

When humans domesticated animals, some adapted genetically to digest milk into adulthood (lactase persistence). The gut microbiomes of people with lactase-persistent genotypes (AA or AG) differ from those with lactase-nonpersistent genotypes (GG) by containing fewer bacteria belonging to the bifidobacteria, a group which contains beneficial species. Here, we asked if the gut microbiomes of adults with GG and AA/AG genotypes differ in the species of bifidobacteria present. In particular, we used a novel technique which allowed us to compare bifidobacteria in adults at the strain level, without the traditional need for culturing. Our results show that the GG genotype enhances the abundance of bifidobacteria regardless of species. We also noted that a person’s specific strains are recoverable several years later, and twins can share the same ones. Given that bifidobacteria are inherited from mother to child, strain stability over time in adulthood suggests long-term, multigenerational inheritance.
KEYWORDS: Bifidobacterium, lactase persistence, microbiome, strain stability, gut microbiome, human microbiome
ABSTRACT
One of the strongest associations between human genetics and the gut microbiome is a greater relative abundance of Bifidobacterium in adults with lactase gene (LCT) single nucleotide polymorphisms (SNPs) associated with lactase nonpersistence (GG genotypes), versus lactase persistence (AA/AG genotypes). To gain a finer-grained phylogenetic resolution of this association, we interrogated 1,680 16S rRNA libraries and 245 metagenomes from gut microbiomes of adults with various lactase persistence genotypes. We further employed a novel genome-capture-based enrichment of Bifidobacterium DNA from a subset of these metagenomes, including monozygotic (MZ) twin pairs, each sampled 2 or 3 times. B. adolescentis and B. longum were the most abundant Bifidobacterium species regardless of host LCT genotype. LCT genotypes could not be discriminated based on relative abundances of Bifidobacterium species or Bifidobacterium community structure. Three distinct metagenomic analysis methods of Bifidobacterium-enriched DNA revealed intraindividual temporal stability of B. longum, B. adolescentis, and B. bifidum strains against the background of a changeable microbiome. Two of our three methods also observed greater strain sharing within MZ twin pairs than within unrelated individuals for B. adolescentis, while no method revealed an effect of host LCT genotype on Bifidobacterium strain composition. Our results support a “rising tide lifts all boats” model for the dominant bifidobacteria in the adult gut: their higher abundance in lactase-nonpersistent than in lactase-persistent individuals results from an expansion at the genus level. Bifidobacterium species are known to be transmitted from mother to child and stable within individuals in infancy and childhood: our results extend this stability into adulthood.
IMPORTANCE When humans domesticated animals, some adapted genetically to digest milk into adulthood (lactase persistence). The gut microbiomes of people with lactase-persistent genotypes (AA or AG) differ from those with lactase-nonpersistent genotypes (GG) by containing fewer bacteria belonging to the bifidobacteria, a group which contains beneficial species. Here, we asked if the gut microbiomes of adults with GG and AA/AG genotypes differ in the species of bifidobacteria present. In particular, we used a novel technique which allowed us to compare bifidobacteria in adults at the strain level, without the traditional need for culturing. Our results show that the GG genotype enhances the abundance of bifidobacteria regardless of species. We also noted that a person’s specific strains are recoverable several years later, and twins can share the same ones. Given that bifidobacteria are inherited from mother to child, strain stability over time in adulthood suggests long-term, multigenerational inheritance.
INTRODUCTION
Lactose tolerance arose in European, African, and Middle Eastern human populations with animal domestication (1–3). The genetic underpinnings of lactose tolerance represent one of the strongest signals of recent selection in the human genome (3). The enzyme lactase metabolizes lactose, the primary carbohydrate in mammalian milk. The gene regulatory region of the lactase gene (LCT) controls the downregulation of lactase after weaning (4). Allelic variants that inhibit downregulation, resulting in lactase persistence, occur in an estimated 35% of humans (1). Lactase persistence allows hydrolyzation of lactose and uptake of the resulting glucose and galactose directly in the small intestine of adults and is linked to lactose tolerance.
In a striking parallel, one of the strongest signals for human genotype effects on the gut microbiome also relates to lactase persistence. In Western populations, individuals with a lactase-persistent genotype harbor a significantly lower relative abundance of Bifidobacterium in their gut microbiomes than do nonpersistent individuals (5–8). The association was found to be stronger when dairy consumption in the lactase-nonpersistent individuals was considered (9). Together, these observations suggest that for lactase-nonpersistent individuals, Bifidobacterium may benefit from the availability of lactose in the gut. Bifidobacterium is a large genus whose members are often metabolic specialists with regard to particular nutrients (10), which suggests that not all Bifidobacterium species may respond equally to lactose availability. However, beyond an overall enrichment of the Bifidobacterium genus, the effects of the lactase persistence genotype on the bifidobacterial community in the gut remain unclear.
Here, we aimed to interrogate the LCT-Bifidobacterium link at a finer phylogenetic resolution. We reexamined public metagenomic (11) and 16S rRNA gene data (5) from a UK twin cohort with both lactase-persistent (AA, AG) and nonpersistent (GG) individuals. We then performed genome-capture enrichment of Bifidobacterium from 11 twin pairs of each genotype across two or three time points per individual and sequenced each metagenome before and after genome capture. With these data, we asked if lactase persistence genotype influenced strain composition, longitudinal stability within an individual, or similarity within families for three species (B. longum, B. adolescentis, and B. bifidum). Our results suggest a proportional increase of the predominant Bifidobacterium species in the gut microbiomes of the lactase-persistent compared to nonpersistent individuals. We observed strong strain stability within individuals, and sharing of some strains between MZ twins, independent of LCT genotype group.
RESULTS
Our analysis of 16S rRNA gene sequence variants (SVs) confirmed our previous operational taxonomic unit (OTU)-based report (5) of significantly greater mean relative abundance of Bifidobacterium in lactase-nonpersistent (GG) versus persistent (AA/AG) individuals (mean AA/AG = 0.96% ± 0.05% and mean GG = 3.22% ± 0.4% [standard error {SE}], linear mixed-model P < 4.5e−16) (Fig. 1A; see also Table S1, tab 1, in the supplemental material). This analysis revealed 13 Bifidobacterium SVs which occurred in at least two of 1,680 samples. Five of the 13 SVs were unambiguously identified to the species level, while two fell within a broader B. longum/B. breve clade. Both host genotype groups (GG versus AA/AG) were dominated, in order of abundance, by B. adolescentis, B. longum/breve, B. pseudocatenulatum, B. animalis, and B. bifidum. Together these taxa represented over 98.6% of all sequences assigned to the Bifidobacterium genus, although only 1.1% of all sequences across all taxa (Table S1, tab 1). These five species had similar proportions of the total Bifidobacterium community in both genotype groups and almost identical rank orders (GG, 93%; AA/AG, 91%) (Fig. 1B; Table S1, tab 1). Based on SVs, B. animalis and B. dentium were the only taxa with species-level designations that did not have significantly greater relative abundance in GG than in AA/AG genotypes.
FIG 1.
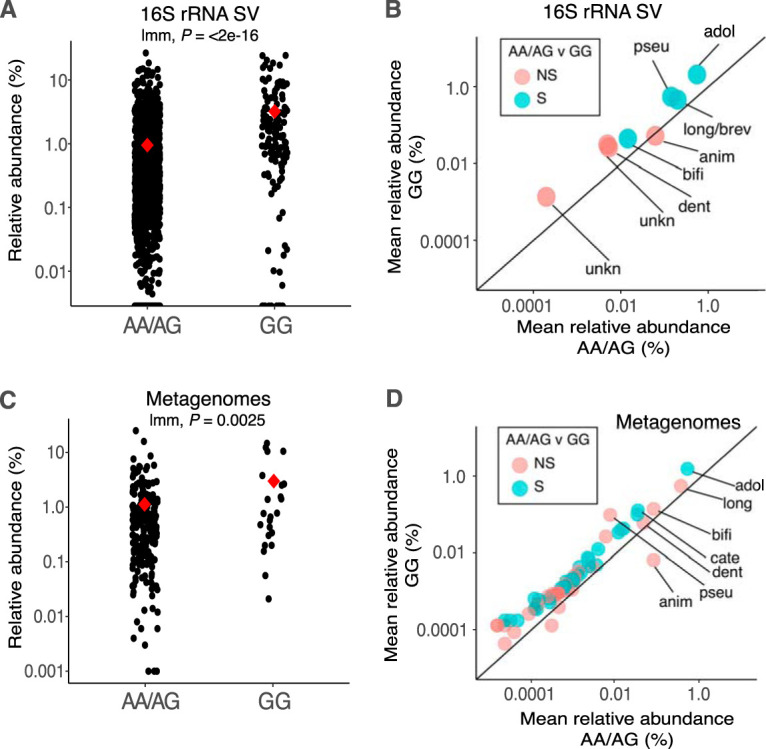
Lactase-persistent genotype enriches most human associated Bifidobacterium species. (A) Relative abundance of reads annotated as Bifidobacterium at the genus level from lactase-persistent (AA/AG) and nonpersistent (GG) individuals based on 16S rRNA SV taxonomic annotations (n: AA/AG = 1,549, GG = 131). (B) Mean relative abundance of Bifidobacterium species in AA/AG (x axis) and GG (y axis) individuals based on 16S rRNA SV taxonomic annotations. Colors indicate significant enrichment in GG. The 1:1 line designates equal proportion in each genotype, and points above the line therefore indicate enrichment in GG and vice versa. Taxa that occurred in only one genotype are not shown. Species are B. adolescentis (adol), B. longum (long), B. pseudocatenulatum (pseu), B. animalis (anim), B. bifidum (bifi), B. dentium (dent), and B. catenulatum (cate; appears only in panel D). Unknown (unkn) indicates SVs that cannot be resolved beyond genus level. NS/S represents nonsignificant or significant enrichment in GG, respectively, according to bootstrapped Wilcoxon rank sum tests. (C) Relative abundance of reads annotated as Bifidobacterium at the genus level from lactase-persistent (AA/AG) and nonpersistent (GG) individuals’ metagenomic annotations (n: AA/AG = 222, GG = 23). Red diamonds indicate means and P values from linear mixed models. (D) Mean relative abundance of Bifidobacterium species in AA/AG (x axis) and GG (y axis) individuals as revealed by metagenomic annotations. For clarity, only the 7 most abundant taxa with species-level annotations are labeled (full list available in Table S1). Colors are as in panel B.
