

ABSTRACT
Histidine is a dietary essential amino acid because it cannot be synthesized in humans. The WHO/FAO requirement for adults for histidine is 10 mg · kg body weight−1 · d−1. Histidine is required for synthesis of proteins. It plays particularly important roles in the active site of enzymes, such as serine proteases (e.g., trypsin) where it is a member of the catalytic triad. Excess histidine may be converted to trans-urocanate by histidine ammonia lyase (histidase) in liver and skin. UV light in skin converts the trans form to cis-urocanate which plays an important protective role in skin. Liver is capable of complete catabolism of histidine by a pathway which requires folic acid for the last step, in which glutamate formiminotransferase converts the intermediate N-formiminoglutamate to glutamate, 5,10 methenyl-tetrahydrofolate, and ammonia. Inborn errors have been recognized in all of the catabolic enzymes of histidine. Histidine is required as a precursor of carnosine in human muscle and parts of the brain where carnosine appears to play an important role as a buffer and antioxidant. It is synthesized in the tissue by carnosine synthase from histidine and β-alanine, at the expense of ATP hydrolysis. Histidine can be decarboxylated to histamine by histidine decarboxylase. This reaction occurs in the enterochromaffin-like cells of the stomach, in the mast cells of the immune system, and in various regions of the brain where histamine may serve as a neurotransmitter.
Keywords: carnosine, histamine, histidase, formiminoglutamate, urocanate, 3-methylhistidine
Introduction
Histidine was first isolated from salmon protamine by Albrecht Kossel in 1896 (1). He chose the name histidine from the Greek word histion meaning “tissue.” It is a basic amino acid with an imidazole side chain. The pK for the side chain of the free amino acid is 6.0 so that both the neutral and protonated forms are present at physiological pH. The imidazolium side chain of histidine provides functions which are unavailable to other amino acids, such as general base catalysis in the catalytic triad of serine proteases (2). The proximal and distal histidines of the β-globin chains of hemoglobin also play essential roles in the oxygenation, rather than oxidation, of hemoglobin under physiological conditions (3).
The content of histidine in different proteins can vary from 73% of total amino acids in the histidine-rich protein of Plasmodium lophurae (4) to virtually no histidine in some mammalian elastins (5). Histidine is one of the least abundant amino acids in whole body protein in humans. Tessari (6) calculated the total body protein content of various amino acids in humans. The most abundant were proline (1328 g) and glycine (1247 g), both important in structural proteins, whereas there were only 245 g histidine, second only to tryptophan at 88 g.
In addition to free and protein-bound histidine in the diet, histidine can be obtained from proteolysis of endogenous protein and from hydrolysis of histidine-containing peptides in the diet (Figure 1). Besides its role in protein synthesis, it can be converted to histamine or to carnosine and excess can be catabolized. All of these pathways of histidine will be reviewed.
FIGURE 1.
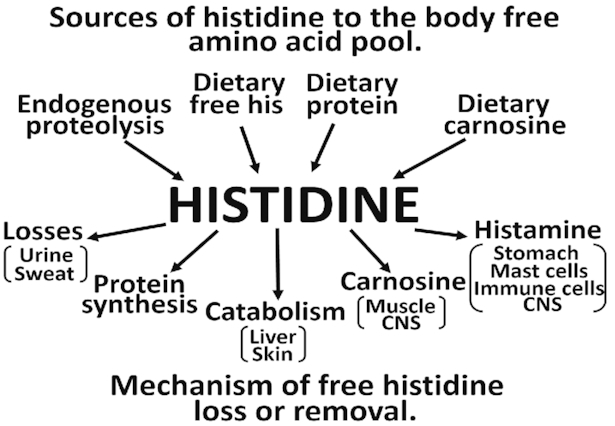
Fates of histidine in the human body. CNS, central nervous system.
Histidine Catabolism
Figure 2 shows the metabolic pathway for histidine metabolism. Histidase (histidine ammonia lyase) is the first and principal regulatory enzyme in the pathway, producing ammonia and trans-urocanate. It is a cytosolic enzyme, principally found in skin and liver, with a Km for histidine in the 1–4 mM range (7). Liver and skin histidases are expressed from the same gene (8). Histidase contains an unusual modified amino acid, dehydroalanine, which is produced from serine (9). Trans-urocanate is nonenzymically converted to cis-urocanate in skin by UV light (270–320 nm), which has led to the suggestion it may serve as a natural protector against sunlight (10). Alternatively, it may play a role in UV-induced immunosuppression (11). A report suggests that cis-urocanate acts on human keratinocytes by generating reactive oxygen species, which in turn result in a transient modulation of epidermal growth factor receptor signaling, followed by induction of PGE2 synthesis and increased apoptotic cell death (12).
