

An innovative imaging tool enables in vivo high-resolution visualization of deep brain structures and functions over large volume.
Abstract
Optical deep-brain imaging in vivo at high resolution has remained a great challenge over the decades. Two-photon endomicroscopy provides a minimally invasive approach to image buried brain structures, once it is integrated with a gradient refractive index (GRIN) lens embedded in the brain. However, its imaging resolution and field of view are compromised by the intrinsic aberrations of the GRIN lens. Here, we develop a two-photon endomicroscopy by adding adaptive optics based on direct wavefront sensing, which enables recovery of diffraction-limited resolution in deep-brain imaging. A new precompensation strategy plays a critical role to correct aberrations over large volumes and achieve rapid random-access multiplane imaging. We investigate the neuronal plasticity in the hippocampus, a critical deep brain structure, and reveal the relationship between the somatic and dendritic activity of pyramidal neurons.
INTRODUCTION
Advances in two-photon microscopy have greatly propelled the studies of neural circuits and brain functions in the past decades, by enabling high-resolution morphological and functional imaging in the living brain. In conjunction with various fluorescent proteins and indicators, two-photon microscopy allows for direct visualization of fine neuronal structures and continuous monitoring of dynamic neural activities at spatiotemporal scales spanning orders of magnitude (1, 2). However, imaging has been restricted to the superficial brain regions because of the severe attenuation of both excitation and emission photons at depth caused by tissue scattering. Although a longer excitation wavelength or red-shifted fluorescence labeling can alleviate the scattering effect, the imaging depth is still limited to 1 to 2 mm in the mouse brain, and the imaging quality degrades rapidly with increasing depth (3, 4).
To image deeper subcortical structures beyond this limit, endomicroscopy that relies on implanting a miniature gradient refractive index (GRIN) lens in the brain has been adopted (5–7). The rod-like GRIN lens acts as a relay between the microscope objective and the sample below. This lens has been integrated with the single-photon epifluorescence microscope to image various neurons lying deep in the brain that are beyond the reach of conventional microscopy (8–10). However, the optical resolution for such a miniature single-photon microscope has been limited to the cellular level, with the image contrast reduced by the out-of-focus fluorescence background. Two-photon endomicroscopy incorporating a high–numerical aperture (NA) GRIN lens has enabled resolving subcellular structures at high resolution (5, 11, 12). However, the imaging field of view (FOV) is restricted to tens of microns in diameter due to the severe off-axis aberrations of the GRIN lens. Moreover, for three-dimensional (3D) imaging, because the GRIN lens is embedded in the biological sample, the focal plane is tuned by changing the distance between the microscope objective and the GRIN lens, which could lead to severe on-axis aberrations when the excitation focus deviates axially from the designed optimum (5). Therefore, the enlarged and distorted point spread function (PSF) resulting from the intrinsic aberrations substantially limits the 3D imaging volume of GRIN lens–based endomicroscopy.
Adaptive optics (AO), an optical technique originally developed for astronomical telescope (13), has recently greatly advanced two-photon imaging by introducing a compensatory wavefront distortion to the excitation laser that cancels the system- or specimen-induced aberrations (14, 15). The wavefront distortion can be determined by either direct (16–21) or indirect (22–24) wavefront sensing. In previous studies, a sensorless AO approach based on pupil segmentation was used to correct the aberrations of the GRIN lens, improving the resolution over an enlarged imaging FOV of 205 μm × 205 μm in a fixed brain slice (25, 26). However, this method is time consuming for wavefront estimation and sensitive to sample motion, which limits its application for in vivo imaging. An unmet challenge is to develop robust AO two-photon endomicroscopy enabling high-resolution imaging over large volumes in the living mouse brain.
Here, we developed an AO two-photon endomicroscope based on direct wavefront sensing for high-resolution deep-brain imaging in vivo (Fig. 1A and fig. S1). Our approach is to use the two-photon excited fluorescence (TPEF) signal as the intrinsic guide star inside biological tissues. The wavefront of the descanned guide star is averaged over a small region (typically 30 μm × 30 μm) and measured with the Shack-Hartmann wavefront sensor (SHWS) consisting of a microlens array and an electron-multiplying charge-coupled device (EMCCD) (27). Because of the low tissue-induced scattering, direct wavefront sensing provides accurate estimation of the aberrations and is well suited for in vivo imaging with a GRIN lens. To achieve fast AO correction over large volume, we first characterized the aberrations of the GRIN lens and developed a lookup table method to precompensate for its intrinsic aberrations before in vivo imaging. Direct wavefront sensing combined with lookup table method permits accurate, rapid, and robust estimation of the optical aberrations during in vivo brain imaging (18). The wavefront information is then used by the deformable mirror (DM) that forms a closed loop with the SHWS to create a compensatory distortion to the excitation light and yield a diffraction-limited focus inside the brain tissues. Using the lookup table and following in situ AO correction, we achieved structural imaging of hippocampal CA1 neurons at synaptic resolution over a large volume. We studied neuronal plasticity of the hippocampus under various pathological conditions. Furthermore, by combining our new endomicroscope with a fast electrically tunable lens (ETL), we demonstrated quasi-simultaneous multiplane calcium imaging of neuronal somata and dendrites at high spatiotemporal resolution.
Fig. 1. Simplified AO two-photon endomicroscope schematic diagram and characterization of the intrinsic aberrations of the GRIN lens.

(A) AO two-photon endomicroscope based on close-loop direct wavefront sensing of the nonlinear fluorescent guide star. A cylindrical coordinate (r, θ, z) was introduced in the sample space with two dots of red and blue circles indicating on- and off-axis positions, respectively. OL, objective lens; Fs laser, femtosecond laser. (B) Spot pattern displayed on SHWS measured from an on-axis position (0, 0, −90 μm) and the corresponding reconstructed aberration wavefront. (C) Lateral and axial PSF measured with 200-nm-diameter fluorescent beads at the same on-axis position as in (B) with only system or full AO correction. The intensity profiles along the yellow dashed line in (C) were plotted for both system and full AO correction. (D and E) Measurement of spot pattern, aberration wavefront, and PSF as in (B) and (C) but from an off-axis position (60 μm, 180, −90 μm). a.u., arbitrary units.