(Tab 1) Taxonomic annotations of 16S rRNA SVs (top, n = 1,680) and metagenomic sequences (bottom, n = 245). (Tab 2) Overview of participant data. (Tab 3) Reference genomes used in the capture array. (Tab 4) Details of StrainPhlAn multilocus sequence alignments for each of the species considered. (Tab 5) Summary of strain-level methods used in the paper and an extension of Table 1 and Table 2 to include P values of significant tests. (Tab 6) P values and comparison numbers for the differences in mean synteny scores between pairwise comparison categories, for each species broken down by genomic region and in aggregate. (Tab 7A) E. coli genomes used for the validation of the synteny-based method. (Tab 7B) Genomic regions in the E. coli genome used for calculation of pairwise synteny scores. Download Table S1, XLSX file, 0.03 MB (34.5KB, xlsx) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
After normalization of Bifidobacterium within each genotype (thereby removing the effect of an overall enrichment of the genus), discriminant analysis using linear discriminant analysis effect size (LEfSe) (12) revealed no discriminant Bifidobacterium SVs between GG and AA/AG individuals (data not shown). This result indicates similar proportional abundances of each Bifidobacterium species within each host genotype group, despite an overall greater relative abundance of all taxa in GG versus AA/AG genotypes. Analysis of similarity (ANOSIM) permutation tests on the Bray-Curtis Dissimilarity (BCD) matrix of within-genotype normalized Bifidobacterium SV matrices also revealed no significant community clustering by host genotype group (ANOSIM P > 0.05), further supporting a proportional increase across most taxa rather than genotypic selection of specific species or strains within the genus.
We also assessed the association between frequency of dairy consumption and Bifidobacterium SVs for the 783 samples for which both data sets were accessible. Linear mixed-effect models, with genotype as a random variable, revealed no overall association between dairy consumption and the relative abundance of the genus (linear mixed model P = 0.113). When SVs were interrogated independently, B. animalis and an unclassifiable bifidobacterium were the only two SVs which showed significant associations (B. animalis, P = 0.0023; B. unknown1, P = 0.014). Interestingly, when each genotype was considered independently, significant associations with levels of dairy consumption were observed for B. animalis within GG individuals, but not in AA/AG individuals (generalized linear model: GG, P = 0.03; AA/AG, P = 0.13).
Our taxonomic annotations of metagenomic reads from existing TwinsUK metagenomes (11) revealed an overall enrichment of the Bifidobacterium genus in lactase-nonpersistent individuals (mean AA/AG = 1.1% ± 0.15% and mean GG = 2.8% ± 0.9%, linear mixed model P = 0.0025) (Fig. 1C; Table S1, tab 1) and a proportional increase across most Bifidobacterium species in GG versus AA/AG individuals (Fig. 1D). Largely concordant with the results of the 16S rRNA SV analysis, Bifidobacterium metagenome annotations from both host genotype groups were dominated by B. adolescentis, B. longum, B. bifidum, and B. animalis, which together represented more than 80% of all Bifidobacterium sequences, though only 1% of the overall community (Fig. 1D; Table S1, tab 1). Taxonomic annotations from metagenomes showed a greater diversity of Bifidobacterium taxa across the full data set compared to the 16S rRNA gene SV-based analysis, despite a lower sample count (245 metagenomes versus 1,680 16S rRNA samples), with 65 species and subdivisions identified (versus 13 SVs).
Functional annotations of the 245 metagenomes revealed no MetaCyc Bifidobacterium metabolic pathways with significantly different relative abundances between the two genotype groups (pairwise Kruskal-Wallis H-tests for mean abundance of each Bifidobacterium pathway in GG versus AA/AG individuals, Bonferroni multiple-comparison correction, implemented in HUMAnN2; data not shown). However, low or zero read coverage for most Bifidobacterium pathways in the metagenomes limited the power to assess differences between host genotype groups at the functional level.
Genome capture enriches Bifidobacterium DNA.
We used genome capture (13, 14) to enrich for Bifidobacterium DNA in metagenome libraries generated from 22 individuals each with 2 or 3 samples obtained at different time points (46 samples total representing 11 GG and 11 AA individuals). Each genotype group also included 4 sets of adult monozygotic twin siblings (Table S1, tab 2). Our custom genome-capture array included a total capture space of >94 Mb and 89k capture targets built from 47 reference Bifidobacterium genomes spanning the entire diversity of the genus (Table S1, tab 3). The capture reaction was largely genus specific, with low levels of enrichment of non-Bifidobacterium Bifidobacteriaceae and other non-Bifidobacterium Actinobacteria (Fig. 2A). The capture reaction increased the mean relative abundance of all bifidobacteria across our metagenomic sample subset from 2.1% (±0.27% [SE]) to 60.2% (±3.7% SE]), representing a nearly 30-fold increase from pre- to postcapture libraries. The mean relative abundance of sequences annotated as a specific Bifidobacterium taxon across all postcapture libraries was proportional to that taxon’s initial relative abundance in the precapture libraries (rho = 0.99, P < 2.2e−16, Fig. 2B), and the mean relative abundance ranking order of the top 5 taxa was the same in pre- and postcapture libraries (data not shown). We note that no analysis method presented below uses post-genome-capture data to make conclusions regarding abundance or relative abundance, given the uncertainties of differential capture between species. Only sequence-composition-based analyses are used (e.g., multilocus sequence alignments [MLSAs], marker single nucleotide polymorphisms [SNPs], and synteny) from postcapture data.
FIG 2.

Bifidobacterium is proportionally enriched using genome capture. (A) Relative abundance of all species-level annotations from the genome capture subset (n = 46), with each point representing the mean relative abundance of a distinct species-level classification before (precapture) and after (postcapture) the Bifidobacterium-DNA capture assay. Points are colored by taxonomic relatedness to Bifidobacterium. Pre- and postcapture libraries from a single sample were shotgun sequenced independently. The dashed 1:1 line represents equal relative abundances in pre- and postcapture libraries, and points below the 1:1 line are enriched in postcapture libraries relative to precapture libraries. The solid black line shows the least-squares regression between pre- and postcapture for the Bifidobacterium annotations. The shallower slope of the Bifidobacterium enrichment line than of the 1:1 line indicates greater enrichment of more common taxa. The red diamond shows the total relative abundance of Bifidobacterium (i.e., the sum of all Bifidobacterium annotations) in each of the pre- and postcapture libraries. (B) Same data as plot A but showing only Bifidobacterium annotations. The blue line shows least-squares regression with 95% confidence intervals; Spearman’s rho is shown along with the significance of the correlation.
Strain-level analysis of Bifidobacterium spp. in postcapture metagenomes.
We compared strains recovered from postcapture metagenomes using three methods: (i) we generated genome-wide strain phylogenies using multilocus sequence alignments (MLSAs) via StrainPhlAn (15), (ii) we used a marker-SNP strain tracking analysis implemented in the Metagenomic Intra-Species Diversity Analysis System (MIDAS) (16), and (iii) we used a custom synteny-based approach. We compared strains in all possible pairwise sample comparisons from our genome-capture subset (n = 46). We then grouped each comparison into one of three categories: intraindividual (comparison between samples from the same person over time), same family (between monozygotic twins within a twinship), and unrelated (between samples from unrelated individuals). These categories allowed us to assess strain stability through time within an individual (intraindividual comparisons) and the influence of monozygotic twins (same-family comparisons). Our postcapture libraries yielded sufficient reads to conduct these analyses in four species, B. longum, B. adolescentis, B. animalis, and B. bifidum, with sample numbers shown in Table 1.
TABLE 1.
Sample number and comparison category overview for StrainPhlAn and MIDAS strain-level analyses
| StrainPhlAn |
MIDAS |
|||||||
|---|---|---|---|---|---|---|---|---|
| No. of samples with sufficient coverage for phylogeny |
No. of comparisons |
No. of samples with sufficient coverage for marker-SNP analysis |
No. of comparisons |
|||||
| Intraindividual | Same family | Unrelated | Intraindividual | Same family | Unrelated | |||
| B. adolescentis | 43 | 23 | 37 | 843 | 37 | 17 | 30 | 619 |
| B. longum | 39 | 20 | 29 | 692 | 36 | 15 | 24 | 591 |
| B. bifidum | 19 | 8 | 8 | 155 | 20 | 6 | 8 | 176 |
| B. animalis | 11 | 3 | 2 | 50 | 15 | 4 | 3 | 98 |
| Total | 54 | 76 | 1,740 | 42 | 65 | 1,484 | ||
MLSAs showed greater similarities of strain sequences in intraindividual than in unrelated comparisons.
