinde V. 2017. Estimates of hospitalization attributable to influenza and RSV in the US during 1997-2009, by age and risk status. BMC Public Health 17:271. doi: 10.1186/s12889-017-4177-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 14.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O’Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian KN, Weisman LE, Rhodes T, Ariagno R, Sanchez PJ, Steichen J, Givner LB, Jennings TL, Top FH Jr, Carlin D, Connor E. 1998. Safety, tolerance and pharmacokinetics of a humanized monoclonal antibody to respiratory syncytial virus in premature infants and infants with bronchopulmonary dysplasia. MEDI-493 Study Group. Pediatr Infect Dis J 17:110–115. doi: 10.1097/00006454-199802000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Johnson S, Oliver C, Prince GA, Hemming VG, Pfarr DS, Wang SC, Dormitzer M, O’Grady J, Koenig S, Tamura JK, Woods R, Bansal G, Couchenour D, Tsao E, Hall WC, Young JF. 1997. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis 176:1215–1224. doi: 10.1086/514115. [DOI] [PubMed] [Google Scholar]
- 17.Battles MB, McLellan JS. 2019. Respiratory syncytial virus entry and how to block it. Nat Rev Microbiol 17:233–245. doi: 10.1038/s41579-019-0149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox RG, Williams JV. 2013. Breaking in: human metapneumovirus fusion and entry. Viruses 5:192–210. doi: 10.3390/v5010192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biacchesi S, Skiadopoulos MH, Yang L, Lamirande EW, Tran KC, Murphy BR, Collins PL, Buchholz UJ. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J Virol 78:12877–12887. doi: 10.1128/JVI.78.23.12877-12887.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Techaarpornkul S, Barretto N, Peeples ME. 2001. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J Virol 75:6825–6834. doi: 10.1128/JVI.75.15.6825-6834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bukreyev A, Whitehead SS, Murphy BR, Collins PL. 1997. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J Virol 71:8973–8982. doi: 10.1128/JVI.71.12.8973-8982.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang A, Masante C, Buchholz UJ, Dutch RE. 2012. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J Virol 86:3230–3243. doi: 10.1128/JVI.06706-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Srivastava M. 2018. Prophylactic and therapeutic approaches for human metapneumovirus. Virusdisease 29:434–444. doi: 10.1007/s13337-018-0498-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villafana T, Falloon J, Griffin MP, Zhu Q, Esser MT. 2017. Passive and active immunization against respiratory syncytial virus for the young and old. Expert Rev Vaccines 16:1–13. doi: 10.1080/14760584.2017.1333425. [DOI] [PubMed] [Google Scholar]
- 25.Mejias A, Garcia-Maurino C, Rodriguez-Fernandez R, Peeples ME, Ramilo O. 2017. Development and clinical applications of novel antibodies for prevention and treatment of respiratory syncytial virus infection. Vaccine 35:496–502. doi: 10.1016/j.vaccine.2016.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen SC, Williams JV. 2015. New approaches for immunization and therapy against human metapneumovirus. Clin Vaccine Immunol 22:858–866. doi: 10.1128/CVI.00230-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rincheval V, Lelek M, Gault E, Bouillier C, Sitterlin D, Blouquit-Laye S, Galloux M, Zimmer C, Eleouet JF, Rameix-Welti MA. 2017. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat Commun 8:563. doi: 10.1038/s41467-017-00655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cifuentes-Munoz N, Branttie J, Slaughter KB, Dutch RE. 2017. Human metapneumovirus induces formation of inclusion bodies for efficient genome replication and transcription. J Virol 91. doi: 10.1128/JVI.01282-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson EV, Schmidt KM, Deflubé LR, Doğanay S, Banadyga L, Olejnik J, Hume AJ, Ryabchikova E, Ebihara H, Kedersha N, Ha T, Mühlberger E. 2016. Ebola virus does not induce stress granule formation during infection and sequesters stress granule proteins within viral inclusions. J Virol 90:7268–7284. doi: 10.1128/JVI.00459-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, Feldmann H. 2012. Inclusion bodies are a site of ebolavirus replication. J Virol 86:11779–11788. doi: 10.1128/JVI.