

An NMR-based strategy was established to improve protein druggability by stabilizing cryptic binding sites in an equilibrium.
Abstract
Cryptic ligand binding sites, which are not evident in the unligated structures, are beneficial in tackling with difficult but attractive drug targets, such as protein-protein interactions (PPIs). However, cryptic sites have thus far not been rationally pursued in the early stages of drug development. Here, we demonstrated by nuclear magnetic resonance that the cryptic site in Bcl-xL exists in a conformational equilibrium between the open and closed conformations under the unligated condition. While the fraction of the open conformation in the unligated wild-type Bcl-xL is estimated to be low, F143W mutation that is distal from the ligand binding site can substantially elevate the population. The F143W mutant showed a higher hit rate in a phage-display peptide screening, and the hit peptide bound to the cryptic site of the wild-type Bcl-xL. Therefore, by controlling the conformational equilibrium in the cryptic site, the opportunity to identify a PPI inhibitor could be improved.
INTRODUCTION
Protein-protein interactions (PPIs) are attractive but difficult targets for therapeutic intervention. The importance of targeting PPIs is evident from their fundamental roles as hubs for many biological processes. It has been shown that there are 130,000 to 650,000 PPIs in the human interactome, and many of those are relevant to human diseases (1, 2). More than 30 PPIs have now been targeted with small molecules, and some of those molecules are clinically used or under clinical trials (3–6). However, the difficulties in targeting PPIs still exist (7, 8). Large and flat interfaces, which are characteristic of PPIs, are a reason for the difficulty. The structural feature is in stark contrast to the deep cavities of enzymes or receptors that are amenable to the binding of small molecules. Although tremendous efforts have been made to target PPIs (9–14), development of PPI inhibitors is still challenging, and a strategy that is orthogonal to the currently available strategies would be needed.
It has been increasingly recognized that the conformational flexibility of proteins contributes to the affinity and specificity of PPIs and their inhibitors (10, 15–18). Among those dynamic PPI interfaces, ligand-binding pockets that seem to be absent in the ligand-free structures but evident in the ligand-complex structures have been called cryptic sites (19, 20). Cryptic sites provide additional interface to the ligands for higher affinity and specificity; thus, successful PPI inhibitors are often associated with cryptic sites. ABT-737, which binds to the antiapoptotic proteins Bcl-xL and Bcl-2, is one of the hallmark examples of PPI inhibitors that target cryptic sites. ABT-737 competitively inhibits the interaction of the BH3 domain–containing proapoptotic proteins, such as BAK and BAD, to Bcl-xL (21). The conformation of Bcl-xL in the ABT-737–bound state is substantially different from the unligated state. Using the α3-α4 loop as the hinge, a cryptic site between α3 and α4 helices is widely open upon the binding to the ligand (Fig. 1). The opening of cryptic site increases the surface exposure area of Bcl-xL from 6875 to 7525 Å2, and the closest Cα-Cα distance between α3 and α4 helices in unligated structure (Leu112 and Val126; 6.3 Å) was substantially expanded to 10.2 Å. With this additional interface, ABT-737 shows the most potent Ki (inhibition constant) value of 0.6 nM, which is comparable to that of the proteins with BH3 domain. Therefore, the benefit in targeting the cryptic site is evident, and the utilization of the cryptic site reportedly expands the druggable human proteome almost twofold (20). However, the cryptic sites have thus far been mostly discovered by chance, after the determination of the complex structure of a ligand. Otherwise, time-consuming experimental and/or computational approaches that are specialized for each target need to be developed (19, 22, 23). Therefore, a general strategy to experimentally identify cryptic sites in the absence of ligands, along with a strategy to rationally target the identified cryptic sites, would be required to develop active ligands for difficult-to-target systems, such as PPIs.
Fig. 1. Structures of Bcl-xL in unligated (top) and ABT-737–bound (bottom) states.
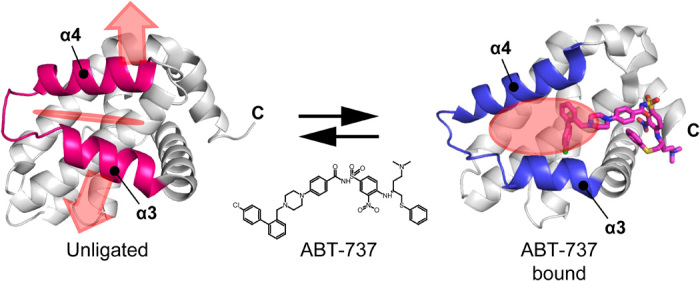
The structure of bound ABT-737 is shown in the magenta stick representation. Cryptic site on Bcl-xL is indicated by a red shadow. Apo structure, 1maz; ABT-737 complex, 2yxj.
Here, we developed a strategy to identify and stabilize the open cryptic site conformation by nuclear magnetic resonance (NMR) dynamics information and semi-rationally designed allosteric mutations. The temperature-dependent chemical shift perturbation (CSP) and the NMR dynamics analysis using relaxation dispersion (RD) experiment identified a lowly populated conformational state in the cryptic site of Bcl-xL under unligated conditions. On the basis of the distribution of the residues that showed the temperature-dependent chemical shift changes, a mutant, F143W, that allosterically stabilizes the cryptic site was developed, and the utilization of the mutant substantially enhanced the efficiency to enrich the Bcl-xL interaction sequences in a phage-display peptide screening. Furthermore, the identified peptide bound to the cryptic site of the wild-type (WT) Bcl-xL. These results indicate that protein druggability can be improved by analyzing and controlling the dynamics of proteins, which would represent a versatile and complement strategy for PPI inhibitor development.