FIGURE 2.
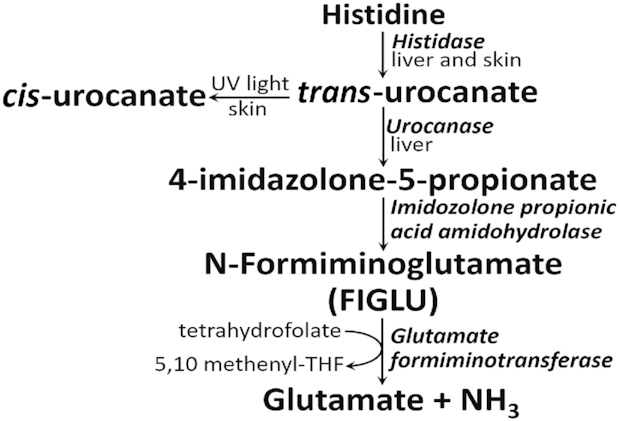
Histidine catabolism in skin and liver. FIGLU, N-formiminoglutamate; THF, tetrahydrofolate.
Patients with histidinemia have blood histidine concentrations which range from 290 to 1420 μM, compared with control subjects who have 70–120 μM histidine (8). When the plasma and tissue histidine concentrations become supraphysiological, histidine can be transaminated to imidazolepyruvic acid which can be detected in the urine of these patients (13). It was thought that there was a specific histidine transaminase, but it has not been possible to isolate such an enzyme; the activity has been reported to copurify with glutamine transaminase in kidney (14) and with serine transaminase in liver (15). Products of histidine transamination are only detected in urine if the histidine concentration is very high, such as in patients with histidinemia.
Trans-urocanate is hydrolyzed in the liver by urocanase to give 4-imidozolone-5-proprionate which, in turn, is converted to formiminoglutamate (FIGLU). Urocanase has a high affinity for its substrate; the human enzyme has a Km of ∼2.2 μM for urocanate (7). The next step involves one-carbon metabolism as the formimino group of FIGLU is transferred to tetrahydrofolate (THF) to produce 5’,10’-methenyl-THF, glutamate, and ammonia. This step couples histidine catabolism to one-carbon metabolism because the 5,10-methenyl-THF formed by histidine catabolism may be metabolized to a variety of products. 5,10-methenyl-THF may be reduced to 5,10-methylene-THF which may be used for synthesis of thymidine or which, in turn, may be reduced to 5-methyl-THF which may methylate homocysteine to methionine. This methionine may be converted to S-adenosylmethionine, the body's principal donor of methyl groups for transmethylation reactions. 5,10-methenyl-THF may also be oxidized to 10-formyl-THF which is used directly for synthesis of purines, such as ATP and GTP (16). The requirement of THF as a substrate for glutamate formiminotransferase implies that folate deficiency could limit histidine catabolism. Evidence for this idea is provided by the increased urinary FIGLU excretion that is found in folate-deficient individuals (17). Glutamate, the other product of glutamate formiminotransferase, is used for many functions, including gluconeogenesis.
In common with many enzymes of amino acid catabolism (18, 19), liver histidase is hormonally regulated. Histidase activity is not apparent in rat liver until 4 d postpartum; thereafter it increases until puberty (8). Sexual maturation in female rats is accompanied by an estrogen-driven doubling in hepatic enzyme activity. Both glucocorticoids and glucagon induce the synthesis of liver histidase (20), as does a high-protein intake or a histidine load (8).
Some thoughts on the relative importance of histidine to one-carbon metabolism
The total flux of one-carbon groups into the canonical 3 products of one-carbon metabolism has been estimated to be ∼26 mmol/d (purines, 5.5; thymidylate, 6.2; methyl groups, 14.5 mmol/d) in humans (16). By comparison, the daily histidine intake is ∼19 and ∼13 mmol/d in men and women, respectively. In the steady state, the flux of amino acids to protein equals that of proteolysis and, assuming that flux of histidine to other products is relatively minor, it might be tempting to conclude that histidine catabolism could provide ≤75% of the necessary one-carbon groups. There are a number of other sources of one-carbon groups, however, of which serine is thought to be the most significant (21). The one-carbon pool should be viewed as being continuously filled from a variety of sources and used for metabolic purposes, with excess one-carbon units being oxidized to carbon dioxide (16). Thus it is likely that histidine contributes a relatively small proportion of the one-carbon groups that go to homocysteine remethylation and synthesis of purines and thymidylate.