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolesnikova L, Muhlberger E, Ryabchikova E, Becker S. 2000. Ultrastructural organization of recombinant Marburg virus nucleoprotein: comparison with Marburg virus inclusions. J Virol 74:3899–3904. doi: 10.1128/jvi.74.8.3899-3904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikolic J, Civas A, Lama Z, Lagaudriere-Gesbert C, Blondel D. 2016. Rabies virus infection induces the formation of stress granules closely connected to the viral factories. PLoS Pathog 12:e1005942. doi: 10.1371/journal.ppat.1005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog 6:e1000958. doi: 10.1371/journal.ppat.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang S, Jiang Y, Cheng Q, Zhong Y, Qin Y, Chen M. 2017. Inclusion body fusion of human parainfluenza virus type 3 regulated by acetylated alpha-tubulin enhances viral replication. J Virol 91:e01802-16. doi: 10.1128/JVI.01802-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlos TS, Young DF, Schneider M, Simas JP, Randall RE. 2009. Parainfluenza virus 5 genomes are located in viral cytoplasmic bodies whilst the virus dismantles the interferon-induced antiviral state of cells. J Gen Virol 90:2147–2156. doi: 10.1099/vir.0.012047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schudt G, Dolnik O, Kolesnikova L, Biedenkopf N, Herwig A, Becker S. 2015. Transport of ebolavirus nucleocapsids is dependent on actin polymerization: live-cell imaging analysis of ebolavirus-infected cells. J Infect Dis 212(Suppl 2):S160–S166. doi: 10.1093/infdis/jiv083. [DOI] [PubMed] [Google Scholar]
- 38.Dietzel E, Kolesnikova L, Maisner A. 2013. Actin filaments disruption and stabilization affect measles virus maturation by different mechanisms. Virol J 10:249–249. doi: 10.1186/1743-422X-10-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santangelo PJ, Bao G. 2007. Dynamics of filamentous viral RNPs prior to egress. Nucleic Acids Res 35:3602–3611. doi: 10.1093/nar/gkm246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh BK, Pfaller CK, Cattaneo R, Sinn PL. 2019. Measles virus ribonucleoprotein complexes rapidly spread across well-differentiated primary human airway epithelial cells along F-actin rings. mBio 10:e02434-19. doi: 10.1128/mBio.02434-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cifuentes-Muñoz N, Ellis Dutch R. 2019. To assemble or not to assemble: the changing rules of pneumovirus transmission. Virus Res 265:68–73. doi: 10.1016/j.virusres.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol 6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- 43.Neilson KA, Yunis EJ. 1990. Demonstration of respiratory syncytial virus in an autopsy series. Pediatr Pathol 10:491–502. doi: 10.3109/15513819009067138. [DOI] [PubMed] [Google Scholar]
- 44.El Najjar F, Cifuentes-Muñoz N, Chen J, Zhu H, Buchholz UJ, Moncman CL, Dutch RE. 2016. Human metapneumovirus induces reorganization of the actin cytoskeleton for direct cell-to-cell spread. PLoS Pathog 12:e1005922. doi: 10.1371/journal.ppat.1005922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehedi M, McCarty T, Martin SE, Le Nouen C, Buehler E, Chen YC, Smelkinson M, Ganesan S, Fischer ER, Brock LG, Liang B, Munir S, Collins PL, Buchholz UJ. 2016. Actin-related protein 2 (ARP2) and virus-induced filopodia facilitate human respiratory syncytial virus spread. PLoS Pathog 12:e1006062. doi: 10.1371/journal.ppat.1006062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liljeroos L, Krzyzaniak MA, Helenius A, Butcher SJ. 2013. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc Natl Acad Sci U S A 110:11133–11138. doi: 10.1073/pnas.1309070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bächi T, Howe C. 1973. Morphogenesis and ultrastructure of respiratory syncytial virus. J Virol 12:1173–1180. doi: 10.1128/JVI.12.5.1173-1180.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peret TC, Boivin G, Li Y, Couillard M, Humphrey C, Osterhaus AD, Erdman DD, Anderson LJ. 2002. Characterization of human metapneumoviruses isolated from patients in North America. J Infect Dis 185:1660–1663. doi: 10.1086/340518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loo LH, Jumat MR, Fu Y, Ayi TC, Wong PS, Tee NWS, Tan BH, Sugrue RJ. 2013. Evidence for the interaction of the human metapneumovirus G and F proteins during virus-like particle formation. Virol J 10:294. doi: 10.1186/1743-422X-10-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dubois J, Cavanagh MH, Terrier O, Hamelin ME, Lina B, Shi R, Rosa-Calatrava M, Boivin G. 2017. Mutations in the fusion protein heptad repeat domains of human metapneumovirus impact on the formation of syncytia. J Gen Virol 98:1174–1180. doi: 10.1099/jgv.0.000796. [DOI] [PubMed] [Google Scholar]