RESULTS
NMR characterization of ABT-737 binding to Bcl-xL
To characterize the ligand-induced opening of the cryptic site in the Bcl-xL protein, we titrated ABT-737 to methyl 13C–labeled Bcl-xL (Fig. 2). The substantial CSPs in slow exchange fashion were observed for those residues that were in close proximity to the compound, such as Leu108, Leu126, Ile130, and Val141, in the α3, α4, and α5 helices. The CSPs are also distributed in those sites that are distal (>7 Å) from the bound compound. These allosteric CSPs were distributed in the residues in the α2 helix (Leu90), the α3-α4 loop (Leu112 and Ile114), the residues in the α5 and α6 helices that form the interface to the α4 helix (Ile140, Leu150, Ile166, and Leu178), and the α8 helix (Leu194). The distribution suggests that the allosteric CSPs induced by ABT-737 binding reflect conformational changes associated with the opening of the cryptic site (Figs. 1 and 2). It should be noted that the resonance originating from Leu112 is weak in the unligated Bcl-xL spectrum, indicating the presence of a conformational exchange in the α3-α4 loop that served as a hinge in the opening of the cryptic site.
Fig. 2. NMR analysis of the interaction between Bcl-xL and ABT-737.
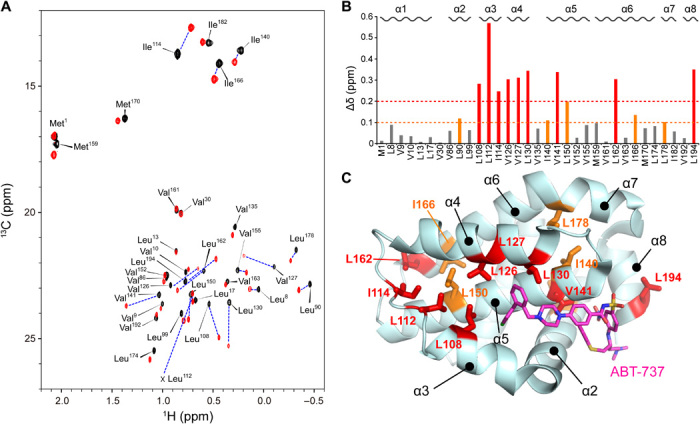
(A) Heteronuclear multiple quantum correlation (HMQC) spectrum of 100 μM [Ile-δ1, Met-ε, Leu-δ2, Val-γ2 methyl-1H13C] Bcl-xL in unligated (black) and excess amount of ABT-737 (120 μM, red). (B) Normalized CSPs for each residue upon binding to ABT-737. (C) Mapping of substantially perturbed residues (Δδ > 0.2 ppm, red; Δδ > 0.1 ppm, orange) on the ribbon representation of Bcl-xL in ABT-737–bound conformation. ABT-737 is shown in a magenta stick representation.
Presence of the open cryptic site conformation in the unligated Bcl-xL
To determine if the opening of the cryptic site can only be induced by compound binding (induced-fit) or the open conformation can be sampled in the unligated state in a conformational selection manner, we analyzed the temperature dependence of the methyl resonances. Reflecting the temperature change from 288 to 308 K, some of the resonances from the unligated Bcl-xL showed the temperature-dependent chemical shift changes (Fig. 3A) (24–27). Most of the residues that showed temperature-dependent CSPs are distributed in the region where the allosteric chemical shift changes upon the binding to ABT-737 were observed (Fig. 3B). Therefore, a conformational equilibrium seems to be present in the cryptic site of the unligated Bcl-xL. In the temperature range, a linear van’t Hoff’s relation was observed, suggesting that the observed conformational equilibrium is between two conformational states (Fig. 3C), presumably between the open and closed conformations of the cryptic site. Nevertheless, the temperature variation induced only small 13C chemical shift changes to the unligated Bcl-xL resonances, as compared to those by the ABT-737 binding (fig. S1). Thus, the effect of temperature-dependent changes on the conformational equilibrium in the cryptic site should be limited and the cryptic site of the unligated Bcl-xL should primarily exist in the closed conformation, as expected.
Fig. 3. Presence of the open cryptic site conformation in unligated Bcl-xL.
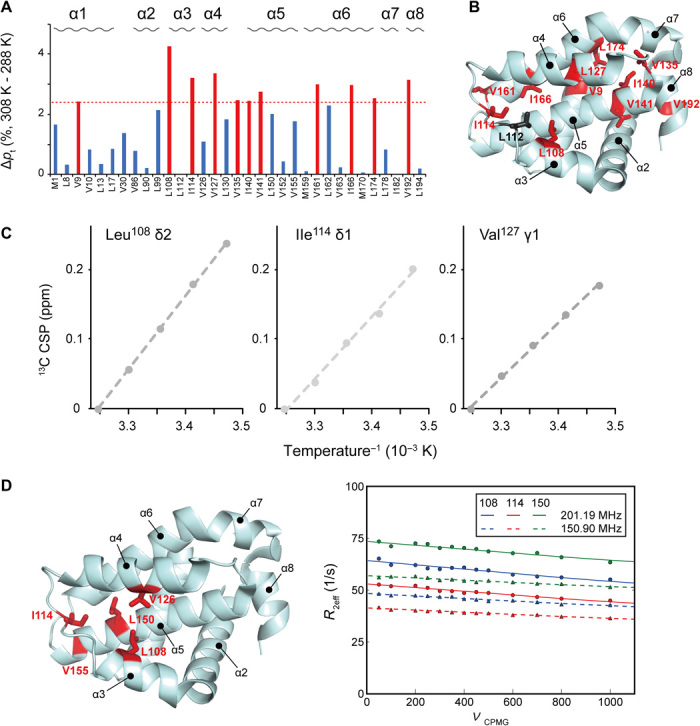
(A) Temperature-dependent population changes in the Ile χ2, Leu χ2, Met χ3, and Val χ1 rotameric states between 288 and 308 K in apo Bcl-xL. The populations in the rotameric states were calculated from the equations shown in Materials and Methods. (B) Mapping of residues that showed substantial temperature dependence of their rotameric states. The methyl moieties with >2 SDs of rotameric population shift in response to the temperature variation are highlighted in red. (C) Van’t Hoff plot of three resonances that showed the largest temperature-dependent 13C chemical shift changes. (D) Methyl CPMG RD experiments of unligated Bcl-xL. The residues that showed substantial Rex (>5 Hz) contributions to the transverse relaxation of the 13C methyl moiety are mapped on the structure of Bcl-xL. For those residues in the cryptic site (Leu108, Ile114, and Leu150), the RD plot and their global fitting are shown.