Inborn errors of histidine catabolism
Genetic mutations have been reported in 3 enzymes of the histidine catabolic pathway in liver: histidase, urocanase, and glutamate formiminotransferase. All 3 disorders are thought to be relatively benign, although many of the patients have been reported to have mental retardation which may be independent of the enzyme defects.
Histidinemia is the most frequent inborn metabolic error in Japan with an incidence of 1:8400 (22). It is characterized by increased concentrations of histidine in the blood and urine and decreased concentrations of urocanate in blood and skin. It results from decreased activity of the histidase protein. The initial characterization of the condition included mental retardation and speech impairment but it is now apparent that these are diverse phenotypes of this disease, ranging from a benign phenotype in the majority of subjects to classical features, including mental retardation, in the minority of subjects. The original subjects were identified by newborn screening. However, screening for histidinemia is no longer carried out in many countries, including Japan (23), which means that “benign” histidinemia is now very rarely identified (24). Kawai et al. (22) investigated a group of subjects who were identified before screening for histidinemia was eliminated. They identified a number of mutations, including 4 missense mutations and 2 exonic and 2 intronic polymorphisms, in the histidase gene.
Urocanic aciduria is caused by a defect in the enzyme urocanase, which is coded for by the gene urocanate hydratase 1 (UROC1). Kalafatic et al. (25) reported on 2 sisters with normal hepatic histidase activity but who demonstrated urocanase deficiency. The few early cases that were reported all showed mental retardation of unknown etiology (26). Recently, Glinton et al. (27) employed untargeted metabolomics to identify adult siblings with urocanic aciduria; in this case there was no sign of mental retardation. Both subjects were found to be compound heterozygotes for missense variants in UROC1. These authors suggest that urocanase deficiency is a benign disease, unrelated to developmental delay.
Formiminoglutamic aciduria is characterized by increased excretion of FIGLU, due to deficiency of glutamate formiminotransferase (28), coded for by the formimidoyltransferase cyclodeaminase (FTCD) gene. This enzyme is a bifunctional protein (formiminotransferase-cyclodeaminase) in which the first domain releases glutamate and the second domain produces ammonia and 5,10-methenyl-THF. Patients present with various degrees of severity, ranging from mental and physical retardation (29) to relatively mild outcomes (30). Hilton et al. (30) observed 2 missense mutations (R135C; R299P) in the FTCD gene in each of 2 siblings; the mutations both occurred in the first domain of the enzyme. The resultant enzyme activity was ∼60%, which effectively blocked the histidine catabolic pathway and resulted in FIGLU excretion in the urine, even in the absence of supplemental histidine. Because THF is a required cofactor for glutamate formiminotransferase, its activity is very low in patients with folate deficiency and FIGLU consequently also appears in the urine.
In all of these inborn errors, a histidine-deficient diet was tried but the diets did not seem to matter for the relatively benign disorders. In addition, the diets were difficult for caregivers to manage so no dietary treatment is recommended (22). The lack of a specific treatment of histidinemia is the reason newborn screening was stopped some years ago (23).
Methylation of Histidine Residues in Peptide Linkage
The imidazole side chain of histidine in proteins, such as actin, may be methylated in the N-3 position, using S-adenosylmethionine as methyl donor. Recently, Wilkinson et al. (31) isolated an enzyme, SET domain-containing protein 3, which can carry out this methylation and showed that this posttranslational modification was important for smooth muscle contraction. Until now, no function was known for 3-methylhistidine residues. When the modified protein is degraded, the 3-methylhistidine is released intact and is excreted in the urine, providing a useful estimation of muscle protein degradation (32).
