

Cryo-EM structure of a peptide toxin exporter reveals molecular insights into Staphylococcus aureus pathogenicity and persistence.
Abstract
Staphylococcus aureus is a major human pathogen that has acquired alarming broad-spectrum antibiotic resistance. One group of secreted toxins with key roles during infection is the phenol-soluble modulins (PSMs). PSMs are amphipathic, membrane-destructive cytolytic peptides that are exported to the host-cell environment by a designated adenosine 5′-triphosphate (ATP)–binding cassette (ABC) transporter, the PSM transporter (PmtABCD). Here, we demonstrate that the minimal Pmt unit necessary for PSM export is PmtCD and provide its first atomic characterization by single-particle cryo-EM and x-ray crystallography. We have captured the transporter in the ATP-bound state at near atomic resolution, revealing a type II ABC exporter fold, with an additional cytosolic domain. Comparison to a lower-resolution nucleotide-free map displaying an “open” conformation and putative hydrophobic inner chamber of a size able to accommodate the binding of two PSM peptides provides mechanistic insight and sets the foundation for therapeutic design.
INTRODUCTION
Antibiotic resistance is a major threat to prevention and treatment of infectious disease. A major human pathogen that has acquired alarming broad-spectrum resistance to many of the commonly used antibiotics, including β-lactams, is Staphylococcus aureus. The Gram-positive S. aureus causes hospital- and community-associated infections responsible for significant morbidity and death (1). Other staphylococcal species, including Staphylococcus epidermidis, Staphylococcus lugdunensis, and Staphylococcus haemolyticus, are also opportunistic pathogens that often cause disease in predisposed patients (2, 3). New therapeutics targeting crucial virulence determinants of these notorious pathogens hold promise for alternative treatment (4). Staphylococcal infections are mediated by a large array of secreted toxins including the phenol-soluble modulins (PSMs) (2, 5). PSMs are amphipathic, α-helical peptides, with pronounced surfactant-like, membrane-destructive properties that have multiple key roles in pathogenesis, including cytolysis of red and white blood cells, abscess formation, biofilm development, and triggering of receptor-mediated inflammatory response (6–8). At high concentrations in vitro, the PSM peptides have been shown to form polymerized filaments (9), although nonpolymerizing PSM variants also exist and have been characterized (10). A specialized adenosine 5′-triphosphate (ATP)–binding cassette (ABC) transport system exports these PSMs to the extracellular environment and is essential for bacterial growth by providing immunity against self-expressed PSMs (11). Compellingly, recent work has also shown a direct correlation of PSM transport (Pmt) with increased resistance to host-produced antimicrobial peptides, the latter share features of amphipathicity, helicity, and membranolytic nature with the PSMs (12), suggesting a potential role of Pmt in their export and subversion of the innate immune system.
ABC transporters are diverse multicomponent systems that use energy derived from ATP hydrolysis to translocate cargo across membranes (13). The typical architecture of ABC transporters consists of a dyad of multispan transmembrane domains (TMDs) coupled to a dyad of nucleotide-binding domains (NBDs). These individual domains can be encoded as separate polypeptide chains or fused to each other in multiple combinations (13). On the basis of their TMD features, ABC exporters have been divided into type I and type II classes. Although both share a common fold composed of six transmembrane helices (TMH), the type I exporters display extended TMHs that protrude into the cytoplasm, while the type II exporters have a more compact fold with typically little or no cytoplasmic extensions. In terms of transport mechanism, the type I exporters undergo an interdomain TMH swap at the dimerization interface during the transport cycle, while the type II exporters remain as two distinctly folded TMDs (14–16). The staphylococcal Pmt system is transcribed from a single operon composed of five genes, with sequence analysis predicting that the corresponding protein products PmtA and PmtC contain the characteristic Walker signature motifs (13) as NBDs, PmtB and PmtD as TMDs, as well as the soluble transcription regulatory protein PmtR that senses intracellular PSMs (17). Sequence analysis with prior known ATP importers (18) and the few known characterized exporter structures (14, 16, 19, 20) showed little conservation, in the range of 8 to 10% sequence identity with the latter. On the basis of this extreme sequence variability, the diversity of transported cargo, and alongside the limited structural and mechanistic insights available for ABC exporters in general, analysis of the Pmt system is essential for molecular characterization of the transport machinery components involved and the as yet uncharacterized and unprecedented amphipathic peptide–specific transport function.
RESULTS AND DISCUSSION
Cellular and in vitro biochemical characterization of the minimal transporter unit
We elucidated the transporter’s minimal composition and domain arrangement through expression of pairwise Pmt NBD and TMD gene combinations in an S. aureus cell–based PSM secretion assay, which demonstrated that PmtAB and PmtCD are two distinctive homodimeric complexes (fig. S1, A and B). However, PmtCD is the minimal unit necessary and sufficient for restoration of PSMα secretion (Fig. 1A). The observation that the PmtCD dyad is a functional stand-alone ABC exporter was further supported by the PmtC “Walker B” ATPase motif (E145Q) cellular point mutant that failed to secrete PSMs (Fig. 1A). Within the staphylococcus genus, pmtA and pmtC co-occur with pmtB and pmtD, respectively, but only pmtC and pmtD are conserved throughout (fig. S1C) (21). After extensive trials in various Escherichia coli recombinant expression systems, the PmtCD transporter was subsequently expressed and purified to homogeneity from the native membrane environment of our engineered S. aureus cells as a stable C2D2 heterotetramer. Functional ATPase assays confirmed that purified PmtCD in liposomes was able to hydrolyze ATP. In addition, microscale thermophoresis (MST) experiments demonstrated that purified PmtCD can bind at least two copies of the potent peptide toxin PSMα3F3A cooperatively (Fig. 1, B and C). The mutated PSMα3F3A peptide was chosen for functional studies as it maintains cytotoxicity and a relevant inflammatory response but does not spontaneously polymerize into large fibrils in vitro as with other PSMs (9, 10). Reduced rates of ATP hydrolysis were observed upon the addition of PSMα3F3A cargo; however (Fig. 1B), we believe the result of the still inherent “membrane” disruptive activity of the amphipathic PSMα3F3A peptide cargo with the liposome bilayers, detergent micelles, or membrane-mimetic peptidiscs used in the ATPase analysis. We note that similar unexplained behavior has been previously reported for ATP activity assays of the peptidase-containing ABC exporter PCAT1 (22).
Fig. 1. Pmt exporter functional characterization and near-atomic cryogenic electron microscopy (cryo-EM) atomic structure of the adenosine 5′-[γ-thio]triphosphate (ATPγS) bound state.