RESULTS
To evaluate the efficacy of our approach, we first characterized the intrinsic aberrations of the GRIN lens using an in vitro setup. The GRIN lens was embedded in a rhodamine/agarose mixture that contains sparsely distributed and immobilized fluorescent beads (200-nm diameter). Because the rhodamine was uniform among the FOV, TPEF of rhodamine was used as the guide star for direct wavefront measurement of the aberrations of the GRIN lens. In addition, the PSF of the endomicroscope was evaluated by imaging the individual fluorescent bead. To achieve the optimal imaging performance, the GRIN lens was precisely aligned to share the same optical axis with the objective. Considering the cylindrical symmetry of the GRIN lens, the cylindrical coordinates (r, θ, z) were adopted to describe the imaging location in the sample where z = 0 corresponds to the designed focal plane of the GRIN lens (Fig. 1A). The representative on- and off-axis aberrations of the GRIN lens at various depths are shown in Fig. 1 (B to E), which are primarily dominated by spherical aberration and astigmatism respectively, consistent with a previous study (26). As can be seen from the figures, despite the aberrations increasing rapidly and the PSF becoming severely distorted when the imaging position deviates from the center point, AO correction can effectively increase the fluorescence signal and recover the diffraction-limited performance at various depths (figs. S2 and S3).
We next applied the AO approach to the in vivo imaging of the mouse hippocampus, in which a subset of pyramidal neurons expresses green fluorescent protein (GFP) under thy1 promoter (Thy1-GFP mice). The CA1 pyramidal neurons are vertically oriented with a typical laminar organization spanning hundreds of microns in depth (28). As can be seen in Fig. 2A and fig. S4, with system correction alone, the neuronal somata and dendrites are severely blurred. After full AO correction and subsequent deconvolution from the local PSF, however, not only is the fluorescence intensity drastically improved, the dendrites and spines can also be clearly visualized owing to the substantially enhanced resolutions (Fig. 2B and fig. S4). The corrective wavefront primarily contains astigmatism as previously characterized for the off-axis scenario, implying that the aberrations mainly came from the imperfections of the GRIN lens (Fig. 2C). The spectral power shows that the higher spatial frequency can be notably recovered by applying full AO correction with subsequent deconvolution (Fig. 2D). Quantitative measurement from individual dendritic spines shows that both lateral and axial resolutions approached diffraction-limited performance for the GRIN lens with NA of 0.8 (Fig. 2, E and F). Furthermore, even for the on-axis scenario where the GRIN lens was designed to yield the best performance, we still achieved tremendous improvement in resolution after full AO correction, with dendritic spines as deep as 300 μm clearly resolved (fig. S5). These results demonstrate that AO based on direct wavefront sensing can effectively correct the aberrations of the GRIN lens and recover the diffraction-limited resolution during in vivo imaging.
Fig. 2. Direct wavefront sensing AO effectively restores diffraction-limited resolution at depth during in vivo brain imaging.

(A) 3D reconstruction of a column (center located at r = 60 μm) of hippocampal CA1 pyramidal neurons in Thy1-GFP mice imaged with our two-photon endomicroscope with system correction only (left) and with full correction plus subsequent deconvolution (right). Full AO correction is performed every 30 μm of depth. (B) Depth color-coded xy maximum intensity projection (MIP) of the stack images (left column: 30 to 70 μm; right column: 160 to 200 μm) from 3D images in Fig. 1A. The images with system correction have been digitally enhanced four- and threefold as indicated for better visualization. Scale bar, 5 μm. (C) Corrective wavefronts of the DM used for full AO correction of the stack images in (B). (D) Spectral power in spatial frequency space (kx, ky) for the images in (B). (E and F) Magnified views of the dendritic spines corresponding to the boxed regions (i and ii) in (B). The spines are shown in lateral (xy) and axial (xz) views. The axial view is shown through the plane defined by the yellow dashed line. Intensity profiles along the blue and red lines are plotted with the curve fitted by a Gaussian function. Scale bars, 2 μm. FWHM, full width at half maximum.
Next, we sought to investigate the extent to which our AO approach can enlarge the imaging FOV in vivo. Because the aberrations vary from site to site, we performed AO correction in a number of subregions of 50 μm × 50 μm each separated by a distance of 30 μm laterally and stitched these subregions together to form a high-resolution image of large FOV. Considering that the photon budget and fluorescence may be too weak for wavefront measurement, we propose a lookup table method to precompensate for the aberrations in wavefront measurement (Fig. 3, A and B). We found that the corrective wavefront from the lookup table serves as a close approximation of the optimal wavefront measured in vivo (Fig. 3, C to F). The lookup table method can greatly boost the fluorescence signal above and beyond system correction and thus drastically decrease the exposure time required for direct wavefront measurement. Because of the large off-axis aberrations of the GRIN lens, the imaging FOV with only system correction was restricted to a small central region (<100 μm in diameter) of poor resolution (Fig. 4A and fig. S6A). After full AO correction, the effective FOV was enlarged to 300 μm together with the recovery of synaptic resolution. The neuronal structures including somata, dendrites, and individual spines can be clearly visualized across the entire FOV, covering all layers of the hippocampal CA1 region (Fig. 4B and fig. S6B).
Fig. 3. Lookup table–based precompensation for GRIN lens aberrations.