- 51.Aerts L, Cavanagh M-H, Dubois J, Carbonneau J, Rhéaume C, Lavigne S, Couture C, Hamelin M-È, Boivin G. 2015. Effect of in vitro syncytium formation on the severity of human metapneumovirus disease in a murine model. PLoS One 10:e0120283. doi: 10.1371/journal.pone.0120283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hotard AL, Lee S, Currier MG, Crowe JE Jr, Sakamoto K, Newcomb DC, Peebles RS Jr, Plemper RK, Moore ML. 2015. Identification of residues in the human respiratory syncytial virus fusion protein that modulate fusion activity and pathogenesis. J Virol 89:512–522. doi: 10.1128/JVI.02472-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villenave R, Shields MD, Power UF. 2013. Respiratory syncytial virus interaction with human airway epithelium. Trends Microbiol 21:238–244. doi: 10.1016/j.tim.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 54.Ravi M, Paramesh V, Kaviya SR, Anuradha E, Solomon FD. 2015. 3D cell culture systems: advantages and applications. J Cell Physiol 230:16–26. doi: 10.1002/jcp.24683. [DOI] [PubMed] [Google Scholar]
- 55.Mertens TCJ, Karmouty-Quintana H, Taube C, Hiemstra PS. 2017. Use of airway epithelial cell culture to unravel the pathogenesis and study treatment in obstructive airway diseases. Pulm Pharmacol Ther 45:101–113. doi: 10.1016/j.pupt.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 56.Hasan S, Sebo P, Osicka R. 2018. A guide to polarized airway epithelial models for studies of host-pathogen interactions. FEBS J 285:4343–4358. doi: 10.1111/febs.14582. [DOI] [PubMed] [Google Scholar]
- 57.Bhowmick R, Derakhshan T, Liang Y, Ritchey J, Liu L, Gappa-Fahlenkamp H. 2018. A three-dimensional human tissue-engineered lung model to study influenza A infection. Tissue Eng Part A 24:1468–1480. doi: 10.1089/ten.TEA.2017.0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pezzulo AA, Starner TD, Scheetz TE, Traver GL, Tilley AE, Harvey BG, Crystal RG, McCray PB Jr, Zabner J. 2011. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am J Physiol Lung Cell Mol Physiol 300:L25–L31. doi: 10.1152/ajplung.00256.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parker J, Sarlang S, Thavagnanam S, Williamson G, O’Donoghue D, Villenave R, Power U, Shields M, Heaney L, Skibinski G. 2010. A 3-D well-differentiated model of pediatric bronchial epithelium demonstrates unstimulated morphological differences between asthmatic and nonasthmatic cells. Pediatr Res 67:17–22. doi: 10.1203/PDR.0b013e3181c0b200. [DOI] [PubMed] [Google Scholar]
- 60.Gray TE, Guzman K, Davis CW, Abdullah LH, Nettesheim P. 1996. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am J Respir Cell Mol Biol 14:104–112. doi: 10.1165/ajrcmb.14.1.8534481. [DOI] [PubMed] [Google Scholar]
- 61.Whitcutt MJ, Adler KB, Wu R. 1988. A biphasic chamber system for maintaining polarity of differentiation of cultured respiratory tract epithelial cells. In Vitro Cell Dev Biol 24:420–428. doi: 10.1007/BF02628493. [DOI] [PubMed] [Google Scholar]
- 62.Zhang L, Collins PL, Lamb RA, Pickles RJ. 2011. Comparison of differing cytopathic effects in human airway epithelium of parainfluenza virus 5 (W3A), parainfluenza virus type 3, and respiratory syncytial virus. Virology 421:67–77. doi: 10.1016/j.virol.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Graaf M, Herfst S, Aarbiou J, Burgers PC, Zaaraoui-Boutahar F, Bijl M, van Ijcken W, Schrauwen EJA, Osterhaus ADME, Luider TM, Scholte BJ, Fouchier RAM, Andeweg AC. 2013. Small hydrophobic protein of human metapneumovirus does not affect virus replication and host gene expression in vitro. PLoS One 8:e58572. doi: 10.1371/journal.pone.0058572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol 76:5654–5666. doi: 10.1128/jvi.