To further confirm the presence of the conformational equilibrium in the cryptic site of the unligated Bcl-xL, we performed a Carr-Purcell-Meiboom-Gill RD (CPMG RD) experiment, which used a variable pulse frequency, νCPMG, to measure the effective transverse (R2) relaxation rates. In the 13C methyl CPMG RD experiment, we observed substantial Rex (>5 Hz) contribution to the R2 of the 13C methyl moiety in the α3-α4 hinge (Leu108 and Ile114), the α4 helix (Val126), and the α5 helix (Leu150 and Val155) (Fig. 3 and fig. S2). All of these residues, except Val155, showed large allosteric CSPs in the 13C dimension upon ABT-737 binding (Fig. 2), indicating the presence of a slow (~ms) conformational equilibrium in the cryptic site of the unligated Bcl-xL.
Development of an allosteric mutant that stabilizes open cryptic site conformation
The presence of conformational equilibrium led us to search for a mutant that allosterically stabilizes the open cryptic site conformation. Such an allosteric mutation elevates the population of the open cryptic site conformation in the equilibrium, without modifying the structure of the binding interface. Thus, the binding affinity of weak ligands, if any, is expected to improve, as the binding-potent open cryptic site conformation is populated more in mutant than in WT.
Upon the ABT-737 binding to the WT Bcl-xL, allosteric CSPs were observed for the residues on the interface between the α5 and α6 helices (the α5/α6 interface; Ile140, Ile166, and Leu176). The α5/α6 interface is not a part of the ligand-binding site. However, as the α5/α6 interface interacts with the α4 helix that experiences a large conformational rearrangement upon the binding to the compound (fig. S3), the opening of the cryptic site would affect the conformation of the interface and, thus, induce the allosteric CSPs. We hypothesized that a mutation in the α5/α6 interface residue might affect the conformational equilibrium of the cryptic site and vice versa. Among the interface residues, Phe143 is the largest and located directly below the α4 helix; thus, we selected the residue and analyzed if there is any substitution that causes similar chemical shift changes to Bcl-xL, as the ABT-737 binding.
Figure 4 shows the overlaid spectra of the F143W mutant in the unligated state, with those of the WT Bcl-xL in the presence and absence of ABT-737. Among residues that showed CSPs upon binding to ABT-737 (Fig. 2), those that are also far from the mutation sites are shown (Leu90, Ile114, Leu150, Ile166, and Leu194). For these residues, the chemical shifts are expected to reflect the conformational equilibrium of the cryptic site, but not the direct effect from the ABT-737 binding or the substitution of Phe143 to Trp. The signals originating from the F143W mutant were observed in the position between those from the unligated and ABT-737–bound WT Bcl-xL. This indicates that the F143W mutation shifted the conformational equilibrium more toward the open cryptic site conformation. Assuming that the unligated and ABT-737–bound signals predominantly reflect the close and open states of the cryptic site, respectively, the F143W mutant is estimated to have 33 ± 24% of the open conformation in the unligated state. The variations in the populations of the open state could originate from the local conformational differences in the cryptic site between the F143W mutant and WT Bcl-xL. However, the overall conformation might be retained, as the signals did not largely deviate from the line connecting the unligated and ABT-737–bound WT Bcl-xL signals (Fig. 4A).
Fig. 4. Structural characterization of open cryptic site mutant F143W.
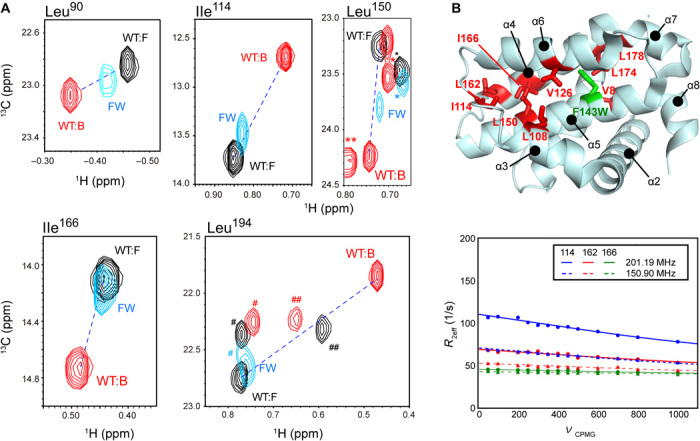
(A) Overlaid spectra of the unligated F143W mutant (cyan) and the unligated (black) and ABT-737–bound (red) WT Bcl-xL. Residues with substantial allosteric CSPs upon ABT-737 binding are shown. The peak positions of the residues that were not subjected to the analysis are indicated by *, **, #, and ## for Leu17, Leu90, Val10, and Leu162, respectively. Unligated and ABT-737–bound peaks are connected by a dashed line. (B) Methyl RD experiments with the unligated F143W mutant. The residues with substantial Rex (>5 Hz) contributions to the 13C methyl R2 were mapped on the structure of Bcl-xL, along with the mutation site. For the residues in the core of the cryptic site (Ile114, Leu162, and Leu166), the RD plots and their global fittings are shown.