Using StrainPhlAn, we created MLSAs across nearly 200 strain-resolving marker genes derived from a species’ core genome (genes shared by all strains in the species). Of the postcapture shotgun metagenomes, B. adolescentis, B. longum, B. bifidum, and B. animalis had sufficient coverage for 43, 39, 19, and 11 samples, respectively (minimum of 2× coverage across entire alignment) (Table 1). The marker gene MLSAs ranged from 36 to 83 kb in length depending on species (Table S1, tab 4). The mean number of polymorphic sites within a sample across an alignment ranged from 0.39% to 0.92%, while the dominant allele at each site ranged upward from 76%, suggesting low strain diversity within samples (Table S1, tab 4, and Fig. S1).
StrainPhlAn alignment statistics show low intrasample strain diversity. (A) The mean percentage of polymorphic sites along our MLSA for each of the four species considered. (B) The mean dominant allele frequency across all polymorphic sites within an MLSA. Boxplots show median and quartiles. Species: adol, B. adolescentis; anim, B. animalis; bifm, B. bifidum; long, B. longum. Significance levels are Wilcoxon rank sum tests. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Nonsignificant comparisons are not indicated. Download FIG S1, PDF file, 0.04 MB (45KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mean patristic distance (i.e., branch length) was significantly less in intraindividual than in unrelated comparisons in phylogenies of B. adolescentis, B. longum, and B. bifidum strains (bootstrapped Wilcoxon rank sum P < 0.05 in all three cases, Fig. 3). This result indicates strain stability: an individual’s strains are more similar for the same person sampled over time compared to strains from a different person. We did observe some instances of large patristic distance between samples of the same individual (Fig. 4), and in both cases where three samples per individual were included, one of the three samples had a large patristic distance more typical of unrelated comparisons (Fig. 4). These large interindividual patristic distances could result from detection errors (i.e., a strain was present in both samples but failed to be detected in one), or from differences in the relative abundance of strains between time points (this analysis picks the most abundant), or from loss and regain of strains.
FIG 3.
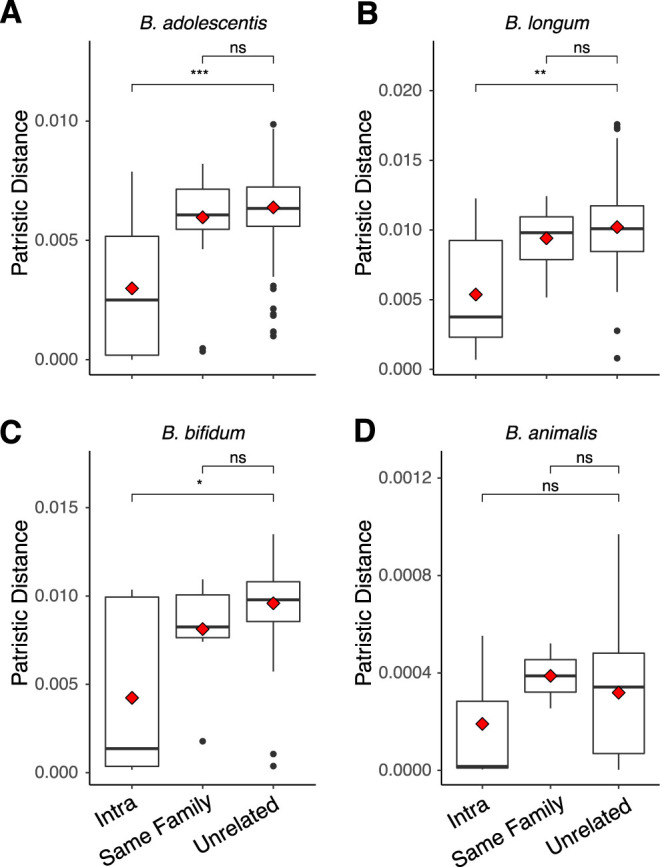
Patristic distances between samples from RAxML phylogenies of four strains show intraindividual strain similarity. Patristic distances (branch lengths) between all samples within our StrainPhlAn phylogenies, with each point representing the distance between two samples on a single phylogeny. Distances are separated by the category of comparison: intraindividual (same person over time), same family (comparisons between twin siblings), and unrelated (comparisons between unrelated people). Species are B. adolescentis (A), B. longum (B), B. bifidum (C), and B. animalis (D). Boxplots show median and quartiles, while red diamonds show means. Significance levels are median Wilcoxon rank sum tests after 999 bootstraps to smallest group size. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
FIG 4.
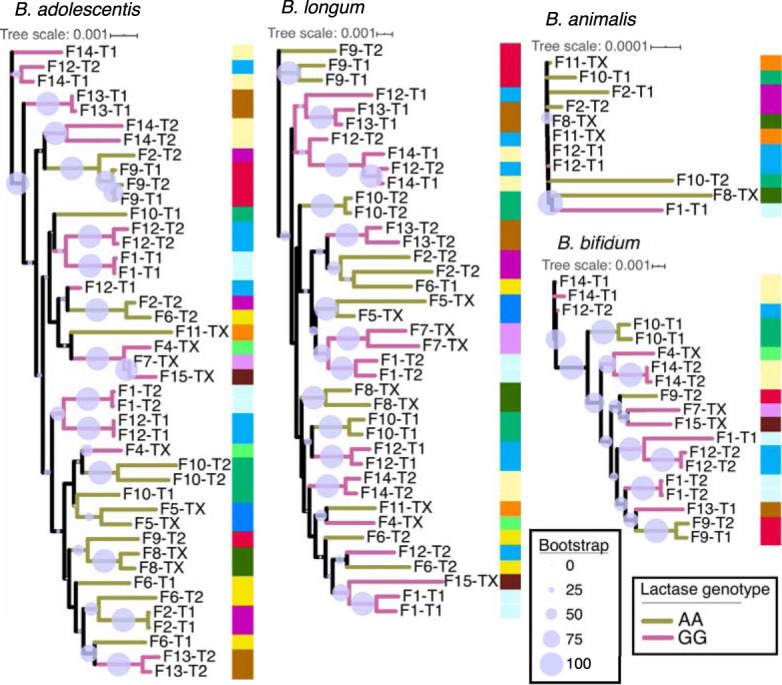
Strain phylogenies show longitudinal intraindividual strain similarities. RAxML phylogenies of StrainPhlAn MLSAs for each of the four species considered. Tree branches are colored by lactase persistence genotype. Columns of colors at each node reference family IDs, with multiple samples from the same individual and their twin having the same color. Samples are labeled by family ID, then by twin ID within the family. Identical labels imply samples from different time points of the same individual (i.e., intraindividual sample). Twin IDs marked with “X” imply that only a single twin had representation on the tree or that the individual had no twin in the data set. Scales show patristic distances while circle sizes represent RAxML bootstrap support of each division.
Mean patristic distance of same-family comparisons was not significantly different from that of unrelated comparisons for any of the four taxa examined (Wilcoxon rank sum of mean distances in intertwin versus unrelated, P > 0.05 in all cases, Fig. 3). Thus, the MLSA-based RAxML phylogenies did not show that co-twins harbored similar strains of these 4 species. Furthermore, this analysis did not reveal any significant grouping of strains according to lactase genotype (ANOSIM permutations of phylogenetic distance matrix P > 0.05, Fig. 4) and longitudinal intraindividual comparisons were not significantly different between genotype groups for any of the four species we examined either individually (Fig. S2) or in aggregate (Fig. S3) (Wilcoxon rank sum test of mean intraindividual distance, AA versus GG, P > 0.05; Table 2A and Table S1, tab 5). The time interval between repeated samples from the same individual and patristic distance did not reveal any relationship for any species (Spearman’s rho nonsignificant in all cases, Fig. S4), supporting strain stability with time.
TABLE 2.