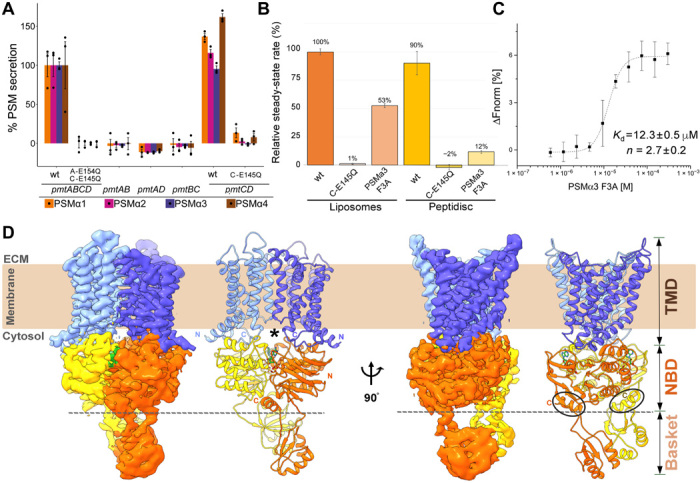
(A) Cell-based PSMα1–4 secretion measured by high-performance liquid chromatography/mass spectrometry (HPLC/MS) of culture supernatants from S. aureus cells encoding pairwise combinations of Pmt NBD and TMD genes. Expression of PSMα1–4 was induced by a xylose-inducible promoter. NBD Walker B motif mutants are indicated as A-E154Q and C-E145Q for PmtA and PmtC, respectively. Secretion of each PSM is normalized to cells encoding all four Pmt ABCD transporter genes. (B) Normalized ATPase activities of purified wild-type (wt) PmtCD, the Walker B inactive mutant, and in the presence of the PSMαa3F3A peptide cargo in both proteoliposomes and the peptidisc membrane mimetic system. Both the peptidisc monomer and the PSMα3 cargo peptide share a similar amphipathic helical nature (fig. S7C), and therefore, it is perhaps not unexpected that we observed loss of integrity of the peptidisc solubilized PmtCD upon addition of free PSMα3 toxin. (C) PmtCD can bind PSMαa3F3A peptide cargo in a cooperative manner. MST measurements of PSMαa3F3A and Alexa Fluor–labeled PmtCD. Titrations are plotted against changes in the normalized fluorescence (ΔFnorm). The corresponding maximum velocity and Hill coefficient are listed. (D) The 4.0-Å cryo-EM C1 reconstruction and atomic model of PmtCD in the ATPγS bound state (EMDB-22211/PDB- 6XJI), colored by subunits in two views; PmtC in yellow/orange, PmtD in light blue/purple, and ATPγS in green; N and C termini are labeled. Dashed gray line distinguishes between the NBDs and the basket domain of the PmtC protomers. PmtC C-terminal “capping” helices that fold back to interact with NBD are marked by gray ellipses. A possible membrane access point located at the intersection of the NBDs and TMD dyads is marked by an asterisk.
Peptidisc reconstitution and PmtCD structure determination by cryogenic electron microscopy
The PmtCD exporter was reconstituted into various membrane mimetics for subsequent structural analysis, with the recently developed detergent-free peptidisc system (23) providing the best resolution in our hands (and with comparable rates of ATPase activity as shown in Fig. 1, B and C). Peptidisc-reconstituted PmtCD was subjected to single particle cryogenic electron microscopy (cryo-EM). Representative alternating transport state datasets, including a nucleotide-free apo-state and an adenosine 5′-[γ-thio]triphosphate (ATPγS) substrate analog inhibitor bound form, were collected and analyzed with the latter reconstructed to near-atomic resolution (fig. S2).
PmtCD ATPγS bound state at near-atomic resolution
Data processing resulted in an overall reconstruction of PmtCD in peptidisc to 4.3-Å resolution (fig. S2, A and B). Signal subtraction of the region of the map corresponding to the peptidisc membrane mimetic improved the resolution to 4.0 Å (fig. S2, B to D). The TMDs and NBDs in this C1 map with no symmetry applied are clearly resolved with higher local resolution (Fig. 1D and fig. S2D). In contrast, the small additional cytosol-facing domain just following the canonical NBDs, akin to the basket of a hot air balloon (residues 210 to 275), was resolved to lower resolution, most likely due to the inherent flexibility of its NBD linkage, but with secondary structure features still observed (Fig. 1D and fig. S2D). Unexpectedly, the typical twofold symmetry of the ABC transporter perpendicular to the membrane bilayer is disrupted at this cytosolic-most basket domain, which, as a dimeric pair, is consistently tilted ~25° to the plane of the membrane. Signal subtraction of this region followed by refinement imposing C2 symmetry on the remaining TMDs and NBDs, resulted in an improved reconstruction to 3.6-Å resolution, revealing clear density for side chains and substrate analog ATPγS bound in both active sites. This reconstruction was used for model building and refinement of the TMDs and NBDs.
High-resolution structural information of the basket—PmtC210–275 spanning the isolated domain—was obtained by x-ray crystallography. Purified PmtC210–275 was crystallized in two different crystal forms diffracting to 1.4- and 2.1-Å resolution, respectively, with distinct crystal packing but an identical dimerization interface that produced a dimeric form that fit readily to the cryo-EM reconstructed map (fig. S3).
The overall PmtCD structure was generated by rigid body refinement of the models for the TMDs and NBDs (traced from the C2 3.6-Å reconstruction) and basket domains (from the highest-resolution x-ray structure) into the overall C1 reconstruction at 4-Å resolution with the map allowing the connecting loops between the NBD and basket domains to be traced. Together, the ATP-bound state of the PmtCD exporter forms a heterotetramer of three distinct regions, two TMDs formed by two parallel PmtD protomers (six TMs from each), core NBDs formed by two parallel PmtC protomers (residues 1 to 209 and 276 to 292), and an additional cytosolic basket domain (residues 210 to 275) (Fig. 1D).
PmtD transmembrane domains
The S. aureus (Gram-positive) PmtCD adopts the ABC type II exporter fold, previously only observed in human variants and duodermic Gram-negative bacteria, presenting shorter TMH in keeping with the Gram-positive bilayer span and intracellular loops that allow the NBDs to be located in closer proximity to the membrane (Fig. 2A and fig. S4). As observed with other type II exporters, the TMD interface does not involve a domain swap between TMHs from the two protomers and is mainly supported by symmetrical interactions along opposing TMH5a with additional capping interactions between the extracellular-facing regions of opposing TMH5b,c and TMH1b (Fig. 2, A and B). In the ATP-bound state, captured using the hydrolysis-impaired substrate analog ATPγS, the TMDs display a closed conformation with no classical ABC exporter outward-facing cavity at the dimer interface, but rather two symmetrical deep surface grooves that can be accessed via the inner-leaflet and/or acyl core of the membrane bilayer (shown as dark gray volumes in Fig. 2B). These large, predominantly hydrophobic cavities with multiple aromatic side chains are formed by TMH1b and TMH2 from one protomer and TMH5a,b of the opposing protomer. The cavity dimensions, approximately ~28-Å vertical length by ~13-Å maximal diameter, readily accommodate the docking of an α-helical peptide with a sequence length of a typical PSMα3 toxin, suggesting a potential role of these cavities in the transport trajectory (Fig. 2B). Although spanning nearly across the membrane bilayer, the two cavities are capped within the extremities of the outer leaflet by their respective flexibly linked TMH5b,c, which, we speculate, could serve as an upper gate during the transport cycle (Fig. 2B). Point mutations of conserved structural residues at the TMD had only mild effects, a ~2- to 3-fold decrease, on PSMα1–4 secretion in vivo (Fig. 2C and fig. S5). In keeping with a hydrophobic interface’s reduced susceptibility to uncharged point mutations, we suggest that the mild deleterious effects observed with our generally conservative substitutions are related to the ability of the transporter to accommodate and export multiple variants of PSMα peptides (and potentially amphipathic antimicrobial peptides) of diverse sequence (four PSMαs reported so far in S. aureus with their sequence similarity ranging between 40 and 85%; see fig. S1D). Therefore, the importance of individual interfacial residue specificity and essentiality in the scope of the entire transporter is diluted by the need for substrate promiscuity.
Fig. 2. PmtD in the ATPγS bound state based on the PmtCD C2 reconstruction at 3.6-Å resolution (EMDB-22210/PDB-6XJH).