With the improved the population of open cryptic site conformation, in the F143W mutation, we tested if the F143W mutant showed improved affinities to the known ligands. Although the isothermal titration calorimetry (ITC) experiment cannot be performed with ABT-737 due to the poor solubility of the compound, the F143W mutant showed elevated affinity to the Bak BH3 peptide, which also binds to the cryptic site. While the KA value of the Bak BH3 peptide to the WT Bcl-xL was 1.1 ± 0.2 × 106 M−1, the F143W mutant showed twofold higher affinity (2.2 ± 0.3 × 106 M−1) to the peptide (fig. S4). The affinity improvement was somewhat smaller than expected. This might be due to the difference in the local conformations of the cryptic site between the F143W mutant and the WT Bcl-xL, as discussed above. Nevertheless, the elevated population of the open cryptic site conformation in the F143W mutant was supported by the accelerated R2 and the larger Rex values in the RD experiments (fig. S2). The residues that showed Rex values >5 Hz are distributed near the mutation sites as well as in the cryptic site, indicating that the F143W mutation allosterically affected the conformational equilibrium in the cryptic site (Fig. 4B). These data indicate that the F143W mutation allosterically shifted the conformational equilibrium to a larger population of the open cryptic site conformation, thereby improving the affinity to the peptide.
The open cryptic site mutant showed improved druggability
As the F143W mutant showed the improved affinity to the ligand, we thought that the open cryptic site mutant might have improved hit rate in ligand screening than that of the WT. To test this, we compared the efficiency in enriching the Bcl-xL binding peptides in a phage display. A phage library that displayed random 10–amino acid foreign peptides on the N terminus of the major coat protein (gVIII protein) of bacteriophage M13 was constructed (28), and three rounds of panning were carried out against the same amount of the immobilized WT Bcl-xL or F143W mutant. The ratio of colony number before and after the panning was compared as the enrichment factor. In the three rounds of panning, the F143W mutant enriched the hit sequence more efficiently than that of WT (Fig. 5A). A linear peptide with an enriched sequence, peptide P1, YPLGSPWLRLHS, showed a clear CSP in the cryptic site (Fig. 5, B and C). In addition, the peptide-induced 13C CSPs showed a substantial correlation with the allosteric 13C CSP upon binding to ABT-737 (Fig. 5D). These results indicate that the obtained hit peptide binds to the WT Bcl-xL with the expected binding mode, and the druggability of Bcl-xL was improved by the allosteric mutation that has more open cryptic site conformation in the unligated condition.
Fig. 5. Efficient screening of the cryptic site–bound peptide by the F143W mutant.
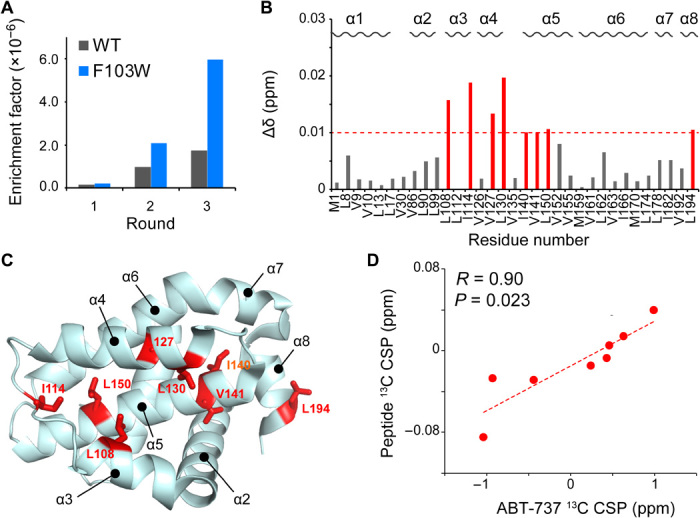
(A) Comparison of the enrichment factors in each round of phage-display screening. The ratio of hit clone against the whole clone cfu was shown for each panning round. (B) Normalized CSP induced by the hit peptide; 160 μM peptide was added to 100 μM WT Bcl-xL. (C) Mapping of the residue that experienced normalized CSP > 0.01 ppm. (D) Correlation plot of the ABT-737–induced allosteric 13C CSPs against the peptide-induced 13C CSPs.
DISCUSSION
Here, we showed a straightforward strategy to experimentally identify the cryptic site using NMR. Because the cryptic sites have thus far been identified only after the determination of the complex structure, the interactions to cryptic site have been thought to happen in an induced-fit manner. However, as we showed here, the open cryptic site conformation is already present in the conformational equilibrium in unligated Bcl-xL. Thus, ligand binding to the cryptic sites can also be achieved in a conformational selection mechanism and an NMR dynamics analysis of the unligated protein would be of importance to identify the presence of the open cryptic site conformation under unligated conditions. This would especially be true for the conformational equilibrium that is slow in its time scale (milliseconds in Bcl-xL). The strength of NMR is in its ability to identify a site that is distal to the cryptic site and change its conformation along with the opening of the cryptic site. Such a site can be identified by the temperature-dependent chemical shift changes and the RD experiments as shown here. Some of the cryptic sites can be identified by molecular dynamics simulations (20), and literatures already pointed out the presence of the dynamics in the BH3 peptide binding interface of Bcl-xL (29). However, experimental evidence is always warranted, and NMR would improve the chance to identify the lowly populated and slowly exchanging cryptic sites.