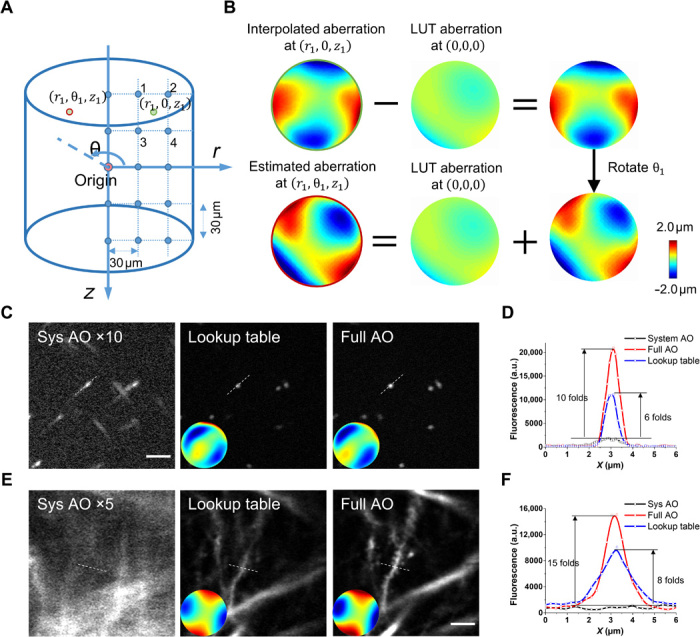
(A) Calibration of the lookup table. The cylindrical coordinate system was used to describe the imaging location. The origin is defined as the center located at the designed working distance of the GRIN lens. The entire imaging FOV had a radius of 150 μm and a depth of 300 μm. We measured the intrinsic aberration of the GRIN lens in the θ = 0 subplane at 30-μm intervals, shown as blue dots. (B) Estimation of GRIN lens–induced aberration using the lookup table. To find the aberration at location (r1, θ1, z1) [red circle in (A)], we first estimate the aberration at the rotational symmetrical point (r1, 0, z1) using linear interpolation of the aberrations nearby (the blue dots labeled 1 to 4). Then, the aberration at the origin is subtracted from the interpolated wavefront distortion, and the resulting wavefront is rotated by the angle θ1 and added back to the aberration at the origin. LUT, lookup table. (C and D) In vitro imaging using fluorescent beads of 200 nm in diameter. (C) Fluorescence images with system AO, lookup table, and full AO correction. The insets show the corresponding corrective wavefront. (D) Intensity profile along the dashed lines in (C). (E and F) In vivo imaging of hippocampal neurons in mice. (E) Fluorescence images with system AO, lookup table, and full AO correction. Scale bar, 5 μm. (F) Intensity profile along the dashed lines in (E).
Fig. 4. AO two-photon endomicroscope enables in vivo imaging of the mouse hippocampus at synaptic resolution over a large FOV.
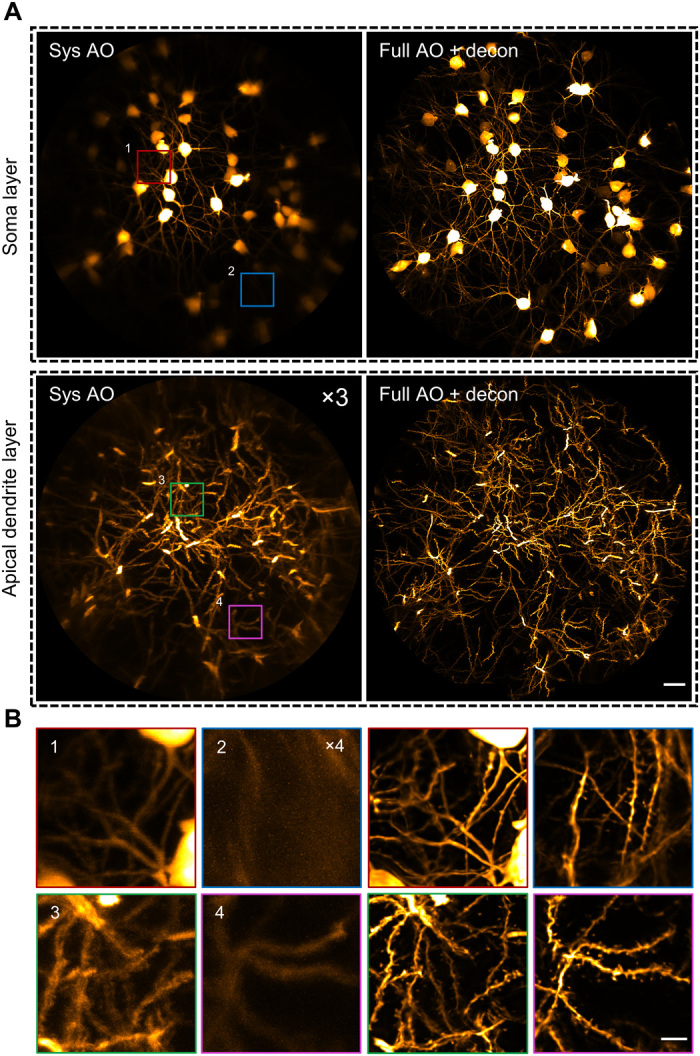
(A) MIP images of different layers of hippocampal CA1 pyramidal neurons in Thy1-GFP mice with only system correction (left column) and with full correction plus subsequent deconvolution (right column). Depth range of projection for soma layer: 90 to 120 μm; apical dendrite layer: 160 to 190 μm. The images with system correction were enhanced for better visualization. Scale bar, 20 μm. (B) Left: Four magnified views of the subregions indicated by the numbered boxes in (A). Right: Views of corresponding subregions with full AO corrections. Scale bar, 5 μm.
Taking advantage of our approach, we further investigated the neuronal plasticity of the hippocampus under various pathological conditions via in vivo time-lapse imaging of the dendritic and spine dynamics. We first demonstrated the dendritic alteration in response to the neuronal injury induced by a laser-mediated microsurgery. We found that micro-lesion of single dendritic shaft will trigger recurrent spinogenesis in the neighboring regions, whereas cutting the dendritic branches with laser will lead to prolonged degeneration of the injured dendrites (fig. S7). In addition, we also monitored the spine dynamics in mice with kainic acid–induced epileptic seizure. In contrast to the dynamic neuronal changes induced by laser microsurgery, our findings show that the changes in spine stability caused by kainic acid–induced epileptic seizure are insignificant, which is in agreement with another study of the hippocampus of mice experiencing pilocarpine-induced epileptic seizure (29). These results shed light on the neuronal mechanisms underlying neurodegenerative disorders.