Overview of analyses for each of the four species examined at the strain level and for all species combined in aggregatea
| Adol | Long | Bifi | Anim | In aggregate | |||
|---|---|---|---|---|---|---|---|
| A | Marker gene-based phylogeny (StrainPhlAn) |
Longitudinal stability | Yes | Yes | Yes | No | Yes |
| Stability GG > stability AA/AG | No | No | NA | NA | No | ||
| Twin effect | No | No | No | No | No | ||
| B | % of shared marker-SNPs (MIDAS) |
Longitudinal stability | Yes | Yes | Yes | No | Yes |
| Stability GG > stability AA/AG | Yes | Yes | NA | NA | Yes | ||
| Twin effect | Yes | No | No | No | No | ||
| C | Synteny (all regions for each species in aggregate) |
No. of gene regions | 7 | 10 | 4 | 0 | 21 |
| Longitudinal stability | 6/7 | 4/10 | 2/4 | NA | Yes | ||
| Stability GG > stability AA/AG | 0/2 | 0/8 | 0/0 | NA | No | ||
| Twin effect | 4/7 | 0/8 | 1/4 | NA | No | ||
| D | Enriched in GG vs AA/AG (16S rRNA SVs) | Yes | Yes | Yes | No | Yes | |
Panels A to C show significance for our three strain-level approaches. StrainPhlAn results are based on best tree values from the RAxML hill-climbing algorithm. Within each panel, “longitudinal stability” refers to significantly greater similarity of Intra (same person over time) versus Unrelated (between unrelated people) comparison categories. “Stability GG > stability AA/AG” refers to greater mean “Intra” values for GG individuals than for AA/AG individuals (i.e., is there greater stability within lactase-nonpersistent than lactase-persistent individuals). Finally, “Twin effect” refers to greater similarity of Same Family (between twin siblings) than Unrelated comparison categories. Panel D shows significance of genotypic enrichment from 16S rRNA SV data across the broad TwinsUK data set (n = 1,680). In each panel, “yes” and “no” refer to significance of the statistical test as described in the main text, while “NA” indicates that 3 or fewer samples were available in at least one category. In panel C (synteny), the number of significant regions out of the total number of regions with sufficient comparisons is shown. P values are shown in Table S1, tabs 5 and 6. Species abbreviations are as follows: Adol, B. adolescentis; Long, B. longum; Bifi, B. bifidum; Anim, B. animalis.
Host genotype effects on intraindividual longitudinal strain stability. All intraindividual longitudinal comparisons for each of B. longum, B. adolescentis, and B. bifidum are plotted according to host genotype for patristic distance (branch lengths) between samples across our RAxML strain phylogenies (A) and the percent marker-SNPs shared between samples (B). Significance levels are Wilcoxon rank sum tests. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Red diamonds represent mean values. Download FIG S2, PDF file, 0.1 MB (81.7KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Host genotype effects on longitudinal strain stability and strain similarities between monozygotic twins across all four species in aggregate. All three metrics of strain comparisons are shown, with each separated by lactase persistence genotype and the category of comparison: Intra (same person over time), same family (comparisons between twin siblings), and unrelated (comparisons between unrelated people). Plots represent combined data for B. longum, B. bifidum, and B. adolescentis. The metrics shown for each comparison category are patristic distance (branch lengths) between samples across our RAxML strain phylogenies (A), the percent marker-SNPs shared (B), and synteny scores across the phylogenies (C). Significance levels are median Wilcoxon rank sum tests after 999 bootstraps to smallest group size. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Download FIG S3, PDF file, 0.2 MB (159.6KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Temporal distance between intraindividual samples is not correlated with patristic distance across MLSAs. All unique intrapersonal comparisons are plotted, with time between samples on the x axis and StrainPhlAn MLSA patristic distance on the y axis. Blue line shows least-squares regression with 95% confidence intervals, while Spearman’s rho and the significance of the correlation are shown. Download FIG S4, PDF file, 0.1 MB (87KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Marker-SNPs are shared at higher percentages in intraindividual than in unrelated comparisons.
To further interrogate Bifidobacterium strain stability within individuals over time, we employed a “strain tracking” feature, implemented in the MIDAS software, to identify rare marker-SNPs highly discriminant for a specific strain in an individual at a single time point. We had sufficient coverage to employ this approach for each of B. longum, B. adolescentis, B. animalis, and B. bifidum species (Table 1). Our results show that microbe marker-SNPs of an individual are far more likely to be found in the same individual at a different time point than in an unrelated individual in three of the four species examined (bootstrapped Wilcoxon rank sum of mean percent shared SNPs, intraindividual versus unrelated, P < 0.0001 in all cases, Fig. 5). B. animalis was the one exception, where SNP sharing was not significantly higher in intraindividual than in unrelated comparisons (bootstrapped Wilcoxon rank sum P = 0.32; Fig. 5, Table 2B, and Table S1, tab 5).
FIG 5.

Marker-SNP sharing over time and between individuals shows strain stability and twin sharing in three of four species. Percentage of marker-SNPs shared for all pairwise comparisons, separated by the category of comparison: intra (same person over time), same family (comparisons between twin siblings), and unrelated (comparisons between unrelated people). Shown are the four taxa for which sufficient coverage existed to calculate marker-SNPs. Species are B. adolescentis (A), B. longum (B), B. bifidum (C), and B. animalis (D). Boxplots show median and quartiles, while red diamonds show means. Significance levels are median Wilcoxon rank sum tests after 999 bootstraps to smallest group size. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
We observed greater mean intraindividual sharing of B. adolescentis and B. longum in GG than in AA individuals (Fig. S2), as well as when all species were considered in aggregate (Wilcoxon rank sum of mean intraindividual percent shared SNPs, GG versus AA individuals, P = 3.3e−9; Fig. S3). This pattern was driven by B. longum and B. adolescentis, the two most abundant species (Fig. S2).
The marker-SNP analysis revealed that high stability in either B. longum, B. bifidum, or B. adolescentis was a good predictor of stability in the other two species within that same individual (Fig. 6). This suggests some individuals carry stable strains, while others witness strain replacement, across all three species together. We detected higher marker-SNP sharing within families (i.e., a twin effect) for B. adolescentis (bootstrapped Wilcoxon rank sum P = 0.021) but not the other three species examined, possibly due to a lower power to detect this pattern in B. bifidum (for which synteny analysis shows a twin effect [see below]). The percentage of shared marker-SNPs for a given intraindividual comparison was not correlated with time between sample collection, nor with other metrics of overall microbiome similarity (Fig. S5), meaning the percentage of shared marker-SNPs between samples does not decrease with time, and the patterns observed here are therefore not a function of time-induced SNP accumulation.
FIG 6.

Intraindividual marker-SNP sharing across different Bifidobacterium species. Each plot shows the percentage of marker-SNPs shared within an individual over time for two different Bifidobacterium species; each of three comparison possibilities is shown across plots A, B, and C. Each point represents a comparison between longitudinal samples from the same individual. Points are colored by the Simka Bray-Curtis Dissimilarity (BCD) value between the two samples, as measured from precapture metagenomes, and is therefore a measure of overall microbiome dissimilarity over time within an individual. Blue line shows least-squares regression with 95% confidence intervals, and Spearman’s rho along with the significance of the correlation is shown.
Temporal distance between intraindividual samples is not correlated with overall microbiome community similarity, synteny, or mean marker-SNP sharing. All unique intrapersonal comparisons are plotted, with time between samples on the x axis and a metric of stability of similarity on the y axis. The metrics are as follows: Bray-Curtis Dissimilarity (BCD) based on SIMKA k-mer spectra of precapture metagenomes (A), BCDs based on metabolic pathways of precapture metagenomes (B), mean synteny scores across all regions examined (C), and mean percentage of marker-SNPs shared across all four taxa (D). Blue line shows least-squares regression with 95% confidence intervals, while Spearman’s rho and the significance of the correlation are shown. Download FIG S5, PDF file, 0.1 MB (89.1KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Notably, Bifidobacterium strains exhibited intraindividual longitudinal stability despite high variability in overall microbiome communities around them. Only a very weak, nonsignificant association was observed between an individual’s overall microbiome stability (assessed using Bray-Curtis Dissimilarity [BCD] of precapture metagenomes) and the number of shared marker-SNPs for a given Bifidobacterium strain (Fig. 6 and Fig. S6). This pattern held when microbiome community dissimilarity was assessed using either Simka BCD values, a k-mer-based annotation-independent method, or BCD values for annotations of metabolic pathways of precapture metagenomes (Fig. S6). These results suggest Bifidobacterium stability may be independent from the dynamics of microbiome communities in which the bacteria exist.
Intraindividual marker-SNP sharing of Bifidobacterium species does not correlate with overall community dissimilarity. Percentage of Bifidobacterium marker-SNPs shared between samples from the same individual over time, plotted against the overall community dissimilarity from those samples, based on precapture metagenomes (i.e., standard metagenome libraries). BCD metrics used to assess overall community dissimilarity are Simka, a k-mer-based dissimilarity metric between the two samples (A), and BCD values for MetaCyc annotations of metabolic pathways generated in HUMAnN2 (B). Blue line shows least-squares regression with 95% confidence intervals, and Spearman’s rho along with the significance of the correlation is shown. Download FIG S6, PDF file, 0.1 MB (53.8KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Synteny analysis reveals sharing of strains within twin pairs and strain persistence in individuals over time.
The analyses described above are based on single nucleotide variations and gene-content comparisons. Gene synteny is defined as the conservation of gene order between chromosomes, along an evolutionary gradient (17), and was previously used as a measure of the distance between genomes (18, 19). Here, we identify syntenic blocks, regions of conserved DNA sequence occurring in the same order in two different chromosomes, to determine the relatedness of Bifidobacterium strains. We created a pipeline to compare the synteny of different genomic regions in three Bifidobacterium species for which we had sufficient coverage from captured metagenomes: B. longum, B. adolescentis, and B. bifidum (see Materials and Methods). Briefly, we performed a de novo assembly of postcapture metagenomes in each sample and used a subset of randomly selected genes to perform a BLAST search against the metagenomic assemblies. Next, we performed a pairwise synteny comparison between all genomic regions, which included the gene used as BLAST query and its flanking regions (3.5 kbp upstream and downstream of the gene). Finally, we defined a synteny score and performed the same pairwise comparisons as outlined above for regions which were identified in >15 single-individual assemblies. In summary, we analyzed 10, 7, and 4 regions for B. longum, B. adolescentis, and B. bifidum, respectively.