On the basis of the NMR information, a mutant that allosterically stabilizes the open cryptic site conformation can be established. Such a mutant would improve the ligand hit rate in a ligand screening. The strength of NMR in identifying and establishing a mutant that changes the conformational equilibrium of the distal site is already described in previous studies (30, 31). It should be noted that the allosteric mutation does not introduce any modification in the ligand binding interface per se. Therefore, ligands that bind to the open cryptic site mutants are expected to bind to the WT protein, as shown here for the peptide ligand established with the F143W mutant. It has been shown that there is a correlation between the experimental hit rate and the ability to identify high-affinity ligands (32); thus, improvement of hit rate would contribute to improve the protein druggability. Although we only discussed the F143W mutant in the results section, the type of residues that is appropriate for Phe143 substitution is not known a priori. We also made the F143L mutant, which makes the residue smaller as compared to the bulkier Trp residue. Unlike the Phe to Trp substitution, the Phe to Leu substitution did not affect the population of the cryptic site (fig. S5). In general, the availability of the x-ray structure of the ABT-737 complex helped us to identify the distal site to be substituted; however, as discussed above, such a site can also be identified by using NMR information. In the case where we already have a compound or an endogenous ligand, the discrimination of allosteric versus orthosteric chemical shift changes can be established by other NMR methods, such as intermolecular nuclear Overhauser effects and cross-saturation (33–35), which only reflects the direct binding events. It should also be noted that, by using NMR, the distal site to be mutated can be identified without compounds. In the case of Bcl-xL, the temperature-dependent chemical shift changes spread outside of the cryptic site and include the interface between the α5 and α6 helices, where Phe143 is located. Such information might not be obtained from the x-ray structure of the unligated Bcl-xL.
The identified peptide sequence (P1), YPLGSPLWRLHS, is similar to the canonical consensus sequence of the peptide BH3 peptide, which has amphiphilic helix characters (fig. S6) (36, 37). Although the canonical sequence has four hydrophobic residues (H1-H4) separated by two to three residues, the P1 peptide has only three of them (the corresponding amino acid is underlined in the sequence), missing the small and acidic residues after the H3 position (fig. S6). Thus, the P1 peptide seems to have a room to improve affinity. It should also be pointed out that the H1 and H2 sites are the part of the motif that binds to the cryptic site in the Bcl-xL. Thus, the peptides that bind to the cryptic site seem to be selected with the F143W mutant in the phage-display screening. CSPs, upon binding to the P1 peptide, correlate well with those of the ABT-737 binding (Fig. 5). These results indicate that protein druggability can be improved by analyzing and modulating the dynamics of the protein interface, which would represent a versatile strategy that could be implemented in the currently available strategies for the development of PPI inhibitors. It is clear that the strategy provided here not only is applicable to identify a small-molecule ligand but also contributes to find a ligand from emerging modalities such as middle-sized molecules.
MATERIALS AND METHODS
Expression and purification of human Bcl-xL
The deletion mutant of human Bcl-xL, which lacks the putative C-terminal transmembrane region (residues 210 to 233) and a flexible loop (residues 45 to 84), was used in the NMR study according to the previous literature (38). The DNA fragment (5′-gccagatctcatatggaaaacctgtacttccaaggtatgagccagagtaatcgggaattggtcgtcgactttctgtcctacaagctgtcgcagaaagggtactcttggtcacagttcagcgatgtggaggaaaatcgcactgaagcgccagaaggcaccgaaagtgaagccgtgaaacaggccttacgtgaagctggggatgaattcgagttacgctatcgtcgtgcgttttcggatctgacctctcaactgcacatcacaccgggtaccgcgtatcagtcctttgaacaggttgtaaacgagctgtttcgcgatggtgtgaactggggacgtatcgttgcgttcttcagctttggtggcgcattgtgcgtagagtcagtggacaaagagatgcaagtgctggtttcgcgcattgcagcctggatggcgacgtatctgaacgaccatcttgaaccgtggattcaggagaatggaggctgggatacgtttgtcgaactctatggcaataacgccgcagctgaaagccgcaaaggccaagaacgctaatag-3′), which encodes the deletion mutant of Bcl-xL (we call this deletion protein as WT Bcl-xL throughout the paper), was synthesized (Operon) and ligated into the pET15b vector (Novagen) at the Xho I restriction sites. Residue numbers correspond to the full-length Bcl-xL (UniProtKB, Q07817). The resultant vector expresses Bcl-xL with an N-terminal His6-tag with thrombin and tobacco etch virus (TEV) protease restriction sites. Bcl-xL mutants were constructed by the QuikChange strategy (Agilent Technology). Escherichia coli strain BL21(DE3) was transformed with the plasmid. The BL21(DE3) harboring the Bcl-xL plasmid was inoculated in 10 ml of LB medium containing kanamycin (50 μg/ml) and cultured at 37°C for 4 hours. The cells collected by centrifugation were further inoculated into 1 liter of kanamycin-M9 medium, supplemented with 2H d-glucose (3 g/liter) and 15NH4Cl (1 g/liter), as carbon and nitrogen sources. At an optical density of 0.8 and at a 600-nm wavelength (OD600), 0.6 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce Bcl-xL expression. The induced culture was further incubated at 32°C for 16 hours. For selective Ile-δ1, Met-ε, Leu-δ2, Val-γ2 methyl-1H13C labeling of Bcl-xL, a QLAM-Iδ1MεLVproS labeling kit (NMR Bio) that contains [4-13C-methyl, 3,3-2H2]-α-ketobutyric acid, [ε-methyl-13C]-methione, and [2-13C-methyl, 4-2H3]acetolactate was supplemented into the medium according to the manufacturer’s protocol (39). The cells were harvested by centrifugation and frozen at −80°C before purification.