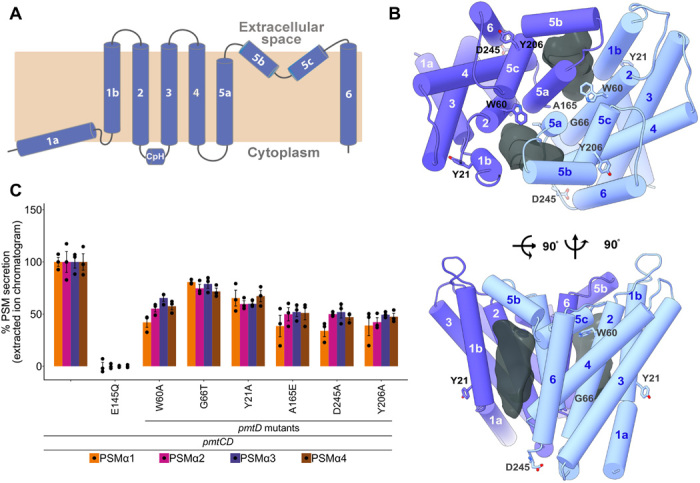
(A) Transmembrane topology of PmtD. PmtD is composed of six TMH with the cytosolic TMH2/3 loop forming the coupling helix (CpH). The large extracellular loop of PmtD is composed of two short gating helices, TMH5b-c. (B) Extracellular and side views of the TMD dimer. Conserved residues that were mutated appear in stick representation, and lateral, extended hydrophobic membrane cavities are delineated as dark gray volumes. (C) Cell-based PSMα1–4 secretion by PmtD mutants measured by HPLC/MS of culture supernatants from S. aureus cells encoding PmtCD. Expression of PSMα1–4 was induced by a xylose-inducible promoter. Secretion of each PSM is normalized to cells encoding the wild-type PmtCD sequence.
PmtC nucleotide-binding domains
As generally observed with other ABC exporters, a conserved interaction interface between individual NBDs and TMDs likely mediates conformationally driven ATP-dependent translocation across the membrane (14, 16, 19). For PmtCD, this allosteric network involves the coupling helix (CpH) located in the loop between TMH2 and TMH3 as well as the TMH1a connecting helix (CnH), both packed against the short α2 helix in the NBD (Fig. 3, A and B). Two bound ATPγS substrate analogs can be clearly identified in the reconstructed maps (Fig. 3C and fig. S2G). Also observed, albeit weakly, are the requisite magnesium ions that are readily modeled in both of the symmetrical binding sites of the C2 reconstructed density map and are in keeping with typical binding ligands and distances observed in previously characterized ABC transport systems (Fig. 3C and fig. S2G). Each NBD adopts a RecA-like fold that mediates the binding of each ATPγS substrate analog by the highly conserved residues of the Walker A motif (“33GKNGVGK39”) of one NBD and an unusual modification of the typical ABC signature sequence deriving from the opposing NBD (Fig. 3C). Notably, PmtC does not have the ABC signature sequence (“LSGGQ”) shown in previous structures to be involved in nucleotide binding (24), but rather a “120YSMGM124” sequence, conserved in and thus far unique to PmtC and PmtA (fig. S6). In our structure, the common serine (S121) of the signature motif acts as a direct ligand for the α-phosphate of bound ATPγS, and the common glycine (G123) provides φ/ϕ values of ~92° and −5° to introduce the needed sharp main chain turn to pack around the substrate nucleotide, similar to previous structures. The remaining unique residues of the “120YSMGM124” motif, Y120 and dual methionines, stabilize the signature motif itself (via aromatic stacking with the side chain of Y112) and provide additional intimate hydrophobic packing directly around the ATP analog (Fig. 3C). Clustering of methionine side chains has been observed at pivot points of conformational change in EF-hand calcium binding proteins such as calmodulin, with the possibility of regulation by oxidation (25). Additional conserved motifs located at the binding sites in PmtC are associated with nucleotide stabilization and catalysis; these include the aromatic residue (Y10) of the A-loop that stacks against the adenine ring, the catalytic glutamate (E145) of the Walker B motif, and the histidine switch (H178) stabilizing γ-phosphate (or thiotriphospate in our case) and magnesium binding. The overall NBD dimer interface reveals a closed “head-to-tail” conformation with a visible cavity/possible inner leaflet access point at the intersection of the NBD and TMD dyads (marked by an asterisk in Fig. 1D) that resembles most closely an analogous opening proposed for the human multidrug exporter ABCG2 (19).
Fig. 3. PmtC and interaction interfaces of the ATPγS bound state based on the PmtCD C2 reconstruction at 3.6-Å resolution (EMDB-22210/PDB-6XJH) and basket domain crystal structure (PDB-6XFU).
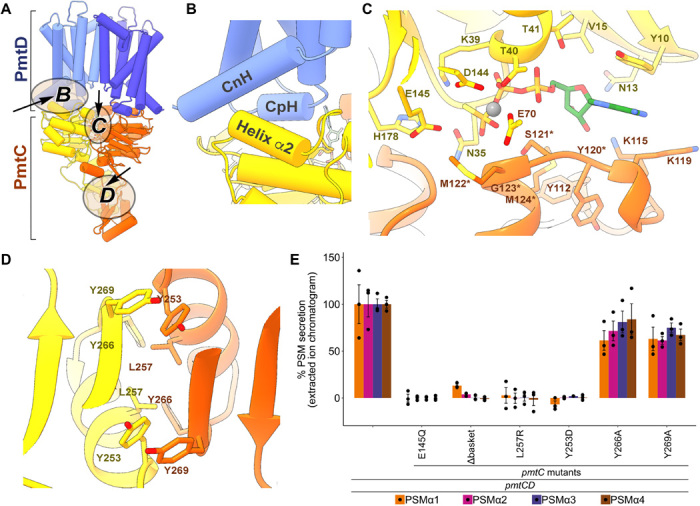
(A) Overall architecture of the PmtCD heterotetramer; ellipsoids highlight structural features presented in respective panels. (B) A conserved interface typical of ABC type II exporters, located between individual NBD and TMD pairs, is likely to allosterically mediate ATP-dependent translocation across the membrane. This allosteric network involves the CpH and CnH of PmtD, both packed against the short α2 helix of PmtC. (C) ATP-binding interface formed by the NBD dimer. PmtC monomers are in yellow and orange, and ATPγS is in stick representation with a magnesium ion in a gray sphere. ATP binding is mediated by conserved residues of the Walker A and B motifs from one monomer and the conserved and unique “YSMGM” sequence from the adjacent monomer. Residues of the “YSMGM” sequence are labeled with an asterisk. (D) Basket domain dimerization interface and central side chains therein are shown in stick representation. (E) Cell-based PSMα1–4 secretion by PmtC mutants measured by HPLC/MS of culture supernatants from S. aureus cells encoding PmtCD. Expression of PSMα1–4 was induced by a xylose-inducible promoter. Secretion of each PSM is normalized to cells encoding the wild-type PmtCD sequence. Δbasket: PmtC basket domain residues 209 to 275 were replaced with a “GSG” linker to the C-terminal α helix, which interacts with the NBD.