For all three species, intraindividual comparisons yielded significantly higher synteny scores than comparisons of unrelated individuals (Fig. 7), although this pattern varied somewhat by region; 6/7 genomic regions for B. adolescentis, 2/4 regions for B. bifidum, and 4/10 regions for B. longum (results are summarized in Table 2C and P values are shown in Table S1, tab 6). These results indicate that the same strains persist in the gut over time. Comparisons of synteny between individuals within a twin pair yielded significantly higher scores (relative to comparisons of unrelated individuals) in 4 regions in B. adolescentis and in one region in B. bifidum. In B. longum, although not statistically significant, 3 regions showed lower synteny scores for individuals within a pair (P values 0.055 to 0.08, Table S1, tab 6). Finally, LCT genotype did not influence longitudinal strain stability as measured by synteny. No differences were seen in mean intraindividual synteny scores between lactase persistence genotypes (Wilcoxon rank sum test of mean intraindividual synteny, AA versus GG, P > 0.05), and each genotype independently showed significant differences between intraindividual and unrelated comparisons (bootstrapped Wilcoxon rank sum test, P = 0.0005 for AAs and P = 6.1e−5 for GGs, Fig. S3). Together, these results support strain-sharing events within the families and maintenance of strains over time within individuals.
FIG 7.
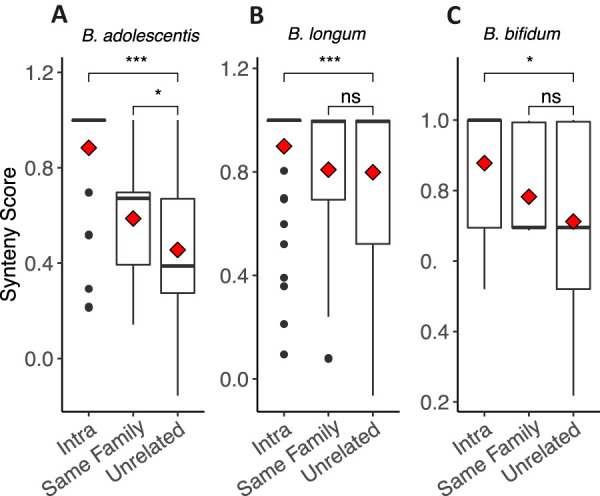
Within-species synteny scores over time and between individuals. Pairwise synteny scores for genomic regions of the three Bifidobacterium species for which sufficient read depth was generated for synteny analyses. Plots show all pairwise comparisons, separated by the category of comparison: intra (same person over time), same family (comparisons between twin siblings), and unrelated (comparisons between unrelated people). Species are B. adolescentis (A), B. longum (B), and B. bifidum (C). Boxplots show median and quartiles, while red diamond shows mean. For each species, all regions were combined into a single analysis. Significance levels are Wilcoxon rank sum tests. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
DISCUSSION
One of the strongest signals of host genetic effects on microbiome communities is the association between Bifidobacterium and an SNP in the regulatory region of the lactase gene LCT. Several independent studies have noted a greater relative abundance of the genus Bifidobacterium in the gut microbiomes of lactase-nonpersistent compared to lactase-persistent individuals of European origin (5, 7–9). We did not detect an interaction between dairy consumption, LCT genotype, and levels of Bifidobacterium here, as reported by Bonder et al. (9). However, our results show that compared to the lactase-persistent genotypes (AA/AG), the lactase nonpersistence genotype (GG) enhances the proportion of prevalent and abundant Bifidobacterium species, without bias toward particular species, strains, or genome content. Our results also indicate that strains of certain Bifidobacterium species are shared within twin pairs and persist over time within individuals against a background of a dynamic microbiome community.
Corroborating results of previous reports (20–22), we observed that B. adolescentis and B. longum dominated the Bifidobacterium communities of the adult gut microbiome. Strain comparisons within an individual over time indicated that B. adolescentis, B. longum, and B. bifidum were stable within adults over multiyear timescales as reveled by multiple methods. Strain sharing between twins showed a weaker signal in general, with B. adolescentis as the only species with a significant twin effect across multiple methods. Mechanisms leading these four species to vary in regard to longitudinal stability or twin effects remain to be elucidated. Nevertheless, it is interesting that B. animalis, the one species for which stability was not shown, was also among the few species not enriched in GG versus AA/AG genotype groups from our broader 16S rRNA and metagenomic surveys. These observations may reflect a different ecology of B. animalis compared to the others. B. animalis is commonly isolated from dairy and other sources outside the human microbiome (23) and has a reduced genetic repertoire for carbohydrate metabolism versus other taxa in the genus (24). B. animalis may therefore be a more transient autochthonous member of the adult gut microbiome. It is also important to consider that B. adolescentis and B. longum both showed the highest number of significant patterns across our methods and had the largest sample numbers. The lack of significant findings for B. animalis and B. bifidum must therefore be considered in light of the low power attributable to their analyses. Adding additional samples with future research could confirm that variability in longitudinal stability between species and strains is not due to sample number biases.
A leading hypothesis for the greater relative abundance of Bifidobacterium in lactase-nonpersistent individuals is a greater availability of undigested lactose in the large intestine, which may be preferentially used by bifidobacteria (9, 25). The availability of undigested lactose in the guts of lactase-nonpersistent individuals could in principle result in niche partitioning between the different species. Indeed, many taxa within the Bifidobacterium genus are considered specialists (20, 26), and some devote significant proportions of their genomes to particular host-derived resources including human milk oligosaccharides in Bifidobacterium longum subsp. infantis (27), and mucin glycans in B. bifidum (26). B. longum subsp. infantis, for example, is known to specialize in human milk oligosaccharide metabolism, while Bifidobacterium longum subsp. longum is specialized in metabolism of plant-derived carbohydrates (10, 28). Furthermore, gut Bifidobacterium taxa are known to follow successional patterns with a gradual shift from B. longum subsp. infantis, B. breve, and B. bifidum in infant guts to B. catenulatum, B. adolescentis, and B. longum subsp. longum in adults (21, 22). These shifts are thought to result in part from shifts in diet and are associated with breastfeeding versus formula, weaning, and intake of solid food (29, 30). However, our results instead point to lactose utilization as enhancing all the common Bifidobacterium species, without altering their population structure. This implies that in the adult gut microbiome, common Bifidobacterium species use lactose without competing for it and it does not become a limiting resource. The nondifferentiating effect of host LCT genotype on the Bifidobacterium community results suggests a “rising tide raises all boats” scenario, where a lactose utilization advantage is equally distributed among common Bifidobacterium species.
The strain comparisons in this study relied on enrichment of Bifidobacterium DNA by genome capture. This worked well for the most abundant species, B. adolescentis and B. longum, and allowed a reduced set of comparisons possible for B. bifidum and B. animalis due to lower coverage. To compare strains of the other bifidobacteria present would require deeper sequencing or a more tailored set of probes in the genome capture. To compare strains, we used two published methods based on sequence composition: one using an alignment of marker-genes (StrainPhlAn) and another using individually discriminant marker-SNPs (MIDAS). The novel synteny-based method that we developed allows for the identification of differences in the organization of genomic regions even when the sequence similarity is high. Generally, the two published methods agreed with the synteny approach, with some discrepancies (Table 2). All three methods detected intraindividual longitudinal stability for B. adolescentis, B. bifidum, and B. longum, while no method detected stability in B. animalis. The three methods were divided, however, in their detection of within-family sharing of strains (a twin effect) and on the influence of lactase persistence genotype on strain stability. Both the marker-SNP and synteny approaches detected greater similarity of B. adolescentis strains between twins than between unrelated individuals, while our marker gene alignments did not. Only the synteny approach detected twin sharing in B. longum and B. bifidum. The marker-SNP approach was the only method to reveal any influence of lactase persistence on longitudinal stability and did so only in B. adolescentis and B. longum. Why each method varied in its ability to detect a particular pattern is not clear; however, their agreement with regard to longitudinal stability of B. adolescentis, B. longum, and B. bifidum gives strong support to the conclusion that these species are stable within adults over multiyear timescales.
Temporal stability, along with vertical transmission within families, is expected to be associated with heritability. In addition to temporal stability and within-family sharing of strains shown here, we had previously noted that the genus Bifidobacterium was both heritable and stable over time, based on 16S rRNA gene analysis (31). For the relative abundances of microbial taxa or genes to be stably heritable over time, they must be present every generation and associated with host genetic variation. If the acquisition of microbes from the environment or other individuals is less reliable than acquisition from parents, the heritability of horizontally acquired microbes should be less stable over time than the heritability of vertically acquired microbes. These results, together with previous observations, continue to reveal the bifidobacteria as highly human-adapted members of the microbiome.
B. adolescentis, B. longum, and B. bifidum have also previously been shown to be temporally stable in the gut microbiomes of infants, where they are far more abundant than in the adult gut microbiome. Strains of these species were stable within children for up to 3 years of age across diverse geographic cohorts (30), and B. longum subsp. longum was stable from infancy until 6 years of age (32). Furthermore, B. bifidum and B. longum are transmitted from mother to infant (33, 34). By extending existing evidence of Bifidobacterium strain stability to an adult cohort and demonstrating strain sharing within families for B. adolescentis, our results contribute to an overall picture of this genus, one which suggests long-term maintenance of specific strains within an individual which can be transmitted across generations.