In addition to morphological imaging, we further applied the AO two-photon endomicroscope to the in vivo functional calcium imaging of pyramidal neurons in the hippocampus of awake behaving mice that expressed a calcium indicator (AAV-CaMKII-GCaMP6s). Taking advantage of the large imaging volume provided by AO correction, we developed a random-access multiplane imaging method that can quickly capture arbitrarily selected regions of interest (ROIs) across the effective imaging space (Fig. 5A). The galvanometer scanners were programmed to scan the ROIs sequentially, and an ETL was synchronized with the scanners for fast switching between imaging planes (fig. S8). To recover the optimal imaging performance in all the ROIs, the wavefront distortion in each ROI was measured individually, and the corrective pattern of DM was synchronously updated to compensate for the field-dependent aberrations (fig. S8). After full AO corrections in ROIs, the fine neurites and individual spines contaminated by the neuropil background can be easily distinguished owing to the greatly enhanced resolution and fluorescence intensity (Fig. 5, B and C, and movie S1). Moreover, the imaging system can clearly resolve the tightly packed neurons with the calcium transients accurately recorded and without interference from the neighboring neurons (Fig. 5, D to F, and movie S2). The results demonstrate that our AO approach provides a sensitive and accurate characterization of neuronal activity and enables simultaneous calcium imaging of neuronal somata and dendrites at synaptic resolution.
Fig. 5. Random-access multiplane Ca2+ imaging of hippocampal neurons in vivo.

(A) Schematic diagram of random-access multiplane imaging. Three image planes over the entire imaging volume of 0.3 mm × 0.3 mm × 0.3 mm can be randomly selected and sequentially scanned at 5 Hz with synchronized ETL and xy galvanometer scanners. (B) Multiplane Ca2+ imaging of neuronal somata, dendrites, and spines at various locations with system correction (left) and with full correction (right). The virus AAV-CaMKII-GCaMP6s was injected into the hippocampus CA1 of C57/B6 mice. Images are shown as average-intensity projection of 600 frames. The images with system correction were enhanced as indicated for better visualization. The cylinder coordinates: (μm, degree, μm). Scale bar, 5 μm. (C) Fluorescence traces for the ROIs indicated in (B). (D) Ca2+ imaging of pyramidal neurons in hippocampus CA1 with system and full AO correction. Images are shown as average-intensity projections of 600 frames. Scale bar, 5 μm. (E) Calcium transients (∆F/F) of the selected neurons as shown in (A). (F) Correlation coefficient matrices calculated from ∆F/F traces of all neurons in (B). Colored boxes refer to the three ROIs as shown in (A).
We then applied this AO-assisted functional imaging technology to investigate the relationship between somatic and dendritic activity in mouse hippocampus CA1. The soma and two apical dendrites of single pyramidal neuron were targeted for simultaneous calcium imaging (Fig. 6, A and B, and movie S3). We found that the dendritic and somatic signals are highly correlated, despite that their amplitudes and kinetics are largely different (Fig. 6, C to E). This functional correlation of dendrites and neuronal somata has also been reported in the cortex of awake mouse, in which the persistent coupling of somato-dendritic activity is unchanged by stimuli and mouse locomotion (30). Our further results show that the correlated somato-dendritic activity in mouse hippocampus is brain state dependent. In comparison with full wakefulness, isoflurane-induced light anesthesia will not only weaken the activity of different neuronal compartments but also disrupt the somato-dendritic correlation (fig. S9). In future work, it would be of great interest to extend this method to study the dendritic integration in awake mice performing behavioral tasks (31, 32).
Fig. 6. AO-assisted multiplane Ca2+ imaging of somato-dendritic activity in hippocampus CA1.
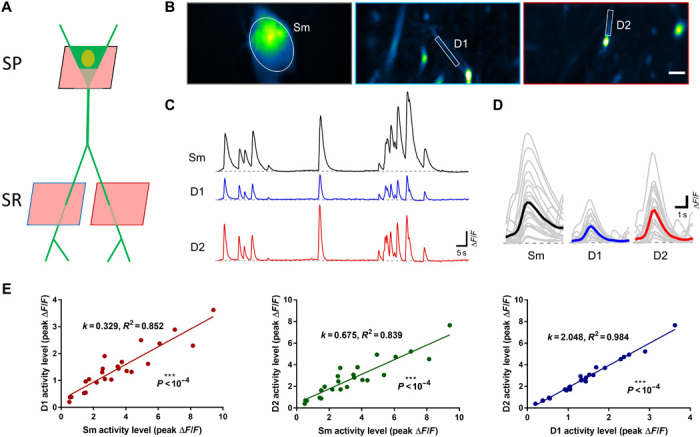
(A) Experimental approach to target the dendrites and soma for single neuron recording in hippocampus CA1. SP, stratum pyramidale; SR, stratum radiatum. (B) Quasi-simultaneous Ca2+ imaging of spontaneous activity from the soma (Sm) and two dendrites (D1 and D2) in awake behaving mice. Images are shown as SD projection of 600 frames. Scale bar, 5 μm. (C) Calcium transients (∆F/F) of the soma and dendrites as shown in (B). (D) Firing events of soma (Sm) and dendrites (D1 and D2) as shown in (B). Gray and colored curves represent the individual and average events, respectively. (E) Relationship between activity strength of the soma-dendrite pairs and dendrite-dendrite pair.
DISCUSSION
In this work, we combined the AO technique based on the direct wavefront sensing of the TPEF guide star with a high-NA endomicroscope, which enables the recovery of diffraction-limited resolution over large imaging volumes. Using this system, we achieved in vivo morphological imaging of pyramidal neurons at synaptic resolution across all layers of mouse hippocampus CA1. Moreover, by integrating the system with a random-access multiplane imaging technique, we demonstrated quasi-simultaneous calcium imaging of separately distributed somata and dendrites in the hippocampus of awake behaving mice. The AO endomicroscope can also benefit the imaging of other deep brain structures such as the striatum, the substantia nigra, and the hypothalamus (33). Note that although brain tissue can be surgically removed and replaced with a cannula window (34) or glass plug (35) to provide direct optical access, the procedure requires removing too much tissue and is not applicable to deep-brain imaging. An endomicroscope based on a miniature GRIN lens provides an approach to deep-brain imaging with minimized trauma but suffers from poor resolutions due to the inherent aberrations of the GRIN lens. One way to solve this problem directly is by adding an extra lens inside the GRIN lens assembly to correct the off-axis aberrations (36). However, the GRIN lens is only optimized at the designed working distance. For in vivo imaging where the GRIN lens remains fixed in the biological samples, 3D imaging is achieved by changing the laser convergence entering the GRIN lens, which could lead to severe aberrations when the focus deviates from the designed working plane. Our AO endomicroscope, on the other hand, provides a versatile approach for correcting the aberrations of various GRIN lenses with suboptimal design. This study demonstrates the great potential of the AO two-photon endomicroscope to facilitate neuroscience research in the deeper regions of the brain.