PmtC cytosolic basket domain
The basket-forming cytosolic-most domains are encoded within the PmtC polypeptide chain between the NBD and the C-terminal helix, the latter is redirected away from the basket domain to pack against the core NBD (Fig. 1D). Unexpectedly, the basket-forming domains adopt the canonical βαββαβ fold of an ACT (aspartate kinase, chorismate mutase, and the prephenate dehydrogenase tryA) domain, found in a variety of metabolic enzymes (26) and suggested to play a key regulatory role associated with amino acid concentration (fig. S3). Typically, a pair of ACT domains forms an eight-stranded antiparallel β sheet and can bind two amino acid ligands near their dimer interface. In PmtC, the essential basket region contributes ~40% of the total dimerization interface between the two monomers, with each ACT domain oriented perpendicularly to the other, leading to the formation of two “crossed” four-stranded β sheets (fig. S3). Although, overall, the basket domain is the least conserved region of PmtC, the residues located at the dimerization interface are highly conserved and essential for translocation activity (Fig. 3, D and E, and fig. S6). Amino acid sequence analysis of PmtA also suggests the existence of an ACT domain with a similar conservation pattern at the predicted dimerization interface. By comparison, based on sequence and existing structural information, few other ABC type II exporters use C-terminal extensions following their core NBDs to increase the dimerization interface (fig. S4A). The only known examples include the human multidrug ABCG2 and sterol ABCG5/G8 transporters, which share a conserved NBD “NPXDF” dimerization motif and the LptB2FG lipopolysaccharide (LPS) transporter, which incorporates an extra C-terminal helix (fig. S4A) (16, 19, 27). In addition, the human lipid exporter ABCA1 NBD contains the “R-domain” of unknown function that displays secondary structural elements resembling the ACT fold (primary sequence and connecting loops were not assigned in that structure because of insufficient resolution) (fig. S4A) (14). Notably, an overall similar arrangement of motifs along the polypeptide chains can be seen for both PmtC and ABCA1 and include the NBD and ACT domain followed by a C-terminal helix that is packed against the core NBD. Although both the R-domain and the basket share the ACT fold, they differ in their dimerization interface with a typical eight-stranded planar β sheet seen in the R-domain compared with the perpendicular ACT twist seen in the PmtCD basket (fig. S4A). It has been suggested that increasing the size of the dimerization interface supports the NBDs dimeric conformation even in the absence of bound nucleotide (14). Furthermore, in addition to the common TMD and NBD fold reported for ABC type II exporters and importers, a notable similarity is observed between the PmtCD basket domain and the regulatory “C2” domains of the MetNI methionine importer (fig. S4A). In the latter, the MetN protein contains the NBD, with a C-terminal ACT-folded C2 domain able to change its conformation in response to methionine binding (28). Previous studies have demonstrated that during a transport cycle, the C2 dimer can rotate ~30° around the importer twofold axis, leading to a register shift in the hydrogen-bonding network between the β sheets comprising the dimerization interface (fig. S4, B and C) (29). C2-domain conformational changes are translated into substantial changes in the distance between the NBDs and subsequent amplified changes in the TMDs. By analogy and based on similarities in the dimerization interface, we hypothesize that the essential ACT-domains could use similar conformational changes to support the transporter shift between nucleotide-bound and nucleotide-free conformational states. Furthermore, we suggest the basket could potentially have a regulatory role during PSM peptide export, although the nature of the regulator remains to be identified. Together, our structures significantly further illuminate our understanding that both ABC importers as well as exporters, as exemplified here in the first characterized Gram-positive bacterial variant, display amazing domain plasticity that can be modulated or exchanged to support their designated function across different subgroups.
The ACT-domain comprising the basket also exhibits a fold similarity to ribosome-binding proteins such as the flexible domain III of the Shwachman-Bodian-Diamond syndrome protein (30). Domain III has a ferredoxin-like fold found to be associated with many RNA binding proteins pointing toward a possible role of the Pmt basket domain as an analogous interaction point and perhaps hinting at a mechanism of cotranslational transport of the potentially self-harming cytotoxic PSMs from bacterial ribosomes directly to the PmtCD transporter.
Visualization of peptidisc membrane mimetic system
Peptidisc membrane mimetics (22) involve multiple noncovalently associated copies of a short amphipathic peptide designed to pack around the hydrophobic membrane-exposed region of a target membrane protein (fig. S7). Earlier fixed-diameter membrane mimetics such as nanodiscs (31) that require lipid incorporation to fill the central lumen of the disk moiety can result in heterogeneous alignment of the disk relative to a smaller protein swimming within and thus a low-resolution, fuzzy disk appearance in cryo-EM reconstructed maps. However, the noncovalent association and, thus, customizable and intimately associated peptidisc subunits (literally acting as molecular Spanx in our structures) have been clearly resolved in our maps, allowing placement of the helical secondary structures and highlighting their customized, intimate association with the TMDs as per their design. Specifically, our three-dimensional (3D)–refined unsubtracted C1 PmtCD ATP analog map revealed defined tubular densities assembled into a curved, continuous disk into which we were able to readily dock 14 copies of the 37–amino acid peptide used (fig. S7). Although the resolution of the peptidisc region itself is not sufficient to unambiguously model side-chain positions, the spacing of the units to that of the transporter TMDs (range of ~3 to 6 Å) suggests a direct intermolecular packing. Notably, the peptidisc does not necessarily displace endogenously bound ordered lipids with an example in keeping with a diacyl phospholipid (or possibly alternate conformation of a monoacyl lipid) bound within a hydrophobic cleft on the TMH surface (fig. S8).
Low-resolution reconstruction of nucleotide-free state
To better understand the ATP-driven conformational changes associated with PSM peptide transport, we sought to compare the PmtCD nucleotide-free and ATPγS bound reconstructions. The nucleotide-free state reconstruction generated with C1 symmetry (fig. S2), although at lower ~8-Å overall resolution, unambiguously showed major generalized differences from that of the ATPγS bound state (Fig. 4). Substantial TMD displacement, with each of the TMHs accounted for and clearly demarcated in our nucleotide-free maps as individual cylinders (Fig. 4, A and B), results in the formation of a large, inner lumen between the two TMD protomers (~10 Å at its most constricted point, ~20 Å at its widest) (Fig. 4). We also observe a concomitant increase of ~25 Å in the peptidisc diameter in the nucleotide-free state to accommodate the observed TMD expansion (Fig. 4E). Collectively, these data support a major rearrangement to presumably accommodate the binding of the relatively large cargo peptides in the nucleotide-free state.
Fig. 4. ATPγS induced conformational changes.
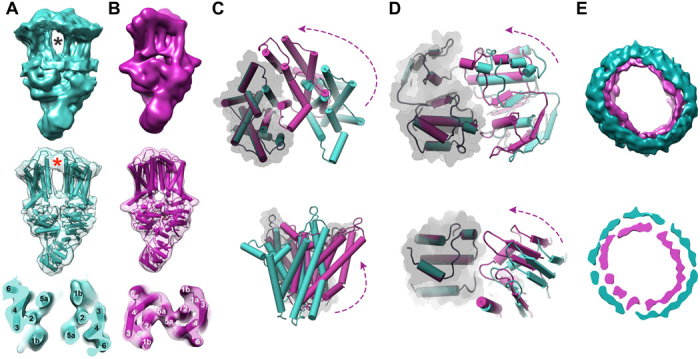
(A) Nucleotide-free state ~8-Å reconstructed cryo-EM map after peptidisc subtraction and the corresponding docked rigid body atomic model in teal (EMDB-20639). Side and top (slabbed, middle) TMD views are presented. Gray asterisk represents the large inner lumen between the two TMDs. Red asterisk represents additional density not related to the predicted atomic model (we suggest this may represent diffuse lipid). (B) ATPγS bound state 8-Å low-pass-filtered reconstructed cryo-EM map (presented at lower resolution to allow clearer comparison with the nucleotide-free state) after peptidisc subtraction and corresponding atomic model in pink. Side and top (slabbed) TMD views are presented. (C) Extracellular and side views of overlaid TMDs of the rigid-body docked nucleotide-free model and the near-atomic-resolution ATPγS bound states. Dimer overlay was performed using a fixed reference of a single TMD and presented in transparent gray surface. (D) Cytosol-facing and side views of the NBDs of overlaid TMDs of the rigid-body docked nucleotide-free model and experimentally determined ATPγS analog bound states. Dimer overlay was performed using a fixed reference of a single NBD and presented in transparent gray surface. (E) Extracellular top and slabbed viewpoints of overlaid peptidiscs of the ATPγS bound and nucleotide-free states in pink and teal, respectively.