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, Pickles RJ. 2005. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J Virol 79:1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kwilas S, Liesman RM, Zhang L, Walsh E, Pickles RJ, Peeples ME. 2009. Respiratory syncytial virus grown in Vero cells contains a truncated attachment protein that alters its infectivity and dependence on glycosaminoglycans. J Virol 83:10710–10718. doi: 10.1128/JVI.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kwilas AR, Yednak MA, Zhang L, Liesman R, Collins PL, Pickles RJ, Peeples ME. 2010. Respiratory syncytial virus engineered to express the cystic fibrosis transmembrane conductance regulator corrects the bioelectric phenotype of human cystic fibrosis airway epithelium in vitro. J Virol 84:7770–7781. doi: 10.1128/JVI.00346-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Corry J, Johnson SM, Cornwell J, Peeples ME. 2016. Preventing cleavage of the respiratory syncytial virus attachment protein in Vero cells rescues the infectivity of progeny virus for primary human airway cultures. J Virol 90:1311–1320. doi: 10.1128/JVI.02351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johnson SM, McNally BA, Ioannidis I, Flano E, Teng MN, Oomens AG, Walsh EE, Peeples ME. 2015. Respiratory syncytial virus uses CX3CR1 as a receptor on primary human airway epithelial cultures. PLoS Pathog 11:e1005318. doi: 10.1371/journal.ppat.1005318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dvorak A, Tilley AE, Shaykhiev R, Wang R, Crystal RG. 2011. Do airway epithelium air-liquid cultures represent the in vivo airway epithelium transcriptome? Am J Respir Cell Mol Biol 44:465–473. doi: 10.1165/rcmb.2009-0453OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pickles RJ. 2013. Human airway epithelial cell cultures for modeling respiratory syncytial virus infection. Curr Top Microbiol Immunol 372:371–387. doi: 10.1007/978-3-642-38919-1_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Villenave R, Thavagnanam S, Sarlang S, Parker J, Douglas I, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2012. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc Natl Acad Sci U S A 109:5040–5045. doi: 10.1073/pnas.1110203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Broadbent L, Villenave R, Guo-Parke H, Douglas I, Shields MD, Power UF. 2016. In vitro modeling of RSV infection and cytopathogenesis in well-differentiated human primary airway epithelial cells (WD-PAECs). Methods Mol Biol 1442:119–139. doi: 10.1007/978-1-4939-3687-8_9. [DOI] [PubMed] [Google Scholar]
- 74.Wright PF, Ikizler MR, Gonzales RA, Carroll KN, Johnson JE, Werkhaven JA. 2005. Growth of respiratory syncytial virus in primary epithelial cells from the human respiratory tract. J Virol 79:8651–8654. doi: 10.1128/JVI.79.13.8651-8654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klimyte EM, Smith SE, Oreste P, Lembo D, Dutch RE. 2016. Inhibition of human metapneumovirus binding to heparan sulfate blocks infection in human lung cells and airway tissues. J Virol 90:9237–9250. doi: 10.1128/JVI.01362-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nicolas de Lamballerie C, Pizzorno A, Dubois J, Julien T, Padey B, Bouveret M, Traversier A, Legras-Lachuer C, Lina B, Boivin G, Terrier O, Rosa-Calatrava M. 2019. Characterization of cellular transcriptomic signatures induced by different respiratory viruses in human reconstituted airway epithelia. Sci Rep 9:11493. doi: 10.1038/s41598-019-48013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dubois J, Pizzorno A, Cavanagh M-H, Padey B, Nicolas de Lamballerie C, Uyar O, Venable M-C, Carbonneau J, Traversier A, Julien T, Lavigne S, Couture C, Lina B, Hamelin M-È, Terrier O, Rosa-Calatrava M, Boivin G. 2019. Strain-dependent impact of G and SH deletions provide new insights for live-attenuated HMPV vaccine development. Vaccines 7:164. doi: 10.3390/vaccines7040164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu Q, Lu B, McTamney P, Palaszynski S, Diallo S, Ren K, Ulbrandt ND, Kallewaard N, Wang W, Fernandes F, Wong S, Svabek C, Moldt B, Esser MT, Jing H, Suzich JA. 2018. Prevalence and significance of substitutions in the fusion protein of respiratory syncytial virus resulting in neutralization escape from antibody MEDI8897. J Infect Dis 218:572–580. doi: 10.1093/infdis/jiy189. [DOI] [PubMed] [Google Scholar]