MATERIALS AND METHODS
Sample inclusion and access.
All existing samples from the TwinsUK cohort were included assuming they had both (i) metagenomic or 16S rRNA sequence data and (ii) genotype data at the 13910 G/A allele. All work involving the use of these previously collected samples was approved by the Cornell University IRB (protocol ID 1108002388).
16S rRNA gene-based community analyses.
16S rRNA reads were extracted from existing data sets (n = 1,680) and analyzed via the Qiime2 pipeline (35) with minor deviations. Briefly, PCR amplicons for the V4 region of the 16S rRNA gene were generated with primers 515F-806R and were sequenced with the Illumina MiSeq 2 × 250 v2 kit at the Cornell University Institute for Biotechnology as previously described (5). DADA2 (36) was used to call 100% sequence identity sequence variants (SVs, also known as 100% OTUs or ASVs). Taxonomy was assigned to SVs with the QIIME2 q2-feature-classifier (37) using the SILVA database (v119) (38). Taxonomic annotations of 16S rRNA sequence variants (SVs) were improved from the standard DADA2 output by extracting each SV representative sequence and querying it against the NCBI type strain database. In one case all NCBI annotations fell within the B. longum/B. breve clade, and it was thus assigned as “B. longum/breve” in subsequent analyses; in all other cases the original DADA2 assignment was kept.
Statistical testing for the influence of genotype was done using a nonparametric Wilcoxon rank sum test of the total relative abundance of all Bifidobacterium SVs between genotypes. Only SVs that occurred in at least 2 individuals across the data set of 1,680 samples were included. Individuals with either AA or AG at 13910*A (rs4988235) were considered lactase persistent, while only GG was considered lactase nonpersistent. Independent Wilcoxon rank sum tests were run for each Bifidobacterium SV independently across the two genotype groups. Because our data set included replicate samples from the same individual (therefore not independent), we determined the overall influence of genotype on Bifidobacterium SV relative abundance using a linear mixed-effect model, with participant ID as a random effect, in the R package “lmr4”: BifidobacteriumRA ∼ Genotype + (1 | ParticipantID).
To assess the community structure of the genus without the influence of an overall enrichment in GG individuals, all SVs annotated as “bifidobacterium” were extracted into new SV tables and again normalized by 1 within an individual and then input into LEfSe (12) via the Galaxy web interface with default parameters, genotype set as class, and no subclass. Longitudinal analyses of Bifidobacterium SVs narrowed the total sample count to 556 (278 matched pairs), and Spearman’s rho was calculated assigning time 1 and time 2 to one sample or another at random. To overcome unequal sample sizes (GG = 30, AA/AG = 248), a mean Spearman rho was calculated after subsampling to n = 15 for each genotype over 999 permutations. All analyses were done in RStudio (v. 1.0.136).
Dairy intake was assessed as the portion size frequency for dairy for a week, adjusted for energy intake, using food frequency questionnaires developed and described elsewhere (50). Associations with Bifidobacterium SVs were conducted using linear mixed-effect models, with genotype as a random effect, in the R package “lmr4”: BifidobacteriumRA ∼ DairyFrequency + (1 | ParticipantID). Statistical significance was assigned using Satterthwaite’s method in the R package “lmerTest” (39).
Metagenomic community analyses.
Metagenome sequences were used from existing data sets (n = 245) which were extracted as described above with library preparation previously described (40). Taxonomic assignments were made using Kraken2 (41). We assessed the influence of genotype on the relative abundance of Bifidobacterium annotations using a mixed-effect model as described above. Metabolic pathway annotations were generated using the HUMAnN2 pipeline against the MetaCyc database (42), and significantly discriminant gene pathways were revealed using HUMAnN2’s built-in ‘humann2_associate’ script with default parameters at the gene pathway level (42).
Subset for genome capture, strain-level analyses, and discriminant functional pathways.
An additional subset of the TwinsUK cohort was selected for genome capture based on (i) the availability of genotype data at the lactase persistence gene (rs4988235) and (ii) at least 2 longitudinal samples between 8 months and 4 years apart with a body mass index (BMI) change of less than 3. We note that variation in temporal distance between sampling events had no impact on stability, community composition, or synteny values (see Fig. S4 and S5 in the supplemental material). In total 20 individuals with 2 time points and 2 individuals with 3 time points were selected (a total of 46 samples across 11 GG individuals/11 AA individuals). Each genotype also contained 4 sets of twin siblings (Table S1, tab 2). DNA samples were brought through metagenome creation, capture reaction, and sequencing according to NimbleGen (Madison, WI, USA) SeqCap EZ HyperCap Workflow v.1.0. Briefly, genomic DNA (gDNA) underwent enzymatic fragmentation and adapter-mediated PCR using KAPA HyperPlus library preparation, followed by a 16-h hybridization with a custom set of biotinylated long oligonucleotide probes (the “probe array”), followed by a final reamplification. The probe array was designed and manufactured by NimbleGen and included overlapping coverage of a total capture space of >94 Mb and 89k capture targets which covered 47 type strain Bifidobacterium genomes (Table S1, tab 3). Precapture and postcapture metagenomes were then sequenced across two Illumina paired-end 300-cycle sequencers (HiSeq 3000).
Analyses on each pre- and postcapture library from our 46-sample longitudinal subset (92 metagenomes total) were conducted with Simka, an annotation-independent k-mer-based metric (43), and the annotation-based HUMAnN2 pipeline against the MetaCyc database (42). Simka Bray-Curtis Dissimilarity (BCD) matrices were calculated directly within the software. HUMAnN2 BCD matrices were generated from gene pathway relative abundance tables, after removal of collapsed pathway stratifications, using vegdist in the Vegan R package (V. 1.0.136). Each pairwise comparison within the BCD matrices was then classified into one of three categories: intraindividual (same person over time), same family (comparisons between twin siblings), and unrelated (comparisons between unrelated people). Wilcoxon rank sum tests were used to determine differences between comparison categories. To overcome issues of nonindependence related to multiple samples from the same individual within the data set, we ran 9,999 permutations of Wilcoxon rank sum tests using subsets of individuals equal to the smallest number of samples from either category and reported the mean value. Finally, ANOSIM tests of BCD hierarchical clustering were conducted using the ‘anosim’ script part of the ‘vegan’ R package with 9,999 permutations. ANOSIM permutation tests were run on intraindividual clusters, unrelated clusters, and same-family clusters.
MIDAS marker-SNPs and StrainPhlAn MLSAs.
Strain-level assessments were made using the Metagenomic Intra-Species Diversity Analysis System (MIDAS) (16) and StrainPhlAn (15). MIDAS’s ‘strain_straintracker.py’ program was used with default settings to identify marker-SNPs as previously described (16). Briefly, MIDAS initially identifies abundant species by mapping unassembled metagenomic reads to a database of 30,000 reference genomes and then identifies per-species SNPs for each abundant species within a sample. To identify the SNPs used in our analyses, we had MIDAS identify rare, sample-discriminatory SNPs from a pool of 1 sample per individual (i.e., SNPs that were unique to that individual). We term these sample-discriminatory SNPs “marker-SNPs.” Each individual’s additional sample(s) was then added back into the pool, and marker-SNP overlap was assessed between sets of two samples in all possible pairwise comparisons across the data set. The percentage of shared marker-SNPs was the number of marker-SNPs shared between two samples, divided by the total number across both individuals. Bootstrapped Wilcoxon rank sum tests were performed on the mean percentage of marker-SNPs shared in each comparison category described above (i.e., intraindividual, same family, and unrelated). Bootstrapping was done down to the lowest number in a single category for any given comparison and run for 9,999 permutations, and the mean P value was reported.
StrainPhlAn multilocus sequence alignment (MLSA) phylogenies were created using the standard RAxML hill-climbing algorithm, as described elsewhere (15, 33). Bootstrap values were generated using the rapid bootstrapping RAxML algorithm with 100 iterations (44). Resulting phylogenies were uploaded into the ITOL program (45) for graphical display of family and twin ID annotations, and best tree phylogenies from the hill-climbing algorithm were uploaded into Geneious (v 6.1.8) to retrieve patristic distances (branch lengths). Mean patristic distances were compared across comparison categories using bootstrapped Wilcoxon rank sum tests as described above.
All software was run parallelized on a high-performance computing cluster at the Max Planck Institute for Developmental Biology via Snakemake (46).
Synteny analyses.
First, a de novo assembly of postcapture metagenomes was performed for each sample by using the MetaCompass software (47). Next, a set of genes representing different genomic regions was selected randomly (using a custom script) from each of the reference genomes of B. longum, B. adolescentis, B. animalis, and B. bifidum. Each gene was used as a query for a BLASTn (48) search against the assembled metagenomes, with minimal identity of 97% and minimal coverage of 90%. For each of the blast hits in the assembled metagenomes, the gene and flanking sequences (3.5 kb upstream and downstream of the blast hit) were retrieved and used for synteny comparison. Further analysis was carried out only for regions found in >15 samples. If for a given species the final number of suitable regions was lower than 4, the process iterated with a new set of genes, keeping a minimal gap of 5 genes between each two selected genes, to avoid overlap between regions.