MATERIALS AND METHODS
AO two-photon endomicroscopy
The schematic of our homebuilt AO two-photon endomicroscopy system is shown in fig. S1. The beam of a tunable mode-locked femtosecond laser (Coherent, Mira 900) was expanded by a pair of achromatic lens to slightly overfill the aperture of the DM (Alpao, DM97-15). The DM was conjugated to the 5-mm galvanometric x-scanning mirror (Cambridge Technology, 6215H) by a 4f telescope formed by two visible–near infrared (VIS-NIR) achromatic doublets L3 and L4 (Edmunds, 49-365 and 49-794). Two scanning mirrors were mutually conjugated by the lens pair L5 and L6, each of which consisted of two doublets (Edmunds, 49-392). Last, the galvanometric y-scanning mirror was conjugated to the back focal plane of the objective by a 4f relay formed by the scan lens L7 [consisting of two doublets (Edmunds, 49-391)] and the tube lens L8 (Edmunds, 49-393). A 10× air objective (NA = 0.45; Carl Zeiss, Plan-Apochromat) was used to match the image NA of the GRIN lens (image NA = 0.415, object NA = 0.8; GRINTECH GmbH, GT-MO-080-0415-810) and mounted on an encoded translation stage (Thorlabs, LNR50SEK1) for axial sectioning. Precise alignment was carried out to ensure that the objective and the GRIN lens shared the same optical axis.
For two-photon imaging, the fluorescence emission signal collected by the GRIN lens and the objective was directed to the photo-detection unit via dichroic mirror D2 (Semrock, FF705-Di01-25×36). Then, the fluorescence was separated by dichroic mirror D3 (Semrock, FF560-Di01-25×36) into a red and a green channel. The bandpass filters F2 (Semrock, FF01-525/50) and F3 (Semrock, FF01-593/46) were placed before the current photomultiplier tube (PMT) modules (Hamamatsu, H11461-01 and H11461-03) to select a particular wavelength range of detection. The corresponding PMT signal was then fed into a current amplifier (Stanford Research, SR570 and Femto, DLPCA-200) and subsequently into a data acquisition device (National Instruments, PCIe-6353) controlled with custom-written software.
For wavefront sensing, D2 was switched to another dichroic mirror (Semrock, Di02-R488-25×36) using a motorized flipper (Thorlabs, MFF101/M) so that the guide star signal of fluorescence emission can transmit through D2 and be descanned by the galvanometer scanning mirrors. Then, the fluorescence signal was reflected by the DM and separated from the excitation laser by dichroic mirror D1 (Semrock, FF705-Di01-25×36). The DM was conjugated to the microlens array (SUSS MicroOptics, 18-00197) of the SHWS by the lens pair L9 and L10 (Throlabs, AC254-200-A and AC254-100-A). An ultrasensitive EMCCD (Andor, iXon Ultra 888) was placed in the focal plane of the microlens array to capture the spot pattern, which provides direct measurement of the wavefront distortion. Overall, the DM, the galvanometer scanning mirrors, the back focal plane of the objective, and the microlens array of SHWS are all mutually conjugated via the 4f relay system.
Calibration of the DM
The DM was calibrated before it was integrated into the microscope system. Following a previously reported procedure (19), the driving voltage pattern of the DM for the first 65 of Noll’s Zernike modes was measured using a homebuilt Michelson interferometer. In this way, the DM can be controlled to take any desired shape via a linear combination of these Zernike modes.
System AO correction
The aberrations of the microscope system were corrected before any imaging experiment. First, the 10× air objective and the GRIN lens were replaced by another objective with nearly aberration-free performance (25×, 1.05 NA; Olympus, XLPLN25XWMP2) such that the measured aberrations would mainly come from the imperfections of the optical components or alignments of the system. Then, the two-photon fluorescence intensity of a fluorescent dye (rhodamine 6G) solution was used as the feedback to optimize the shape of the DM. Here, a Zernike mode–based sensorless AO algorithm (37) was adopted. Briefly, seven to nine different values of each Zernike mode were applied to the DM, and the corresponding fluorescence intensity was fitted with a Gaussian function to find the optimal value. This procedure was repeated for the first 21 Zernike modes (tip, tilt, and defocus excluded), and the system aberration Zsys could be measured and compensated.
Calibration of the wavefront sensor
The SHWS was calibrated with respect to the DM in the microscope system. The femtosecond laser was focused by the 25× water objective to excite a fluorescent dye (rhodamine 6G) solution, which created a nonlinear fluorescent guide star. The light of the guide star was shaped by the DM and then sent to the SHWS, which formed the closed-loop AO system. The influence matrix Msz of the DM on the SHWS was calibrated by sequentially applying the first 65 Zernike modes to the DM and recording the corresponding spot displacement in the SHWS. The rows of Msz represent the shift in spot position on the SHWS in response to each Zernike mode and formed the basis on which the in vivo wavefront measurement can be decomposed. In this work, we chose the modal wavefront reconstruction algorithm because (i) the aberration is averaged over a small volume and thus is mainly of low order; (ii) this algorithm is robust to the noise and missing spots in the in vivo wavefront measurement (19); and (iii) each Zernike mode has well defined physically meanings such as astigmatism, coma, and spherical aberration, making the interpretation of GRIN lens aberrations explicit.