- 79.Zhu Q, McLellan JS, Kallewaard NL, Ulbrandt ND, Palaszynski S, Zhang J, Moldt B, Khan A, Svabek C, McAuliffe JM, Wrapp D, Patel NK, Cook KE, Richter BWM, Ryan PC, Yuan AQ, Suzich JA. 2017. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med 9:eaaj1928. doi: 10.1126/scitranslmed.aaj1928. [DOI] [PubMed] [Google Scholar]
- 80.Griffin MP, Khan AA, Esser MT, Jensen K, Takas T, Kankam MK, Villafana T, Dubovsky F. 2017. Safety, tolerability, and pharmacokinetics of MEDI8897, the respiratory syncytial virus prefusion F-targeting monoclonal antibody with an extended half-life, in healthy adults. Antimicrob Agents Chemother 61:e01714-16. doi: 10.1128/AAC.01714-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Derdowski A, Peters TR, Glover N, Qian R, Utley T, Burnett A, Williams J, Spearman P, Crowe J. 2008. Human metapneumovirus nucleoprotein and phosphoprotein interact and provide the minimal requirements for inclusion body formation. J Gen Virol 89:2698–2708. doi: 10.1099/vir.0.2008/004051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE Jr, Santangelo PJ. 2012. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shaikh FY, Cox RG, Lifland AW, Hotard AL, Williams JV, Moore ML, Santangelo PJ, Crowe JE Jr. 2012. A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles. mBio 3:e00270-11. doi: 10.1128/mBio.00270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cox RG, Mainou BA, Johnson M, Hastings AK, Schuster JE, Dermody TS, Williams JV. 2015. Human metapneumovirus is capable of entering cells by fusion with endosomal membranes. PLoS Pathog 11:e1005303. doi: 10.1371/journal.ppat.1005303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jumat MR, Nguyen Huong T, Wong P, Loo LH, Tan BH, Fenwick F, Toms GL, Sugrue RJ. 2014. Imaging analysis of human metapneumovirus-infected cells provides evidence for the involvement of F-actin and the raft-lipid microdomains in virus morphogenesis. Virol J 11:198. doi: 10.1186/s12985-014-0198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shahriari S, Wei K-J, Ghildyal R. 2018. Respiratory syncytial virus matrix (M) protein interacts with actin in vitro and in cell culture. Viruses 10:535. doi: 10.3390/v10100535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shaikh FY, Utley TJ, Craven RE, Rogers MC, Lapierre LA, Goldenring JR, Crowe JE Jr. 2012. Respiratory syncytial virus assembles into structured filamentous virion particles independently of host cytoskeleton and related proteins. PLoS One 7:e40826. doi: 10.1371/journal.pone.0040826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gower TL, Peeples ME, Collins PL, Graham BS. 2001. RhoA is activated during respiratory syncytial virus infection. Virology 283:188–196. doi: 10.1006/viro.2001.0891. [DOI] [PubMed] [Google Scholar]
- 89.Burke E, Mahoney NM, Almo SC, Barik S. 2000. Profilin is required for optimal actin-dependent transcription of respiratory syncytial virus genome RNA. J Virol 74:669–675. doi: 10.1128/jvi.74.2.669-675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ulloa L, Serra R, Asenjo A, Villanueva N. 1998. Interactions between cellular actin and human respiratory syncytial virus (HRSV). Virus Res 53:13–25. doi: 10.1016/s0168-1702(97)00121-4. [DOI] [PubMed] [Google Scholar]
- 91.Mas V, Herfst S, Osterhaus AD, Fouchier RA, Melero JA. 2011. Residues of the human metapneumovirus fusion (F) protein critical for its strain-related fusion phenotype: implications for the virus replication cycle. J Virol 85:12650–12661. doi: 10.1128/JVI.