Low-resolution rigid-body modeling of PmtCD in the nucleotide-free state
Recent studies have described alterations between transport states of ABCG2 and MetNI exporters as a rigid-body movement of the NBDs and TMDs (19, 29). Keeping in mind the limitations of resolution, we performed rigid-body docking for each chain onto that of the nucleotide-free state map. The resulting nucleotide-free model readily fit to the observed densities of both NBDs and TMDs when each domain, including the cytosolic basket (either as the cryo-EM or crystallographically observed dimer), was treated independently (Fig. 4, A and B). The more open conformation observed for the nucleotide-free model TMD’s placement underscores the resulting hydrophobic lumen and inner volume that is in keeping with the binding of multiple amphipathic cargo peptides simultaneously (Fig. 4A).
Proposed hypothetical Pmt virulence peptide cargo transport mechanism in methicillin-resistant S. aureus
Recent structural analysis of type II exporters with large lipidated cargo such as that for the LptB2FG LPS transporter above and the ABCA1 lipid exporter has hypothesized a lateral access mechanism through the membrane inner leaflet for initiation of substrate passage by the transporter (14, 32, 33). As such, a similar mechanism is also compelling for PSMα peptide secretion here given the amphipathic and membrane-penetrating nature of the substrate as well as the observed opening to the bilayer in the nucleotide-free state. Together with the dimensions and alternate conformations observed for the PmtCD exporter, and in keeping with the observed cooperative mode of cargo binding (Fig. 1C), we suggest a model that involves the accommodation of at least two toxin peptides within the hydrophobic TMD lumen in the nucleotide-free state simultaneously, with their hydrophilic faces packed favorably toward each other and their hydrophobic surfaces oriented to the surrounding hydrophobic lumen. The polar outer surface and dimensions of the earlier reported filamentous form of the isolated peptide created at high concentrations (9) (cross-sectional diameter of ~29 Å) would not fit into the observed cavity/channel in our structures due to substantial size and electrostatic incompatibility.
To prevent the formation of a constantly “open” and potentially deleterious channel in the absence of cargo, we speculate that membrane lipid molecules may diffuse in to block this open passage, similar to the lipid plug mechanism suggested for the LPS O-antigen transporter LptB2FG but at the larger scale needed for the peptide cargo here (Fig. 5) (32). Binding of substrate cargo peptides would presumably displace the lipid plug, but further biochemical analysis will be required to test this possibility. Upon ATP binding, an induced conformational change would move the TMDs to the observed tight conformation that expels the two peptides to the outer leaflet, separately, potentially involving a trajectory via the aforementioned extended hydrophobic grooves located on both outer surfaces of the TMD dimer and exclusive to the high-resolution ATP analog structure (Figs. 2B and 5). Last, similar to other ABC exporters, ATP hydrolysis and ADP + Pi release would result in a conformational change that restarts the cycle (34). In our suggested model, the essential and unique cytosolic basket domain, in absence of the anchoring domain swap found in type I exporters, could potentially serve as a regulator and/or also maintain a rigid limit to the extent of the (non–domain-swapped) TMDs’ drift from each other in the cargo-loaded state.
Fig. 5. Model of PSM peptide translocation by the PmtCD ABC type II exporter.
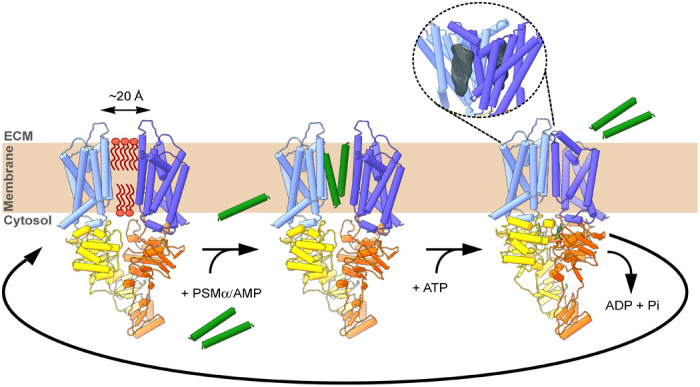
In the resting state, with no nucleotide bound, the TMDs from opposing monomers are separated by up to ~20 Å with a hypothesized internal lipid plug acting to keep the system closed. PSMα helical peptide(s), which are membrane penetrating, could access the inner lumen from the bilayer directly or via a currently unknown facilitated transfer from the cytoplasm. Typical interfacial binding of ATP at the NBD dimer requires their close proximity and results in an inward motion of the cognate TMD pair, as verified by our near-atomic-resolution ATPγS bound reconstruction. This ATP-driven closure would expel the peptides from the central lumen to the extracellular space, potentially via a trajectory involving the two TMD hydrophobic surface cavities observed in the near-atomic-resolution ATPγS state (dark gray volumes in the zoomed-in oval panel compatible with the binding of a single PSM peptide) and potentially driven by the need for solvation of the now exposed polar surfaces of the bound peptides bound therein. ATP hydrolysis and ADP product release provide the energy to allow a conformational change back to the resting state.
Future work will help to distinguish between the compelling lateral access model, analogous with that hypothesized for the lipidated O-antigen polysaccharide transporter and other large, membrane-penetrating cargo type II exporters, and alternative mechanisms. One possibility includes direct uptake of potentially unfolded/inactive PSM cargo from the cytoplasm coupled with unknown cotranslational partner(s) and possibly mediated in some manner by the unusual basket domain and conserved cavity localization at the inner leaflet intersection of TMD and NBD dyads observed here. A combination of these two mechanisms is also possible and lends to exciting future questions in this important methicillin-resistant S. aureus virulence mechanism.
MATERIALS AND METHODS
Construction of expression constructs and mutagenesis
To create the general-use staphylococcal expression plasmid pTX17, we deleted the geh gene and simultaneously introduced a multiple cloning site (MCS) with an embedded ribosome-binding site (RBS) downstream of the xylA promoter by inverse polymerase chain reaction (PCR) of pTX15 (35). An Nde I site was designed to place a start codon in register with the RBS. Extraneous Eco RV and Nde I sites outside the MCS were subsequently removed by inverse PCR. PmtCD was amplified from pRB473 pmtABCD (11) with primers designed to introduce an Nde I site overlapping with the PmtC start codon and an Not I site immediately downstream of the PmtD stop codon. The PCR product was digested with Nde I and Not I-HF (NEB) and subcloned into respective sites of pTX17. A fragment encoding the His10 affinity tag and thrombin cleavage site was introduced after the PmtD start codon by inverse PCR to create the expression construct pTX17-pmtC, His-D.
Pairwise NBD and TMD constructs for the cell-based PSM secretion assay were engineered by inverse PCR of pRB473 pmtABCD. Each missense mutation and the deletion of the PmtC basket domain (residues 209 to 274) with simultaneous insertion of a “GSG” short linker to the C-terminal helix were constructed by inverse PCR of pRB473-pmtCD.