The frozen pellets of Bcl-xL–expressing cells were resuspended in lysis buffer, consisting of 50 mM tris-HCl (pH 8.0), 300 mM NaCl, 10 mM imidazole, and 5 mM β-mercaptoethanol, and disrupted by sonication. Bcl-xL was purified from the supernatant of the cell lysate. The supernatant was applied to a 3-ml COSMOGEL His-Accept column and equilibrated with the lysis buffer. The column was washed with 40 ml of the lysis buffer, and then Bcl-xL was eluted with 20 ml of elution buffer, containing 50 mM tris-HCl (pH 7.5), 300 mM NaCl, 300 mM imidazole, and 5 mM β-mercaptoethanol. The eluate was concentrated by ultrafiltration, using an Amicon Ultra centrifugal filter unit (molecular weight cutoff, 10,000; Millipore) and then passed through a 0.22-μm syringe filter for further purification by size exclusion chromatography. After enzymatic digestion with TEV protease, the sample was applied to a HiLoad Superdex 75 prep grade column (GE Healthcare) and equilibrated with 50 mM sodium phosphate buffer (NaPi; pH 6.8) containing 300 mM NaCl and 2 mM dithiothreitol (DTT). The elution fraction was collected; buffer-exchanged into NMR buffer containing 10 mM NaPi (pH 6.8), 100 mM NaCl, and 2 mM DTT; and stored at −80°C until use.
NMR experiments
All titration experiments were performed at the same field strength using a Bruker Avance 600-MHz spectrometer equipped with a cryogenic triple resonance probe. In contrast, the experiments for assignments were performed using both a 600-MHz spectrometer and an Avance III 800-MHz spectrometer equipped with a cryogenic triple resonance probe. All spectra were recorded using 10 mM NaPi buffer (pH 6.8) and 2 mM DTT, in either 90% H2O/10% D2O or 100% D2O, depending on the experiments. For compound titration experiments, 5% of deuterated dimethyl sulfoxide (DMSO) (D6) was added to the buffer to increase the solubility of the compound. The typical concentration of Bcl-xL was 0.1 mM (subunit concentration). Unless otherwise stated, the experiments were performed at 298 K. Spectra were processed using TOPSPIN (Bruker Biospin) and analyzed using Sparky (40). The assignments in the unligated state were established by referring the reported assignments (BMRB: 18250) and were confirmed by three-dimensional (3D) hCCH-TOCSY, 3D HcCH-TOCSY, 3D NOESY-13C-HMQC (mixing time, 200 ms), and 3D NOESY-15N-HSQC experiments (mixing time, 200 ms). The assignments in the ABT-737–bound state were established by 3D hCCH-TOCSY, 3D HcCH-TOCSY, 3D NOESY-13C-HMQC (mixing time, 200 ms), and 3D NOESY-15N-HSQC experiments (mixing time, 200 ms), referring the crystal structure of the Bcl-xL in complex with ABT-737 [Protein Data Bank (PDB) ID: 2YXJ]. The assignments in the ABT-737–bound state were further confirmed by introducing L112I, V126I, L130I, L162I, and L194I mutations. The assignments of the F143W mutant were established by referring the established assignment of the WT Bcl-xL in the unligated state.
The normalized CSP values (Δδnorm) were calculated as follows:
Δδnorm was used to compare the amplitude of CSPs in each condition. In contrast, 13C CSPs were used to compare the correlation between different conditions, as the 13C chemical shift is known to more directly reflect the local conformation (24).
The rotameric equilibria of the Ile χ2, Leu χ2, Met χ3, and Val χ1 angles were deduced from the Ile (δ1), Leu, Met, and Val methyl 13C chemical shifts, respectively. The 13C chemical shifts of methyl signals are reportedly dependent on the side-chain rotamer, as revealed by theoretical and experimental analyses (24–27). The population of the trans rotameric state (pt) for each residue was calculated according to the chemical shift values of the methyl 13C signals [δobs; parts per million (ppm)] using Eqs. 1 to 4
| (1) (24) |
| (2) (27) |
| (3) (25) |
| (4) (26) |
If the equation yielded a pt value of >1 or <0, then pt was fixed to 1 (all trans) or 0 (all gauche-), respectively.
RD profiles of CH3 groups of Bck-xL are obtained using the reported pulse sequences (41) and analyzed by Nessy software for automated NMR RD data analysis (42).
Construction of phagemid vector and phage library
The phagemid vector containing the DNA sequence for the gVIII signal and gVIII was generated as previously described (28). For construction of the phage library, two rounds of polymerase chain reaction (PCR) were performed. In the first PCR, the resulting phagemid vector was used as a template, and the following primers were used: 5′-AAACAGACCATGGCCATGAAAAAGTCTTTA-3′ (forward; underline indicates the Nco I site) and 5′-GTCACCGGATCCCATNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNGTAAGCGAAAGACAGCATCGG-3′ (backward; underline indicates the Bam HI site). In the second PCR, we used the products from the first PCR as a template, and the following primers were used: 5′-AAACAGACCATGGCCATGAAAAAGTCTTTA-3′ (forward; same as the forward primer of first PCR) and 5′-GTCACCGGATCCCAT-3′ (backward; underline indicates the Bam HI site). The resulting fragments were inserted into the Nco I and Bam HI sites of the phagemid vector. The resulting phagemid encodes a fusion product with the gVIII signal peptide Y-XXXXXXXXXX-M-GSGD and gVIII [Y, detectable residue at 280 nm by high-performance liquid chromatography; XXXXXXXXXX, 10-mer random sequence (peptide library sequence); M, cyanogen bromide (CNBr) cleavage residue; and GSGD, flexible linker]. The phagemid mixture was transformed by electroporation into E. coli XL1-Blue. The transformants were then infected with the KO7 helper phage (Amersham Biosciences). The resulting library had a diversity of 7.0 × 105.