Pairwise synteny comparison between each two DNA sequences was done using the DECIPHER R package (49). Breaks in the synteny were defined as nonhomologous regions longer than 15 bp. To compare the synteny between different types of pairwise comparisons (i.e., intraindividual, twins, and nonrelated individuals) we defined the synteny score (equation 1):
| (1) |
where n is the number of synteny blocks identified in each pairwise comparison, Lseq is the length of the shorter sequence in each pair of compared sequences, and Lsb(n) represents the length of the nth synteny block.
We performed a validation of this synteny method using a set of 15 Escherichia coli genomes classified to two different clades, either K-12 MG1655 or O157:H7 (Table S1, tab 7A). We started our analysis by randomly selecting five genes from the E. coli O157:H7 reference genome (NCBI reference sequence NC_002695.2) and analyzing the genomic regions flanking each target gene (10 kb up- and downstream of the gene). Two genes (mnmC and ubiD) were found in high homology only in O157:H7 genomes while the other genes (polB, hemA, and nupG) were detected in genomes of both groups. Pairwise comparisons of all three regions showed with high significance that the within-clade synteny scores are higher than the between-clade synteny scores (Table S1, tab 7B, and Fig. S7).
Synteny method validation with E. coli genome. Synteny scores of pairwise comparisons, for 3 regions in the E. coli genome (see Materials and Methods). Pairwise comparisons are grouped by the type of the pair, either within clade or between clades. Panel titles denote the gene located at the center of each genomic region. Download FIG S7, PDF file, 0.1 MB (71KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ethics approval and consent to participate.
All work involving the use of these previously collected samples from human subjects was approved by the Cornell University IRB (protocol ID 1108002388).
Availability of data and materials.
Sequences of pre- and postcapture metagenomes (the only new data generated for this paper) are available on the European Nucleotide Archive under accession number PRJEB38000.
ACKNOWLEDGMENTS
We thank the team at NimbleGen and Roche Sequencing Solutions, Nicole Leahy, Christine Kuch, and others, for their help in developing the genome capture array.
TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, Chronic Disease Research Foundation (CDRF), Zoe Global Ltd., and the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. This work was supported by the Max Planck Society and NIH grant no. 2R01DK093595-05A1.
We declare we have no competing interests.
V.S. and R.L. conceived the project. V.S. and N.D.Y. ran the bioinformatics and statistical analyses. H.E. conceived and executed synteny analyses. V.S., R.L., and H.E. wrote the paper. T.S. provided resources and feedback. R.L. supervised the project.
REFERENCES
- 1.Itan Y, Jones BL, Ingram CJE, Swallow DM, Thomas MG. 2010. A worldwide correlation of lactase persistence phenotype and genotypes. BMC Evol Biol 10:36. doi: 10.1186/1471-2148-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranciaro A, Campbell MC, Hirbo JB, Ko WY, Froment A, Anagnostou P, Kotze MJ, Ibrahim M, Nyambo T, Omar SA, Tishkoff SA. 2014. Genetic origins of lactase persistence and the spread of pastoralism in Africa. Am J Hum Genet 94:496–510. doi: 10.1016/j.ajhg.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, Ibrahim M, Omar SA, Lema G, Nyambo TB, Ghori J, Bumpstead S, Pritchard JK, Wray GA, Deloukas P. 2007. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet 39:31–40.doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, International HapMap Consortium, et al. 2007. Genome-wide detection and characterization of positive selection in human populations. Nature 449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, Spector TD, Bell JT, Clark AG, Ley RE. 2016. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19:731–743. doi: 10.1016/j.chom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panaccione R, Otley A, Xu L, Shestopaloff K, Moreno-Hagelsieb G, Paterson AD, Croitoru K, GEM Project Research Consortium. 2016. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet 48:1413–1417. doi: 10.1038/ng.3693. [DOI] [PubMed] [Google Scholar]
- 7.Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT, Spector TD, Keinan A, Ley RE, Gevers D, Clark AG. 2015. Host genetic variation impacts microbiome composition across human body sites. Genome Biol 16:191. doi: 10.1186/s13059-015-0759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, Shilo S, Lador D, Vila AV, Zmora N, Pevsner-Fischer M, Israeli D, Kosower N, Malka G, Wolf BC, Avnit-Sagi T, Lotan-Pompan M, Weinberger A, Halpern Z, Carmi S, Fu J, Wijmenga C, Zhernakova A, Elinav E, Segal E. 2018. Environment dominates over host genetics in shaping human gut microbiota. Nature 555:210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 9.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, Zhernakova DV, Jankipersadsing SA, Jaeger M, Oosting M, Cenit MC, Masclee AAM, Swertz MA, Li Y, Kumar V, Joosten L, Harmsen H, Weersma RK, Franke L, Hofker MH, Xavier RJ, Jonkers D, Netea MG, Wijmenga C, Fu J, Zhernakova A. 2016. The effect of host genetics on the gut microbiome. Nat Genet 48:1407–1412. doi: 10.1038/ng.3663. [DOI] [PubMed] [Google Scholar]
- 10.O’Callaghan A, van Sinderen D. 2016. Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol 7:925. doi: 10.3389/fmicb.2016.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie H, Guo R, Zhong H, Feng Q, Lan Z, Qin B, Ward KJ, Jackson MA, Xia Y, Chen X, Chen B, Xia H, Xu C, Li F, Xu X, Al-Aama JY, Yang H, Wang J, Kristiansen K, Wang J, Steves CJ, Bell JT, Li J, Spector TD, Jia H. 2016. Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome. Cell Syst 3:572–584.e3. doi: 10.1016/j.cels.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C, Segeta N, Izard J, Waldron L, Gevers D, Miroplolsk L, Garrett S, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carpi G, Walter KS, Bent SJ, Hoen AG, Diuk-Wasser M, Caccone A. 2015. Whole genome capture of vector-borne pathogens from mixed DNA samples: a case study of Borrelia burgdorferi. BMC Genomics 16:434. doi: 10.1186/s12864-015-1634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metsky HC, Siddle KJ, Gladden-Young A, Qu J, Yang DK, Brehio P, Goldfarb A, Piantadosi A, Wohl S, Carter A, Lin AE, Barnes KG, Tully DC, Corleis B, Hennigan S, Barbosa-Lima G, Vieira YR, Paul LM, Tan AL, Garcia KF, Parham LA, Odia I, Eromon P, Folarin OA, Goba A, Simon-Lorière E, Hensley L, Balmaseda A, Harris E, Kwon DS, Allen TM, Runstadler JA, Smole S, Bozza FA, Souza TML, Isern S, Michael SF, Lorenzana I, Gehrke L, Bosch I, Ebel G, Grant DS, Happi CT, Park DJ, Gnirke A, Sabeti PC, Matranga CB, Viral Hemorrhagic Fever Consortium. 2019. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat Biotechnol 37:160–168. doi: 10.1038/s41587-018-0006-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. 2017. Microbial strain-level population structure & genetic diversity from metagenomes. Genome Res 27:626–638. doi: 10.1101/gr.216242.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nayfach S, Rodriguez-Mueller B, Garud N, Pollard KS. 2016. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res 26:1612–1625. doi: 10.1101/gr.201863.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engström PG, Sui SJH, Drivenes Ø, Becker TS, Lenhard B. 2007. Genomic regulatory blocks underlie extensive microsynteny conservation in insects. Genome Res 17:1898–1908. doi: 10.1101/gr.6669607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nadeau JH, Taylor BA. 1984. Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci U S A 81:814–818. doi: 10.1073/pnas.81.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sankoff D, Leduc G, Antoine N, Paquin B, Lang BF, Cedergren R. 1992. Gene order comparisons for phylogenetic inference: evolution of the mitochondrial genome. Proc Natl Acad Sci U S A 89:6575–6579. doi: 10.1073/pnas.89.14.6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, Cagnasso P, Bizzarri B, de’Angelis GL, Shanahan F, van Sinderen D, Ventura M. 2009. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl Environ Microbiol 75:1534–1545. doi: 10.1128/AEM.02216-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato K, Odamaki T, Mitsuyama E, Sugahara H, Zhong Xiao J, Osawa R. 2017. Age-related changes in the composition of gut Bifidobacterium species. Curr Microbiol 74:987–995. doi: 10.1007/s00284-017-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arboleya S, Watkins C, Stanton C, Ross RP. 2016. Gut bifidobacteria populations in human health and aging. Front Microbiol 7:1204. doi: 10.3389/fmicb.2016.01204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Z, Zhang W, Guo C, Yang X, Liu W, Wu Y, Song Y, Kwok LY, Cui Y, Menghe B, Yang R, Hu L, Zhang H. 2015. Comparative genomic analysis of 45 type strains of the genus Bifidobacterium: a snapshot of Its genetic diversity and evolution. PLoS One 10:e0117912. doi: 10.1371/journal.pone.0117912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milani C, Lugli GA, Duranti S, Turroni F, Mancabelli L, Ferrario C, Mangifesta M, Hevia A, Viappiani A, Scholz M, Arioli S, Sanchez B, Lane J, Ward DV, Hickey R, Mora D, Segata N, Margolles A, van Sinderen D, Ventura M. 2015. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci Rep 5:15782. doi: 10.1038/srep15782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. 2016. Cross-species comparisons of host genetic associations with the microbiome. Science 352:532–535. doi: 10.1126/science.aad9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duranti S, Milani C, Lugli GA, Turroni F, Mancabelli L, Sanchez B, Ferrario C, Viappiani A, Mangifesta M, Mancino W, Gueimonde M, Margolles A, van Sinderen D, Ventura M. 2015. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ Microbiol 17:2515–2531. doi: 10.1111/1462-2920.12743. [DOI] [PubMed] [Google Scholar]