The truncated pmtC gene (residues 210 to 275) was amplified by PCR from the pTX17-PmtCD plasmid. The primers were designed to introduce an Nde I site at the initiation codon, ATG, and a Bam HI site after the termination codon. The DNA fragments were digested with Nde I and Bam HI (NEB) and cloned into the respective sites of plasmid pET28a (+) to construct the p28-basket plasmid, in which the pmtC truncated gene was fused in-frame to express a 6-His tag at the N terminus of the protein followed by thrombin proteolysis site. All PCR products were amplified with either Phusion (NEB) or Q5 polymerase (NEB), gel purified, treated with T4 PNK (NEB) and T4 DNA ligase (NEB), and transformed by chemical transformation into E. coli DC10B or electroporation into S. aureus RN4220 (36). All constructs were verified by Sanger sequencing. Primers and plasmids used in this study are respectively listed in tables S3 and S4 of the Supplementary Materials.
Recombinant protein expression and purification
S. aureus RN4220 cells, harboring the pTX17-PmtCDhis expression plasmid, were cultivated in tryptic soy broth (TBS) without dextrose medium containing tetracycline (12.5 μg/ml) and induced by 0.5% d-xylose at 37°C for 20 hours. The cells were harvested by centrifugation at 7438g for 15 min at 4°C. The expressing cells were suspended in phosphate-buffered saline (PBS) and incubated with deoxyribonuclease (DNase) I (10 mg/ml) and lysostaphin (1 mg/ml) at 37°C for 20 min. The cells were then sonicated at 50% power for 8 min in ice water, and cell debris was removed by 15,000g centrifugation for 20 min at 4°C. Membranes were fractionated by high-speed centrifugation (200,000g, 45 min, 4°C) and homogenized in PBS buffer with 5% glycerol. Proteins were extracted using 1% n-dodecyl-β-d-maltoside (DDM) for 1 hour at 4°C followed by high-speed centrifugation (200,000g, 30 min, 4°C) to remove insoluble components. Solubilized protein complex was applied onto a gravity Ni-NTA column (Bio-Rad Econo-Column chromatography column, Thermo Scientific HisPur Ni-NTA resin) pre-equilibrated with buffer A [20 mM tris (pH 8.8), 200 mM NaCl, 5% glycerol, 25 mM imidazole, 0.018% DDM]. The protein complex was washed with buffer B [20 mM tris (pH 8.8), 200 mM NaCl, 5% glycerol, 50 mM imidazole, 0.018% DDM] and eluted with buffer C [20 mM tris (pH 8.8), 200 mM NaCl, 5% glycerol, 300 mM imidazole, 0.018% DDM]. The protein complex was then applied onto a size exclusion column (Superose Increase 6 10/30, GE Healthcare Biosciences) pre-equilibrated with buffer D [20 mM tris (pH 8.8), 200 mM NaCl, 5% glycerol, 0.009% DDM]. For on-column peptidisc peptide reconstitution, 10 mg of the peptidisc peptide (23) was solubilized in 50 ml of buffer E [20 mM tris (pH 7.4), 150 mM NaCl, 50 mM imidazole] and applied on the PmtCD bound Ni-NTA beads. The beads were washed with buffer E, and the peptidisc reconstituted protein complex was eluted with buffer F [20 mM tris (pH 7.4), 150 mM NaCl, 500 mM imidazole]. The protein complex was then applied onto a size exclusion column (Superose Increase 6 10/30, GE Healthcare Biosciences) pre-equilibrated with buffer G [20 mM tris (pH 7.4), 150 mM NaCl].
E. coli BL21 cells, harboring the p28 basket domain expression plasmid, were cultivated in LB medium containing kanamycin (50 μg/ml) and induced with 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG) at an A600 of 0.6 at 22°C for 20 hours. The cells were harvested by centrifugation at 7438g for 15 min at 4°C. The expressing cells were suspended in buffer A [60 mM tris (pH 8), 300 mM NaCl, 5% glycerol] incubated with DNase I (10 mg/ml) and protease inhibitor tablets (Roche) at 4°C. The cells were then disrupted by two cycles in a French press pressure cell at 172 MPa. Cell debris was removed by high-speed centrifugation (Beckman, 70Ti rotor, 45k rpm, 45 min, 4°C), and the soluble fraction was applied onto a gravity Ni-NTA column (Bio-Rad Econo-Column chromatography column, Thermo Scientific HisPur Ni-NTA resin) pre-equilibrated with buffer H. The protein was washed with 50 ml of each buffers I [20 mM tris (pH 8), 300 mM NaCl, 5% glycerol, 20 mM imidazole], J [20 mM tris (pH 8), 1 M NaCl, 5% glycerol, 40 mM imidazole], and K [20 mM tris (pH 8), 150 mM NaCl, 5% glycerol, 40 mM imidazole] and eluted with buffer L [20 mM tris (pH 8), 150 mM NaCl, 5% glycerol, 500 mM imidazole]. To remove the 6-His tag, thrombin was added to the eluted protein, and the mixture was dialyzed against buffer M [10 mM tris-HCl (pH 8) and 150 mM NaCl, 5% glycerol] for 16 hours at 4°C. The protein was then applied onto a size exclusion column (Superdex Increase 75 10/30, GE Healthcare Biosciences) pre-equilibrated with buffer F. Purified basket domain was then concentrated to ~10 mg/ml, flash frozen in liquid nitrogen, and stored at −80°C.
Crystallization and structure determination
Basket domain crystals were grown using the sitting drop vapor diffusion method using 2 μl of protein solution (10 mg/ml) for the native crystal form and 0.2 μl of protein solution (5 mg/ml) for the derivatized crystal form used for phasing, mixed with an equal volume of precipitant. Native basket domain was crystalized at 4°C in 0.2 M sodium thiocyanate and 20% polyethylene glycol (PEG) 3350 (pH 7.4) and was flash frozen following the addition of cryoprotectant solution (25% PEG 3350) in liquid nitrogen. Native data were collected at the Advanced Photon Source on Beamline 23ID-B equipped with Eiger 16M detector. Data collection was performed at 100 K, and a total of 1800 frames (360°) were collected with an oscillation range of 1° at 1.0332-Å wavelength. The exposure time was 0.2 s per image, and the crystal-to-detector distance was 250 mm. For phasing, crystal grown in 0.2 M ammonium sulfate, 0.1 M ammonium acetate (pH 4.6), and 28% PEG MME 550 at 25°C was soaked with 1 M NaI for 30 s and harvested after the addition of 50% PEG 400 solution as a cryoprotectant. Heavy atom diffraction data were collected using a home source using an image plate detector system (MAR 345 mm) (X-Ray Research) and MicroMax-007HF (Rigaku) generator. Data collection was performed at 100 K, and a total of 360 frames (360°) were collected with an oscillation range of 1°. The exposure time was 6 min per image, and the crystal-to-detector distance was 150 mm. Data were reduced and scaled using the HKL2000 suite (37). Phases were obtained by CRANK2 (38), and 17 iodine atoms were found in the substructure solution. The model was further built using Coot (39) against high-resolution native data and refined with Phenix refine (40). For the Rfree calculation, 5% of the data were excluded. Structural analysis and figures were prepared with Chimera (41). Data collection and refinement statistics are provided in table S2.