Full AO correction
The full AO correction started with the system aberration corrected and further compensated for the aberrations of the GRIN lens and those induced by biological samples. The reference spot pattern on SHWS Sref = (x1⋯xN, y1⋯yN) was recorded using the fluorescence signal of rhodamine at the FOV center of the endomicroscope with aberration corrected by the sensorless algorithm described above. For in vivo aberration measurement, the femtosecond laser was scanned over a small FOV (30 μm × 30 μm), and the excited fluorescence signal was descanned and integrated at the SHWS. The spot location of the in vivo SHWS measurement Sall was estimated using a Gaussian-fit centroid algorithm (38), and the reliability weight of each spot was determined by its signal-to-background ratio W = Diag(w1⋯wN, w1⋯wN). The spot displacement of the in vivo measurement relative to the reference position was calculated as ∆S = Sall − Sref. Then, the additional corrective pattern of the DM could be computed by minimizing the total aberration as follows
The full AO correction pattern applied to the DM was Zfull = Zsys + ∆Z. Here, all the plotted wavefront distortions represent the aberrations induced by the GRIN lens and biological samples, i.e., ∆Z.
Animal preparation
C57 (C57BL/6J) and Thy1-GFP [Tg(Thy1-EGFP)MJrs/J] mice were obtained from the Jackson laboratory. The mice were housed at the Animal and Plant Care Facility of The Hong Kong University of Science and Technology (HKUST). Mice of the same sex were housed four per cage with a 12-hour light/dark cycle and food and water ad libitum. All animal procedures were conducted in accordance with the Guidelines of the Animal Care Facility of HKUST and approved by the Animal Ethics Committee at HKUST.
Virus injection
The AAV9-CaMKII-GCaMP6f virus was obtained from the Penn Vector Core at the University of Pennsylvania. Virus was diluted 1:10 in phosphate-buffered saline and delivered as a bolus (0.5 μl) at 50 to 100 nl/min via a Hamilton syringe into the hippocampal CA1 region (anteroposterior, −2.00 mm; mediolateral, ±1.50 mm; dorsoventral, −1.4 mm; relative to the bregma) in 3- to 4-month-old C57 mice. After injection, the needle was kept in place for 10 min and then retracted from the brain. The mice were then returned to their home cages to recover for 3 to 4 weeks.
GRIN lens implantation
Dexamethasone (0.2 μg/mg) solution was subcutaneously administered 1 hour before the procedure to prevent brain swelling and reduce inflammatory responses. The mouse was then secured on a stereotaxic instrument and anesthetized with isoflurane (1.5 to 2% in oxygen). After the skull was exposed and cleaned with ethanol (70%), three stainless steel screws were implanted in the skull to form a triangle with the center located over the hippocampal CA1 region for skull stabilization. Then, a 1.6-mm-diameter craniotomy was performed at stereotactic coordinates (2 mm, 1.5 mm) posterior and lateral to the bregma point. Next, the cylindrical column of the exposed cortex was slowly aspirated and removed using a 27-gauge blunt needle until the white matter appeared. The GRIN lens was then positioned over the cranial window and gently inserted into the brain. After a thin layer of adhesive luting cement applied to the skull surrounding the window had dried and hardened, a small amount of dental cement was used to cover the exposed skull surface. A custom-designed rectangular head plate with a round hole of clearance for GRIN lens at the center was then permanently glued to the exposed skull. It would be rigidly mounted the mouse head on a holding device with angle adjusters (NARISHIGE, MAG-2) during in vivo imaging experiments. After the surgery, the mouse was allowed to recover from the anesthesia on a heating blanket before returning to its home cage.
Alignment of the GRIN lens
For in vitro and in vivo experiments, the sample was mounted on the rotational stage MAG-2 and further fixed to a three-axis translation stage, which enabled precise adjustment of the angle and position of the GRIN lens. A visible laser diode was introduced to the endomicroscopy system to assist the alignment. The laser beam was first aligned with the optical axis of the objective. Then, the objective was removed, and the tilt/tip of the GRIN lens was adjusted so that the laser was normally incident on the upper surface of the GRIN lens. This guaranteed that the optical axes of the GRIN lens and the objective were parallel to each other. Next, we looked at the imaging plane of the objective through the eyepiece and translated the GRIN lens until its upper surface came into focus. Then, the objective was lifted 100 μm up so that the GRIN lens could operate at its designed working distance. Last, we switched to the two-photon imaging mode and translated the GRIN lens sideways until the fluorescence image appeared at the center of the FOV.
Lookup table for the aberrations of the GRIN lens
To evaluate the efficacy of our approach, we first characterized the intrinsic aberrations of the GRIN lens that was embedded in a rhodamine/agarose mixture that contains sparsely distributed and immobilized fluorescent beads (200-nm diameter). Its aberration was measured by the wavefront distortion of rhodamine fluorescence, whereas the PSF was determined by imaging the individual fluorescent bead. To achieve the optimal imaging performance, the GRIN lens was precisely aligned to share the same optical axis with the objective. Considering the cylindrical symmetry of the GRIN lens, the cylindrical coordinates (r, θ, z) were adopted to describe the imaging location in the sample where z = 0 corresponds to the designed focal plane of the GRIN lens. The representative on- and off-axis aberrations of the GRIN lens at various depths are shown in figs. S2 and S3, which are primarily dominated by spherical aberration and astigmatism, respectively, consistent with a previous study (26). As can be seen from figs. S2 and S3, despite the aberrations increasing rapidly and the PSF becoming severely distorted when the imaging position deviates from the center point, AO correction can effectively increase the fluorescence signal and recover the diffraction-limited performance at various depths. Because the optical properties of the GRIN lens are determined by the optical design and manufacturing process, we can precalibrate the intrinsic aberrations of the GRIN lens and store them in a lookup table. The lookup table was established using an in vitro setup. In principle, we need to measure all the aberrations at different field locations. Considering that the aberrations of the GRIN lens are cylindrically symmetrical along the optical axis (25, 26), the number of aberration measurements could be greatly reduced. To cover the entire cylindrical FOV with a radius of 150 μm and a depth of 300 μm, we built the lookup table by measuring the aberrations along the r and z directions at 30-μm intervals, resulting in 6 × 11 wavefront distortions in the θ = 0 subplane (Fig. 3A).