- 27.Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, Lapidus A, Rokhsar DS, Lebrilla CB, German JB, Price NP, Richardson PM, Mills DA. 2008. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci U S A 105:18964–18969. doi: 10.1073/pnas.0809584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sela DA, Price N, Mills D. 2010. Metabolism of Bifidobacteria, p 45–71. In Baltasar M, van Sinderen D (ed), Bifidobacteria genomics and molecular aspects. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 29.Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, Muzny D, Gibbs RA, Vatanen T, Huttenhower C, Xavier RJ, Rewers M, Hagopian W, Toppari J, Ziegler A-G, She J-X, Akolkar B, Lernmark A, Hyoty H, Vehik K, Krischer JP, Petrosino JF. 2018. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562:583–588. doi: 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vatanen T, Plichta DR, Somani J, Münch PC, Arthur TD, Hall AB, Rudolf S, Oakeley EJ, Ke X, Young RA, Haiser HJ, Kolde R, Yassour M, Luopajärvi K, Siljander H, Virtanen SM, Ilonen J, Uibo R, Tillmann V, Mokurov S, Dorshakova N, Porter JA, McHardy AC, Lähdesmäki H, Vlamakis H, Huttenhower C, Knip M, Xavier RJ. 2019. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat Microbiol 4:470–479. doi: 10.1038/s41564-018-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. 2014. Human genetics shape the gut microbiome. Cell 159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oki K, Akiyama T, Matsuda K, Gawad A, Makino H, Ishikawa E, Oishi K, Kushiro A, Fujimoto J. 2018. Long-term colonization exceeding six years from early infancy of Bifidobacterium longum subsp. longum in human gut. BMC Microbiol 18:209. doi: 10.1186/s12866-018-1358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asnicar F, Manara S, Zolfo M, Truong DT, Scholz M, Armanini F, Ferretti P, Gorfer V, Pedrotti A, Tett A, Segata N, Pasolli E, Huttenhower C, Segata N. 2017. Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems 2:e00164-16. doi: 10.1128/mSystems.00164-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yassour M, Jason E, Hogstrom LJ, Arthur TD, Tripathi S, Siljander H, Selvenius J, Oikarinen S, Hyöty H, Virtanen SM, Ilonen J, Ferretti P, Pasolli E, Tett A, Asnicar F, Segata N, Vlamakis H, Lander ES, Huttenhower C, Knip M, Xavier RJ. 2018. Strain-level analysis of mother-to-child bacterial transmission during the first few months of life. Cell Host Microbe 24:146–154.e4. doi: 10.1016/j.chom.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang K, Bin Keefe CR, Keim P, Kelley ST, Knights D, Koester I, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J. 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuznetsova A, Brockhoff PB, Christensen RHB. 2017. lmerTest package: tests in linear mixed effects models. J Stat Softw 82(13):1–26. [Google Scholar]
- 40.Karasov TL, Almario J, Friedemann C, Ding W, Giolai M, Heavens D, Kersten S, Lundberg DS, Neumann M, Regalado J, Neher RA, Kemen E, Weigel D. 2018. Arabidopsis thaliana and Pseudomonas pathogens exhibit stable associations over evolutionary timescales. Cell Host Microbe 24:168–179.e4. doi: 10.1016/j.chom.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, Huttenhower C. 2018. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15:962–968. doi: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benoit G, Peterlongo P, Mariadassou M, Drezen E, Schbath S, Lavenier D, Lemaitre C. 2016. Multiple comparative metagenomics using multiset k-mer counting. PeerJ Comput Sci 2:e94. doi: 10.7717/peerj-cs.94. [DOI] [Google Scholar]
- 44.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Letunic I, Bork P. 2011. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Köster J, Rahmann S. 2012. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 28:2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 47.Cepeda V, Liu B, Almeida M, Hill CM, Koren S, Treangen TJ, Pop M. 2017. MetaCompass: reference-guided assembly of metagenomes. bioRxiv 212506. doi: 10.1101/212506. [DOI]
- 48.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 49.Wright E. 2016. Using DECIPHER v2.0 to analyze big biological sequence data in R. R J 8:352–359. doi: 10.32614/RJ-2016-025. [DOI] [Google Scholar]
- 50.Spector TD, Williams F. 2006. The UK Adult Twin Registry (TwinsUK). Twin Res Hum Genet 9:899–906. doi: 10.1375/twin.9.6.899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(Tab 1) Taxonomic annotations of 16S rRNA SVs (top, n = 1,680) and metagenomic sequences (bottom, n = 245). (Tab 2) Overview of participant data. (Tab 3) Reference genomes used in the capture array. (Tab 4) Details of StrainPhlAn multilocus sequence alignments for each of the species considered. (Tab 5) Summary of strain-level methods used in the paper and an extension of Table 1 and Table 2 to include P values of significant tests. (Tab 6) P values and comparison numbers for the differences in mean synteny scores between pairwise comparison categories, for each species broken down by genomic region and in aggregate. (Tab 7A) E. coli genomes used for the validation of the synteny-based method. (Tab 7B) Genomic regions in the E. coli genome used for calculation of pairwise synteny scores. Download Table S1, XLSX file, 0.03 MB (34.5KB, xlsx) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
StrainPhlAn alignment statistics show low intrasample strain diversity. (A) The mean percentage of polymorphic sites along our MLSA for each of the four species considered. (B) The mean dominant allele frequency across all polymorphic sites within an MLSA. Boxplots show median and quartiles. Species: adol, B. adolescentis; anim, B. animalis; bifm, B. bifidum; long, B. longum. Significance levels are Wilcoxon rank sum tests. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Nonsignificant comparisons are not indicated. Download FIG S1, PDF file, 0.04 MB (45KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Host genotype effects on intraindividual longitudinal strain stability. All intraindividual longitudinal comparisons for each of B. longum, B. adolescentis, and B. bifidum are plotted according to host genotype for patristic distance (branch lengths) between samples across our RAxML strain phylogenies (A) and the percent marker-SNPs shared between samples (B). Significance levels are Wilcoxon rank sum tests. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Red diamonds represent mean values. Download FIG S2, PDF file, 0.1 MB (81.7KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Host genotype effects on longitudinal strain stability and strain similarities between monozygotic twins across all four species in aggregate. All three metrics of strain comparisons are shown, with each separated by lactase persistence genotype and the category of comparison: Intra (same person over time), same family (comparisons between twin siblings), and unrelated (comparisons between unrelated people). Plots represent combined data for B. longum, B. bifidum, and B. adolescentis. The metrics shown for each comparison category are patristic distance (branch lengths) between samples across our RAxML strain phylogenies (A), the percent marker-SNPs shared (B), and synteny scores across the phylogenies (C). Significance levels are median Wilcoxon rank sum tests after 999 bootstraps to smallest group size. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Download FIG S3, PDF file, 0.2 MB (159.6KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Temporal distance between intraindividual samples is not correlated with patristic distance across MLSAs. All unique intrapersonal comparisons are plotted, with time between samples on the x axis and StrainPhlAn MLSA patristic distance on the y axis. Blue line shows least-squares regression with 95% confidence intervals, while Spearman’s rho and the significance of the correlation are shown. Download FIG S4, PDF file, 0.1 MB (87KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Temporal distance between intraindividual samples is not correlated with overall microbiome community similarity, synteny, or mean marker-SNP sharing. All unique intrapersonal comparisons are plotted, with time between samples on the x axis and a metric of stability of similarity on the y axis. The metrics are as follows: Bray-Curtis Dissimilarity (BCD) based on SIMKA k-mer spectra of precapture metagenomes (A), BCDs based on metabolic pathways of precapture metagenomes (B), mean synteny scores across all regions examined (C), and mean percentage of marker-SNPs shared across all four taxa (D). Blue line shows least-squares regression with 95% confidence intervals, while Spearman’s rho and the significance of the correlation are shown. Download FIG S5, PDF file, 0.1 MB (89.1KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Intraindividual marker-SNP sharing of Bifidobacterium species does not correlate with overall community dissimilarity. Percentage of Bifidobacterium marker-SNPs shared between samples from the same individual over time, plotted against the overall community dissimilarity from those samples, based on precapture metagenomes (i.e., standard metagenome libraries). BCD metrics used to assess overall community dissimilarity are Simka, a k-mer-based dissimilarity metric between the two samples (A), and BCD values for MetaCyc annotations of metabolic pathways generated in HUMAnN2 (B). Blue line shows least-squares regression with 95% confidence intervals, and Spearman’s rho along with the significance of the correlation is shown. Download FIG S6, PDF file, 0.1 MB (53.8KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Synteny method validation with E. coli genome. Synteny scores of pairwise comparisons, for 3 regions in the E. coli genome (see Materials and Methods). Pairwise comparisons are grouped by the type of the pair, either within clade or between clades. Panel titles denote the gene located at the center of each genomic region. Download FIG S7, PDF file, 0.1 MB (71KB, pdf) .
Copyright © 2020 Schmidt et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Sequences of pre- and postcapture metagenomes (the only new data generated for this paper) are available on the European Nucleotide Archive under accession number PRJEB38000.