Cryo-EM and image processing
Three-microliter aliquots of the peptidisc (1 mg/ml) reconstituted PmtCD were applied to the glow-discharged 400 mesh R1.2/1.3 holey carbon grid (Quantifoil, Germany), blotted for 3 s with blotting force 3 at 4°C and 100% humidity in an FEI Mark IV Vitrobot (Thermo Fisher Scientific) and frozen in liquid ethane. The grids were imaged on an FEI Titan Krios at 300 kV equipped with a Gatan K2 direct detector operating in counting mode. Data were collected via SerialEM (42) at the defocus range of 1.5 to 3.0 μm. For each movie, the total exposure time was 10 s and 50 frames for the ATP state and 40 frames for nucleotide free. The pixel size in counting mode is 0.655 Å per pixel for the ATP state and 0.675 Å per pixel for the nucleotide-free state.
The movies of PmtCD in the ATP analog and nucleotide-free states were binned by 2, motion corrected, and dose weighted using MotionCor2 on Relion 3 (43). The contrast transfer function (CTF) was estimated with CTFFIND4 (44). On cisTEM (computational imaging system for transmission electron microscopy) (45), particles were picked in the box size of 256 × 256 pixels and exported to Cryosparc (46), where 2D classification, 3D classification, and ab initio model modeling with a set of two models were performed. The ab initio models were used as reference for heterogeneous and homogeneous refinement. Given the higher resolution, the particles of the ATP state were further exported to Relion 3 for 3D autorefinement, CTF refinement, Bayesian polishing, and peptidisc subtraction. The overall resolutions in all were estimated on the basis of gold standard Fourier shell correlation (FSC) = 0.143. Cryo-EM data collection information is provided in table S1.
Model building and refinement
For datasets, the density was of sufficient quality to permit manual tracing of most of the C-alpha backbone in Coot Model building, and refinement of the TMDs and NBDs from the PmtCD ATPγS dataset was carried out using the C2-averaged reconstruction to 3.6-Å resolution, which was of sufficient quality to permit manual tracing of most of the C-alpha backbone in Coot (39) and subsequent sequence assignment. Phenix real-space refinement (40) and Rosetta (47) refinement using the reconstruction as a restraint were carried out on the complete atomic model while maintaining strict NCS. For the basket domain, the highest-resolution crystal structure was rigid body refined into the C1 map along with the TMDs and NBDs and connecting loops traced. Molprobity (48) and EMRinger (49) were used to validate, with the final models having good stereochemistry. Structural analysis and figures were prepared with Chimera (41). Cryo-EM data collection information and model refinement are provided in table S1.
Binding assays
Peptide synthesis
PSMα3F3A (NMEAVAKLFKFFKDLLGKFLGNNC) peptide was synthesized by commercial vendor at ~95% purity with an N-terminal formyl group.
Microscale thermophoresis
MST assays were conducted using a Monolith NT.115Pico (NanoTemper). PmtCD was fluorescently labeled using Alexa Fluor 647 NHS Ester (Thermo Fisher Scientific). To evaluate the binding of nonfibrillating yet cytotoxic PSMα3F3A mutant (10) to PmtCD, increasing concentrations of unlabeled PSMα3F3A peptide (439 nM to 1.8 mM) were used to titrate fluorescently labeled PmtCD at a constant concentration (535 nM). Three independent experiment repeats were carried out in buffer D. Data were analyzed using the NanoTemper MO Affinity Analysis software. The data were analyzed using MST. The data were fit to the equation for a binding isotherm with an adjustable Hill coefficient.
Reconstitution in liposomes
PmtCD and mutants were reconstituted into liposomes composed of 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) and cardiolipin in 7:3 (w/w) ratio (Anatrace), respectively, similar to that of the reported S. aureus membrane composition (50) and prior protocol for ABC transporters (51). In brief, purified DDM-solubilized proteins were added to Triton X-100–destabilized liposomes in 1:8 (w/w) ratio and incubated for 30 min at 4°C. For detergent removal, Bio-Beads SM-2 adsorbents (Bio-Rad) were added in three consecutive steps. Proteoliposomes were collected a 270,000g and resuspended in 20 mM tris (pH 7.4) and 150 mM NaCl to final lipid concentration of 3 mg/ml.
EnzChek ATPase activity assays
Steady-state PmtCD ATPase activity was assayed using the EnzChek phosphate detection assay kit (Thermo Fisher Scientific). All assays were performed at 27°C at 0.2 mM ATP with 2.25 μM PmtCD in 20-μl reactions in 384-well plates (Corning, 3540), using a Synergy H4 Microplate Reader (BioTek Instruments). Absorption reads at 360 nm were taken at 30-s intervals, and a linear portion over a 7-min time span was used to calculate relative steady-state reaction rate (n = 4).
Cell-based PSM export assay
PSM secretion was monitored as previously described (11) with the following modifications. LAC 3xKO Δpmt cells (12) were cotransformed with pTX + PSMα1–4 and pRB473 encoding pmt genes as indicated. Overnight starter cultures were diluted to an OD600 (optical density at 600 nm) of 2.00 into 3 ml of tryptic soy broth (TSB) without glucose supplemented with tetracycline (12.5 μg/ml) and chloramphenicol (10 μg/ml). Cells were cultured shaking at 37°C for 2 hours, after which xylose was added to 0.5% and cultured for another hour. Cells were centrifuged at 16,000g for 5 min, and the supernatants were analyzed for PSMs by high-performance liquid chromatography/mass spectrometry as described previously (6). Three biological replicates were performed for each group. The secretion of each PSM was normalized to controls performed in parallel. The mean of the positive control (PmtABCD and PmtCD for Figs. 1A, 2C, and 3E, respectively) was set to 100%, and the mean of the negative control (PmtABCD A-E154Q C-E145Q and PmtCD C-E145Q, for Figs. 1A, 2C, and 3E, respectively) was set to 0%. Graphs were generated using RStudio, with individual points, bars, and error bars denoting biological replicates, means, and SEMs, respectively.
Coordinates
Structures and maps have been submitted to the Protein Data Bank (6XFU, 6U2D, 6XJH, and 6XJI) and to the Electron Microscopy Data Bank (EMDB-22194, EMDB-22210, EMDB-22211, and EMDB-20640).