For in vivo imaging, the photon budget is limited, and the two-photon fluorescence intensity was practically too weak for wavefront measurement, especially in the off-axis region of large aberration. To solve this problem, we precompensated for the aberration of the GRIN lens in the imaging region (r1, θ1, z1) using the lookup table (Fig. 3B). First, the aberration of the GRIN lens at location (r1, 0, z1) was estimated from the linear interpolation of the aberrations nearby stored in the lookup table. Second, the aberration at the center was subtracted from that at (r1, 0, z1). Last, the resulting aberration was rotated by the angle θ1 and added back to the aberration at the center, which was the estimated wavefront correction for the desired location (r1, θ1, z1). The aberration at the field center was removed from the rotational operation, because it was mainly induced by the alignment of the GRIN lens and objective and thus is not cylindrically symmetric (26). As shown in Fig. 3, because the intrinsic aberrations of the GRIN lens were largely corrected by using the lookup table, the fluorescence intensity and imaging resolution were improved tremendously. Then, we were able to perform in vivo AO correction based on direct wavefront sensing to further eliminate the residual aberrations caused by the alignment error of the GRIN lens as well as biological tissues.
In vivo imaging
The imaging experiment was performed 3 weeks after the implantation of the GRIN lens. The mouse was first briefly anesthetized with isoflurane (1 to 2% in oxygen) and mounted on the head-holding device. The GRIN lens was precisely aligned to share the same optical axis with the objective lens. The femtosecond laser tuned at 920 nm was used to excite GFP and GCaMP6s. For wavefront sensing, the aberration of the GRIN lens was first precompensated using the lookup table, and the residual aberration was measured and corrected using the two-photon excited guide star. The laser power was 30 to 50 mW through the objective, and the exposure time was less than 3 s. For most imaging conditions, a single frame of image is acquired per slice. In case of severe animal motion, we increased the pixel rate and applied image registration to several sequential frames for each slice. The registered frames were then averaged to reduce the motion artifact and improve the signal-to-background ratio. The detailed imaging parameters including excitation power, pixel rate, and guide star integration time can be found in table S1. For the morphological imaging, the animals were anesthetized with isoflurane (1% in oxygen) with the head secured on a head-holding stage (NARISHIGE, MAG-2). For calcium imaging, the mice were fully awake or lightly anesthetized with isoflurane and placed on a head-fixation behavioral platform that was based on a rotary treadmill.
Laser-mediated neuronal injury
The field-dependent GRIN lens aberration was first measured and corrected as previously described to enable precise and efficient laser-mediated microsurgery. For laser cutting of the dendritic branch, the 920-nm laser with 160-mW power through the objective was line scanned (1 s per line) across the branch repeatedly for 20 s. For micro-lesion of dendritic shaft, the laser with the same optical power was focused on the shaft center for 5 s. The parameters of optical power and duration for laser microsurgery have been optimized to induce reproducible neuronal injuries.
Multiplane calcium imaging
An ETL (Optotune, EL-16-40-TC-VIS-5D-C) was placed close to the rear stop of the 10× objective lens for quick tuning of the focal plane. We first picked three ROIs and measured the wavefront distortion using the method described above. The three corrective patterns of the DM were stored locally for further use. Next, we conducted near-simultaneously calcium imaging of the three ROIs at a volume rate of 5 Hz. The ETL and DM were synchronized with the scanner, as shown in fig. S8. The driving voltage of the ETL was low-pass filtered to avoid high-order oscillation and improve the temporal response. The premeasured corrective patterns were updated on the DM accordingly to compensate for the field-dependent aberrations.
Image analysis
The images were processed in MATLAB or ImageJ (39). For image deconvolution, the Richardson-Lucy algorithm of the DeconvolutionLab2 plugin (40) in ImageJ was adopted. The PSF used for deconvolution was experimentally measured at different imaging positions. To make the PSF measurement less laborious, we took advantage of the cylindrical symmetry of the GRIN lens and only measured the PSF in the θ = 0 subplane. The PSF in other regions could be estimated using the rotation-based procedure. For the mosaic images, multi-tile subimages were stitched together using either MosaicJ (41) or Grid/Collection Stitching (42) plugin in ImageJ. To cover the entire FOV of 300 μm in diameter, 81 subimages at 30-μm intervals were acquired.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Hong Kong Research Grants Council through grants 662513, 16103215, 16148816, 16102518, T13-607/12R, T13-706/11-1, T13-605/18W, C6002-17GF, C6001-19E, and N_HKUST603/19; the Innovation and Technology Commission (ITCPD/17-9); the Area of Excellence Scheme of the University Grants Committee (AoE/M-604/16, AOE/M-09/12); and the HKUST through grant RPC10EG33. Author contributions: Z.Q., C.C., N.Y.I., and J.Y.Q. conceived the research idea. C.C. and Z.Q. designed and conducted the experiments and data analysis. Z.Q., S.H., and C.C. built the AO two-photon and multiplane imaging system. Y.W. and K.F.T. carried out the surgery of virus injection and GRIN lens implantation. C.C. and Z.Q. took the lead in writing the manuscript with inputs from all other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from J.Y.Q.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabc6521/DC1
REFERENCES AND NOTES
- 1.Helmchen F., Denk W., Deep tissue two-photon microscopy. Nat. Methods 2, 932–940 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Svoboda K., Yasuda R., Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron 50, 823–839 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Horton N. G., Wang K., Kobat D., Clark C. G., Wise F. W., Schaffer C. B., Xu C., In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat. Photonics 7, 205–209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tischbirek C., Birkner A., Jia H., Sakmann B., Konnerth A., Deep two-photon brain imaging with a red-shifted fluorometric Ca2+ indicator. Proc. Natl. Acad. Sci. U.S.A. 112, 11377–11382 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barretto R. P. J., Messerschmidt B., Schnitzer M. J., In vivo fluorescence imaging with high-resolution microlenses. Nat. Methods 6, 511–512 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barretto R. P. J., Ko T. H., Jung J. C., Wang T. J., Capps G., Waters A. C., Ziv Y., Attardo A., Recht L., Schnitzer M. J., Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy. Nat. Med. 17, 223–228 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lecoq J., Savall J., Vučinić D., Grewe B. F., Kim H., Li J. Z., Kitch L. J., Schnitzer M. J., Visualizing mammalian brain area interactions by dual-axis two-photon calcium imaging. Nat. Neurosci. 