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Domachowske JB, Khan AA, Esser MT, Jensen K, Takas T, Villafana T, Dubovsky F, Griffin MP. 2018. Safety, tolerability and pharmacokinetics of MEDI8897, an extended half-life single-dose respiratory syncytial virus prefusion F-targeting monoclonal antibody administered as a single dose to healthy preterm infants. Pediatr Infect Dis J 37:886–892. doi: 10.1097/INF.0000000000001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schuster JE, Cox RG, Hastings AK, Boyd KL, Wadia J, Chen Z, Burton DR, Williamson RA, Williams JV. 2015. A broadly neutralizing human monoclonal antibody exhibits in vivo efficacy against both human metapneumovirus and respiratory syncytial virus. J Infect Dis 211:216–225. doi: 10.1093/infdis/jiu307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stephens LM, Varga SM. 2020. Function and modulation of type I interferons during respiratory syncytial virus infection. Vaccines (Basel) 8:177. doi: 10.3390/vaccines8020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uche IK, Guerrero-Plata A. 2018. Interferon-mediated response to human metapneumovirus infection. Viruses 10:505. doi: 10.3390/v10090505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Soto JA, Gálvez NMS, Benavente FM, Pizarro-Ortega MS, Lay MK, Riedel C, Bueno SM, Gonzalez PA, Kalergis AM. 2018. Human metapneumovirus: mechanisms and molecular targets used by the virus to avoid the immune system. Front Immunol 9:2466–2466. doi: 10.3389/fimmu.2018.02466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marsico S, Caccuri F, Mazzuca P, Apostoli P, Roversi S, Lorenzin G, Zani A, Fiorentini S, Giagulli C, Caruso A. 2018. Human lung epithelial cells support human metapneumovirus persistence by overcoming apoptosis. Pathog Dis 83:fty013. doi: 10.1093/femspd/fty013. [DOI] [PubMed] [Google Scholar]
- 98.Liu Y, Haas DL, Poore S, Isakovic S, Gahan M, Mahalingam S, Fu ZF, Tripp RA. 2009. Human metapneumovirus establishes persistent infection in the lungs of mice and is reactivated by glucocorticoid treatment. J Virol 83:6837–6848. doi: 10.1128/JVI.00379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yacine A, Guy B. 2008. Persistent human metapneumovirus infection in immunocompromised child. Emerging Infectious Dis J 14:854–856. doi: 10.3201/eid1405.071459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Debiaggi M, Canducci F, Sampaolo M, Marinozzi MC, Parea M, Terulla C, Colombo AA, Alessandrino EP, Bragotti LZ, Arghittu M, Goglio A, Migliavacca R, Romero E, Clementi M. 2006. Persistent symptomless human metapneumovirus infection in hematopoietic stem cell transplant recipients. J Infect Dis 194:474–478. doi: 10.1086/505881. [DOI] [PubMed] [Google Scholar]
- 101.Oketch JW, Kamau E, Otieno GP, Otieno JR, Agoti CN, Nokes DJ. 2019. Human metapneumovirus prevalence and patterns of subgroup persistence identified through surveillance of pediatric pneumonia hospital admissions in coastal Kenya, 2007–2016. BMC Infect Dis 19:757. doi: 10.1186/s12879-019-4381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Roberts SR, Compans RW, Wertz GW. 1995. Respiratory syncytial virus matures at the apical surfaces of polarized epithelial cells. J Virol 69:2667–2673. doi: 10.1128/JVI.69.4.2667-2673.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hornsleth A, Loland L, Larsen LB. 2001. Cytokines and chemokines in respiratory secretion and severity of disease in infants with respiratory syncytial virus (RSV) infection. J Clin Virol 21:163–170. doi: 10.1016/s1386-6532(01)00159-7. [DOI] [PubMed] [Google Scholar]
- 104.Malmo J, Moe N, Krokstad S, Ryan L, Loevenich S, Johnsen IB, Espevik T, Nordbø SA, Døllner H, Anthonsen MW. 2016. Cytokine profiles in human metapneumovirus infected children: identification of genes involved in the antiviral response and pathogenesis. PLoS One 11:e0155484. doi: 10.1371/journal.pone.0155484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guerrero-Plata A, Casola A, Garofalo RP. 2005. Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J Virol 79:14992–14997. doi: 10.1128/JVI.79.23.14992-14997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wen X, Mousa JJ, Bates JT, Lamb RA, Crowe JE Jr, Jardetzky TS. 2017. Structural basis for antibody cross-neutralization of respiratory syncytial virus and human metapneumovirus. Nat Microbiol 2:16272. doi: 10.1038/nmicrobiol.2016.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Corti D, Bianchi S, Vanzetta F, Minola A, Perez L, Agatic G, Guarino B, Silacci C, Marcandalli J, Marsland BJ, Piralla A, Percivalle E, Sallusto F, Baldanti F, Lanzavecchia A. 2013. Cross-neutralization of four paramyxoviruses by a human monoclonal antibody. Nature 501:439–443. doi: 10.1038/nature12442. [DOI] [PubMed] [Google Scholar]