Screening of phage library
The phage libraries were cycled through three rounds of binding selection with Bcl-xL. For each round, the WT Bcl-xL or the F143W mutant (5 μg) in phosphate-buffered saline (PBS) was immobilized on the Ni–nitrilotriacetic acid–coated 96-well plates (Pierce) and incubated overnight at 4°C. After, the coating plates were blocked with BlockAce (Dainippon Sumitomo Pharma, Osaka, Japan) at room temperature for 30 min and then washed with 0.1% Tween 20 (Sigma-Aldrich) in PBS (T-PBS). Phage particles were incubated with the plates for 1 hour at room temperature. The beads or plates were washed three times by 20% BlockAce (Dainippon Sumitomo Pharma) and 0.1% Tween 20 (Sigma-Aldrich) in PBS (BA–T-PBS) for 3 min. The bound phage particles were eluted with 0.1 M glycine-HCl (pH 2.0) and immediately neutralized by 1 M tris-HCl (pH 9.0). The eluted sample was used to infect fresh E. coli XL1-Blue cells. After three rounds of binding selection, individual phage particles were isolated and subjected to DNA sequence analysis and the peptide with the resultant sequence, NH3-YPLGSPLWRLHS-COOH, was synthesized (BEX).
ITC measurements
Calorimetric titrations were performed using a VP-ITC microcalorimeter (MicroCal) at 25°C. For the Bcl-xL–Bak peptide interaction, the ITC experiment was performed in 10 mM NaPi buffer (pH 6.8), which is the same as the NMR buffer. Bak BH3 peptide with the sequence NH3-GQVGRQLAIIGDDINR-COOH was chemically synthesized (BEX). Protein samples were extensively dialyzed against ITC buffers before the experiments. The sample cell was filled with 10 μM Bcl-xL, and 100 μM Bak peptide in the syringe was titrated. After a preliminary 3-μl injection, 24 subsequent 10-μl injections were performed. The data were fitted using the one-site binding model embedded in Origin 7.0 (MicroCal).
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of recently deceased colleague, Dr. Yumiko Mizukoshi. Funding: This work was supported by grants from the Japan Agency for Medical Research and Development (grant JP18ae010104 to I.S.) and the Japan Society for the Promotion of Science, KAKENHI (grant JP17H06097 to I.S. and grants JP20H03378 and JP20H04722 to K.T.). Author contributions: Y.M., K.T., Y.T., T.T., H.H., and I.S. designed the project. H.M., M.I., M.F., and H.K. prepared Bcl-xL samples. Y.M., K.T., and Y.T. performed experiments and analyses. Y.M., K.T., Y.T., T.T., H.H., and I.S. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabd0480/DC1
REFERENCES AND NOTES
- 1.Stumpf M. P. H., Thorne T., de Silva E., Stewart R., An H. J., Lappe M., Wiuf C., Estimating the size of the human interactome. Proc. Natl. Acad. Sci. U.S.A. 105, 6959–6964 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venkatesan K., Rual J.-F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T., Hao T., Zenkner M., Xin X., Goh K.-I., Yildirim M. A., Simonis N., Heinzmann K., Gebreab F., Sahalie J. M., Cevik S., Simon C., de Smet A.-S., Dann E., Smolyar A., Vinayagam A., Yu H., Szeto D., Borick H., Dricot A., Klitgord N., Murray R. R., Lin C., Lalowski M., Timm J., Rau K., Boone C., Braun P., Cusick M. E., Roth F. P., Hill D. E., Tavernier J., Wanker E. E., Barabási A.-L., Vidal M., An empirical framework for binary interactome mapping. Nat. Methods 6, 83–90 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morelli X., Bourgeas R., Roche P., Chemical and structural lessons from recent successes in protein–protein interaction inhibition (2P2I). Curr. Opin. Chem. Biol. 15, 475–481 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Basse M.-J., Betzi S., Morelli X., Roche P., 2P2Idb v2: Update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database 2016, baw007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Labbé C. M., Kuenemann M. A., Zarzycka B., Vriend G., Nicolaes G. A. F., Lagorce D., Miteva M. A., Villoutreix B. O., Sperandio O., iPPI-DB: An online database of modulators of protein-protein interactions. Nucleic Acids Res. 44, D542–D547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higueruelo A. P., Jubb H., Blundell T. L., TIMBAL v2: Update of a database holding small molecules modulating protein–protein interactions. Database 2013, bat039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells J. A., McClendon C. L., Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Fuller J. C., Burgoyne N. J., Jackson R. M., Predicting druggable binding sites at the protein–protein interface. Drug Discov. Today 14, 155–161 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Arkin M. R., Tang Y., Wells J. A., Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 21, 1102–1114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makley L. N., Gestwicki J. E., Expanding the number of ’druggable’ targets: Non-enzymes and protein-protein interactions. Chem. Biol. Drug Des. 81, 22–32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Modell A. E., Blosser S. L., Arora P. S., Systematic targeting of protein–protein interactions. Trends Pharmacol. Sci. 37, 702–713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott D. E., Bayly A. R., Abell C., Skidmore J., Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 15, 533–550 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Silvian L., Enyedy I., Kumaravel G., Inhibitors of protein–protein interactions: New methodologies to tackle this challenge. Drug Discov. Today Technol. 