17, 1825–1829 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McHenry J. A., Otis J. M., Rossi M. A., Robinson J. E., Kosyk O., Miller N. W., McElligott Z. A., Budygin E. A., Rubinow D. R., Stuber G. D., Hormonal gain control of a medial preoptic area social reward circuit. Nat. Neurosci. 20, 449–458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamigaki T., Dan Y., Delay activity of specific prefrontal interneuron subtypes modulates memory-guided behavior. Nat. Neurosci. 20, 854–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy D. S., Kitamura T., Okuyama T., Ogawa S. K., Sun C., Obata Y., Yoshiki A., Tonegawa S., Distinct neural circuits for the formation and retrieval of episodic memories. Cell 170, 1000–1012.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Attardo A., Fitzgerald J. E., Schnitzer M. J., Impermanence of dendritic spines in live adult CA1 hippocampus. Nature 523, 592–596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zong W., Wu R., Li M., Hu Y., Li Y., Li J., Rong H., Wu H., Xu Y., Lu Y., Jia H., Fan M., Zhou Z., Zhang Y., Wang A., Chen L., Cheng H., Fast high-resolution miniature two-photon microscopy for brain imaging in freely behaving mice. Nat. Methods 14, 713–719 (2017). [DOI] [PubMed] [Google Scholar]
- 13.J. W. Hardy, Adaptive Optics for Astronomical Telescopes (Oxford Univ. Press, 1998). [Google Scholar]
- 14.Booth M. J., Adaptive optical microscopy: The ongoing quest for a perfect image. Light Sci. Appl. 3, e165 (2014). [Google Scholar]
- 15.Ji N., Adaptive optical fluorescence microscopy. Nat. Methods 14, 374–380 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Aviles-Espinosa R., Andilla J., Porcar-Guezenec R., Olarte O. E., Nieto M., Levecq X., Artigas D., Loza-Alvarez P., Measurement and correction of in vivo sample aberrations employing a nonlinear guide-star in two-photon excited fluorescence microscopy. Biomed. Opt. Express 2, 3135–3149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao X., Azucena O., Fu M., Zuo Y., Chen D. C., Kubby J., Adaptive optics microscopy with direct wavefront sensing using fluorescent protein guide stars. Opt. Lett. 36, 3389–3391 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Wang K., Milkie D. E., Saxena A., Engerer P., Misgeld T., Bronner M. E., Mumm J., Betzig E., Rapid adaptive optical recovery of optimal resolution over large volumes. Nat. Methods 11, 625–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K., Sun W., Richie C. T., Harvey B. K., Betzig E., Ji N., Direct wavefront sensing for high-resolution in vivo imaging in scattering tissue. Nat. Commun. 6, 7276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu R., Li Z., Marvin J. S., Kleinfeld D., Direct wavefront sensing enables functional imaging of infragranular axons and spines. Nat. Methods 16, 615–618 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin Z., He S., Yang C., Yung J. S.-Y., Chen C., Leung C. K.-S., Liu K., Qu J. Y., Adaptive optics two-photon microscopy enables near-diffraction-limited and functional retinal imaging in vivo. Light Sci. Appl. 9, 79 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Débarre D., Botcherby E. J., Watanabe T., Srinivas S., Booth M. J., Wilson T., Image-based adaptive optics for two-photon microscopy. Opt. Lett. 34, 2495–2497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ji N., Milkie D. E., Betzig E., Adaptive optics via pupil segmentation for high-resolution imaging in biological tissues. Nat. Methods 7, 141–147 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Tang J., Germain R. N., Cui M., Superpenetration optical microscopy by iterative multiphoton adaptive compensation technique. Proc. Natl. Acad. Sci. U.S.A. 109, 8434–8439 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C., Ji N., Pupil-segmentation-based adaptive optical correction of a high-numerical-aperture gradient refractive index lens for two-photon fluorescence endoscopy. Opt. Lett. 37, 2001–2003 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Wang C., Ji N., Characterization and improvement of three-dimensional imaging performance of GRIN-lens-based two-photon fluorescence endomicroscopes with adaptive optics. Opt. Express 21, 27142–27154 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Cha J.-W., Ballesta J., So P. T. C., Shack-Hartmann wavefront-sensor-based adaptive optics system for multiphoton microscopy. J. Biomed. Opt. 15, 046022 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neves G., Cooke S. F., Bliss T. V. P., Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Mizrahi A., Crowley J. C., Shtoyerman E., Katz L. C., High-resolution in vivo imaging of hippocampal dendrites and spines. J. Neurosci. 24, 3147–3151 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beaulieu-Laroche L., Toloza E. H. S., Brown N. J., Harnett M. T., Widespread and highly correlated somato-dendritic activity in cortical layer 5 neurons. Neuron 103, 235–241.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stuart G. J., Spruston N., Dendritic integration: 60 years of progress. Nat. Neurosci. 18, 1713–1721 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Sheffield M. E. J., Dombeck D. A., Calcium transient prevalence across the dendritic arbour predicts place field properties. Nature 517, 200–204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bocarsly M. E., Jiang W.-c., Wang C., Dudman J. T., Ji N., Aponte Y., Minimally invasive microendoscopy system for in vivo functional imaging of deep nuclei in the mouse brain. Biomed. Opt. Express 6, 4546–4556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dombeck D. A., Harvey C. D., Tian L., Looger L. L., Tank D. W., Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat. Neurosci. 13, 1433–1440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Velasco M. G. M., Levene M. J., In vivo two-photon microscopy of the hippocampus using glass plugs. Biomed. Opt. Express 5, 1700–1708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matz G., Messerschmidt B., Gross H., Design and evaluation of new color-corrected rigid endomicroscopic high NA GRIN-objectives with a sub-micron resolution and large field of view. Opt. Express 24, 10987–11001 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Park J.-H., Kong L., Zhou Y., Cui M., Large-field-of-view imaging by multi-pupil adaptive optics. Nat. Methods 14, 581–583 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson R. E., Larson D. R., Webb W. W., Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82, 2775–2783 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sage D., Donati L., Soulez F., Fortun D., Schmit G., Seitz A., Guiet R., Vonesch C., Unser M., DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods 115, 28–41 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Thévenaz P., Unser M., User-friendly semiautomated assembly of accurate image mosaics in microscopy. Microsc. Res. Tech. 70, 135–146 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Preibisch S., Saalfeld S., Tomancak P., Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabc6521/DC1