Supplementary Material
Acknowledgments
We thank F. Rossell for advice on MST data collection and analysis. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. In particular, we thank the staff at Beamline 23ID-B for facilitating x-ray crystallographic data collection. Funding: This work was funded by a Banting fellowship to N.Z. from the Canadian Institutes of Health Research (CIHR) and operating grants from the CIHR to N.C.J.S., the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (project number ZIA AI000904 to M.O.) and the National Institute of General Medical Research (grant FI2GM11999101 to S.W.D.), U.S. NIH, and the Howard Hughes International Senior Scholar program to N.C.J.S. Author contributions: N.Z., S.W.D, M.O., and N.C.J.S. designed the research; N.Z., S.W.D., H.T.C., J.H., L.J.W., A.J.A.N, M.L.C., and M.N. performed the research; N.Z., S.W.D., J.H., L.J.W., H.T.C., F.D., M.O., Z.Y., and N.C.J.S. analyzed the data. N.Z. and N.C.J.S. wrote the paper with input from all. Competing interests: N.C.J.S. is a tier I Canada research chair in Antibiotic Discovery. The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabb8219/DC1
REFERENCES AND NOTES
- 1.Klevens R. M., Morrison M. A., Nadle J., Petit S., Gershman K., Ray S., Harrison L. H., Lynfield R., Dumyati G., Townes J. M., Craig A. S., Zell E. R., Fosheim G. E., McDougal L. K., Carey R. B., Fridkin S. K.; Active Bacterial Core surveillance (ABCs) MRSA Investigators , Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298, 1763–1771 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Cheung G. Y. C., Joo H.-S., Chatterjee S. S., Otto M., Phenol-soluble modulins – critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 38, 698–719 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otto M., Staphylococcus epidermidis—The ‘accidental’ pathogen. Nat. Rev. Microbiol. 7, 555–567 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickey S. W., Cheung G. Y. C., Otto M., Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 16, 457–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berube B. J., Wardenburg J. B., Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 5, 1140–1166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R., Braughton K. R., Kretschmer D., Bach T.-H. L., Queck S. Y., Li M., Kennedy A. D., Dorward D. W., Klebanoff S. J., Peschel A., DeLeo F. R., Otto M., Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 13, 1510–1514 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Wang R., Khan B. A., Cheung G. Y. C., Bach T.-H. L., Jameson-Lee M., Kong K.-F., Queck S. Y., Otto M., Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J. Clin. Invest. 121, 238–248 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peschel A., Otto M., Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 11, 667–673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tayeb-fligelman E., Tabachnikov O., Moshe A., Goldshmidt-tran O., Sawaya M. R., Coquelle N., Colletier J.-P., Landau M., The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science 355, 831–833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung G. Y. C., Kretschmer D., Queck S. Y., Joo H.-S., Wang R., Duong A. C., Nguyen T. H., Bach T.-H. L., Porter A. R., DeLeo F. R., Peschel A., Otto M., Insight into structure-function relationship in phenol-soluble modulins using an alanine screen of the phenol-soluble modulin (PSM) α3 peptide. FASEB J. 28, 153–161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee S. S., Joo H.-S., Duong A. C., Dieringer T. D., Tan V. Y., Song Y., Fischer E. R., Cheung G. Y. C., Li M., Otto M., Essential Staphylococcus aureus toxin export system. Nat. Med. 19, 364–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung G. Y. C., Fisher E. L., McCausland J. W., Choi J., Collins J. W. M., Dickey S. W., Otto M., Antimicrobial peptide resistance mechanism contributes to Staphylococcus aureus infection. J. Infect. Dis. 217, 1153–1159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higgins C., ABC transporters: From mircoorganisms to man. Annu. Rev. Cell Biol. 8, 67–113 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Qian H., Zhao X., Cao P., Lei J., Yan N., Gong X., Structure of the human lipid exporter ABCA1. Cell 169, 1228–1239.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Dawson R. J. P., Locher K. P., Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Lee J.-Y., Kinch L. N., Borek D. M., Wang J., Wang J., Urbatsch I. L., Xie X.-S., Grishin N. V., Cohen J. C., Otwinowski Z., Hobbs H. H., Rosenbaum D. M., Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature 533, 561–564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joo H.-S., Chatterjee S. S., Villaruz A. E., Dickey S. W., Tan V. Y., Chen Y., Sturdevant D. E., Ricklefs S. M., Otto M., Mechanism of gene regulation by a Staphylococcus aureus toxin. MBio 7, e01579–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rice A. J., Park A., Pinkett H. W., Diversity in ABC transporters: Type I, II and III importers. Crit. Rev. Biochem. Mol. Biol. 49, 426–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manolaridis I., Jackson S. M., Taylor N. M. I., Kowal J., Stahlberg H., Locher K. P., Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 563, 426–430 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bi Y., Mann E., Whitfield C., Zimmer J., Architecture of a channel-forming O-antigen polysaccharide ABC transporter. Nature 553, 361–365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghebremedhin B., Layer F., König W., König B., Genetic classification and distinguishing of Staphylococcus species based on different partial gap, 16S rRNA, hsp60, rpoB, sodA, and tuf gene sequences. J. Clin. Microbiol. 46, 1019–1025 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin D. Y.-w., Huang S., Chen J., Crystal structures of a polypeptide processing and secretion transporter. Nature 523, 425–430 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Carlson M. L., Young J. W., Zhao Z., Fabre L., Jun D., Li J. J., Li J. J., Dhupar H. S., Wason I., Mills A. T., Beatty J. T., Klassen J. S., Rouiller I., Duong F., The peptidisc, a simple method for stabilizing membrane proteins in detergent-free solution. eLife 7, e34085 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilkens S., Structure and mechanism of ABC transporters. F1000Prime Rep. 7, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy M. R., Thompson A. R., Nitu F., Moen R. J., Olenek M. J., Klein J. C., Thomas D. D., Impact of methionine oxidation on calmodulin structural dynamics. Biochem. Biophys. Res. Commun. 456, 567–572 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lang E. J. M., Cross P. J., Mittelstädt G., Jameson G. B., Parker E. J., Allosteric ACTion: The varied ACT domains regulating enzymes of amino-acid metabolism. Curr. Opin. Struct. Biol. 29, 102–111 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Hicks G., Jia Z., Structural basis for the lipopolysaccharide export activity of the bacterial lipopolysaccharide transport system. Int. J. Mol. Sci. 19, 2680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson E., Nguyen P. T., Yeates T. O., Rees D. C., Inward facing conformations of the MetNI methionine ABC transporter: Implications for the mechanism of transinhibition. Protein Sci. 21, 84–96 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen P. T., Lai J. Y., Lee A. T., Kaiser J. T., Rees D. C., Noncanonical role for the binding protein in substrate uptake by the MetNI methionine ATP Binding Cassette (ABC) transporter. Proc. Natl. Acad. Sci. U.S.A. 115, E10596–E10604 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shammas C., Menne T. F., Hilcenko C., Michell S. R., Goyenechea B., Boocock G. R. B., Durie P. R., Rommens J. M., Warren A. J., Structural and mutational analysis of the SBDS protein family: Insight into the leukemia-associated Shwachman-Diamond Syndrome. J. Biol. Chem. 280, 19221–19229 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Denisov I. G., Sligar S. G., Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 23, 481–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caffalette C. A., Corey R. A., Sansom M. S. P., Stansfeld P. J., Zimmer J., A lipid gating mechanism for the channel-forming O antigen ABC transporter. Nat. Commun. 10, 824 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y., Orlando B. J., Liao M., Structural basis of lipopolysaccharide extraction by the LptB2FGC complex. Nature 567, 486–490 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hofmann S., Januliene D., Mehdipour A. R., Thomas C., Stefan E., Brüchert S., Kuhn B. T., Geertsma E. R., Hummer G., Tampé R., Moeller A., Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571, 580–583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peschel A., Ottenwälder B., Götz F., Inducible production and cellular location of the epidermin biosynthetic enzyme EpiB using an improved staphylococcal expression system. FEMS Microbiol. Lett. 137, 279–284 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Kreiswirth B. N., Löfdahl S., Betley M. J., O’Reilly M., Schlievert P. M., Bergdoll M. S., Novick R. P., The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712 (1983). [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z., Minor W., Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Skubák P., Pannu N. S., Automatic protein structure solution from weak X-ray data. Nat. Commun. 4, 2777 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J. H., Lindahl E., Scheres S. H. W., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant T., Rohou A., Grigorieff N., CisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 47.DiMaio F., Tyka M. D., Baker M. L., Chiu W., Baker D., Refinement of protein structures into low-resolution density maps using Rosetta. J. Mol. Biol. 392, 181–190 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barad B. A., Echols N., Wang R. Y.-R., Cheng Y., DiMaio F., Adams P. D., Fraser J. S., EMRinger: Side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayami M., Okabe A., Kariyama R., Abe M., Kanemasa Y., Lipid composition of Staphylococcus aureus and its derived L-forms. Microbiol. Immunol. 23, 435–442 (1979). [DOI] [PubMed] [Google Scholar]
- 51.Geertsma E. R., Nik Mahmood N. A. B., Schuurman-Wolters G. K., Poolman B., Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc. 3, 256–266 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabb8219/DC1