10, e509–e515 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Valkov E., Sharpe T., Marsh M., Greive S., Hyvönen M., Targeting protein-protein interactions and fragment-based drug discovery. Top. Curr. Chem. 317, 145–179 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Jubb H., Blundell T. L., Ascher D. B., Flexibility and small pockets at protein–protein interfaces: New insights into druggability. Prog. Biophys. Mol. Biol. 119, 2–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stank A., Kokh D. B., Fuller J. C., Wade R. C., Protein binding pocket dynamics. Acc. Chem. Res. 49, 809–815 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Ozlem U., Eyrisch S., Volkhard H., Druggability of dynamic protein-protein interfaces. Curr. Pharm. Des. 18, 4599–4606 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Ostrem J. M. L., Shokat K. M., Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 15, 771–785 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Bowman G. R., Geissler P. L., Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. U.S.A. 109, 11681–11686 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cimermancic P., Weinkam P., Rettenmaier T. J., Bichmann L., Keedy D. A., Woldeyes R. A., Schneidman-Duhovny D., Demerdash O. N., Mitchell J. C., Wells J. A., Fraser J. S., Sali A., CryptoSite: Expanding the druggable proteome by characterization and prediction of cryptic binding sites. J. Mol. Biol. 428, 709–719 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kline M. P., Rajkumar S. V., Timm M. M., Kimlinger T. K., Haug J. L., Lust J. A., Greipp P. R., Kumar S., ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia 21, 1549–1560 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Ostrem J. M., Peters U., Sos M. L., Wells J. A., Shokat K. M., K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bowman G. R., Bolin E. R., Hart K. M., Maguire B. C., Marqusee S., Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. U.S.A. 112, 2734–2739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butterfoss G. L., DeRose E. F., Gabel S. A., Perera L., Krahn J. M., Mueller G. A., Zheng X., London R. E., Conformational dependence of 13C shielding and coupling constants for methionine methyl groups. J. Biomol. NMR 48, 31–47 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen D. F., Neudecker P., Vallurupalli P., Mulder F. A. A., Kay L. E., Determination of Leu side-chain conformations in excited protein states by NMR relaxation dispersion. J. Am. Chem. Soc. 132, 42–43 (2009). [DOI] [PubMed] [Google Scholar]
- 26.London R. E., Wingad B. D., Mueller G. A., Dependence of amino acid side chain 13C shifts on dihedral angle: Application to conformational analysis. J. Am. Chem. Soc. 130, 11097–11105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen D. F., Neudecker P., Kay L. E., Determination of isoleucine side-chain conformations in ground and excited states of proteins from chemical shifts. J. Am. Chem. Soc. 132, 7589–7591 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Mizukoshi Y., Takahashi H., Shimada I., Rapid preparation of stable isotope labeled peptides that bind to target proteins by a phage library system. J. Biomol. NMR 34, 23–30 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Liu X., Beugelsdijk A., Chen J., Dynamics of the BH3-only protein binding interface of Bcl-xL. Biophys. J. 109, 1049–1057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeuchi K., Tokunaga Y., Imai M., Takahashi H., Shimada I., Dynamic multidrug recognition by multidrug transcriptional repressor LmrR. Sci. Rep. 4, 6922 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lisi G. P., Loria J. P., Solution NMR spectroscopy for the study of enzyme allostery. Chem. Rev. 116, 6323–6369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hajduk P. J., Huth J. R., Fesik S. W., Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Shimada I., Ueda T., Matsumoto M., Sakakura M., Osawa M., Takeuchi K., Nishida N., Takahashi H., Cross-saturation and transferred cross-saturation experiments. Prog. Nucl. Magn. Reson. Spectrosc. 54, 123–140 (2009). [Google Scholar]
- 34.Post C. B., Exchange-transferred NOE spectroscopy and bound ligand structure determination. Curr. Opin. Struct. Biol. 13, 581–588 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Takeuchi K., Wagner G., NMR studies of protein interactions. Curr. Opin. Struct. Biol. 16, 109–117 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Lee F. E., Fairlie W. D., The structural biology of Bcl-xL. Int. J. Mol. Sci. 20, 2234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng W., Huang S., Wu H., Zhang M., Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 372, 223–235 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Sattler M., Liang H., Nettesheim D., Meadows R. P., Harlan J. E., Eberstadt M., Yoon H. S., Shuker S. B., Chang B. S., Minn A. J., Thompson C. B., Fesik S. W., Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 275, 983–986 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Gans P., Hamelin O., Sounier R., Ayala I., Durá M. A., Amero C. D., Noirclerc-Savoye M., Franzetti B., Plevin M. J., Boisbouvier J., Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high-molecular-weight proteins. Angew. Chem. Int. Ed. Engl. 49, 1958–1962 (2010). [DOI] [PubMed] [Google Scholar]
- 40.T. D. Goddard, D. G. Kneller, Sparky 3 (University of California, San Francisco, 2002). [Google Scholar]
- 41.Skrynnikov N. R., Mulder F. A., Hon B., Dahlquist F. W., Kay L. E., Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: Application to methionine residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 123, 4556–4566 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Bieri M., Gooley P. R., Automated NMR relaxation dispersion data analysis using NESSY. BMC Bioinformatics 12, 421 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabd0480/DC1