Histidine-Containing Dipeptides
Skeletal muscle of most vertebrates contains significant amounts (1–16 g/kg wet muscle) of ≥1 of the histidine-containing dipeptides (carnosine, anserine, and ophidine/balenine) (33). The only one present in human muscle is carnosine, β-alanyl-l-histidine, but most other mammals also contain 1 of the methylated derivatives, anserine or ophidine/balenine. There are significant quantities of histidine-containing dipeptides in most of the meat or fish that humans eat. Histidine-containing dipeptides are not hydrolyzed by regular (di)peptidases, but are hydrolyzed by their own specific hydrolytic enzymes, the carnosinases. Carnosinase 1 occurs in human serum, but not in serum from most other animals. Carnosinase 2 is located intracellularly in intestinal and kidney cells (33). As Figure 3 shows, dietary carnosine is transported into the apical side of intestinal cells by oligopeptide transporter 1 (PEPT1) where it can remain unchanged and enter the blood or it can be hydrolyzed by carnosinase 2 to give histidine and β-alanine. Transporters on the basolateral side of the membrane must be facilitated (passive) because the sodium gradient works in the wrong direction. The transporter for carnosine is unknown (34); PEPT1 is sodium-dependent and is only present on the apical side of the membrane so it cannot transport carnosine on the basolateral side (35). Amino acid transporters most likely come from the solute carrier family, member 7A, but most amino acids are transported by >1 transporter (36). In the kidney tubule, carnosine is transported by oligopeptide transporter 2 (PEPT2), then it is hydrolyzed by carnosinase 2 and the products enter the blood. Very little histidine or β-alanine escape in the urine. If intact carnosine reaches the blood, it will be rapidly hydrolyzed there by carnosinase 1 so that there is minimal carnosine circulating in the human body (34). Most of the carnosine in the human occurs in skeletal muscle where it is synthesized from β-alanine and histidine by the cytoplasmic enzyme carnosine synthase, at the cost of ATP hydrolysis. It is believed that β-alanine, not histidine, is the limiting substrate for carnosine synthesis (37). There is more carnosine in type II or fast-twitch muscle fibers. Definitive functions of carnosine are not known but it has been suggested that the histidine-containing dipeptides may act as intracellular buffers, metal-ion chelators, antioxidants, and/or free radical scavengers (37). It has been reported that β-alanine supplements will increase carnosine in human muscle and improve exercise performance (38). Thus there is considerable interest in carnosine function in the sports fraternity. Adult men, on average, have 33 kg of muscle (39). Skeletal muscle in humans has ∼1 g carnosine/kg wet muscle (33), so the adult male would contain ∼33 g carnosine in his skeletal muscle, which would represent ∼99% of his total body carnosine (33). There is very little, if any, carnosinase activity in muscle (40) so carnosine would need to be transported out of the muscle to the plasma before it could be hydrolyzed to histidine and β-alanine. Proton-coupled oligopeptide transporter 1, a member of the proton-coupled oligopeptide transporter family, is expressed in human skeletal muscle (40) but it is not yet clear how readily it transports carnosine out of muscle (33). When humans are carnosine loaded, loss of carnosine from muscle is slow (2–4%/wk) (41).
FIGURE 3.
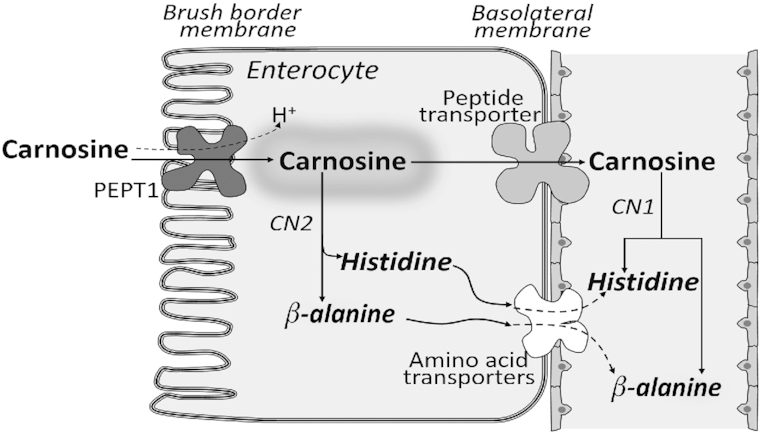
Intestinal handling of dietary histidine-containing dipeptides. CN1, carnosinase 1 in plasma; CN2, carnosinase 2 in cytoplasm of intestinal cells; PEPT1, peptide transporter in brush border of intestine.
Histidine is A Dietary-Essential Amino Acid
It has been reported that rats can grow normally in the absence of dietary histidine if they are supplied with carnosine containing an amount of histidine equimolar to that in a 20%-casein diet (42). Thus histidine can be recovered from carnosine and replace dietary histidine. Histidine was thought to be nonessential in adult humans because it was not possible to show a deficiency in humans who consumed a diet containing purified amino acids for 2 wk, although it was known that infant humans and adults of many other animals do require it (43, 44). It is possible that muscle carnosine gave rise to histidine to allow positive nitrogen retention for the 2 wk of no histidine in the diet. It is now known that histidine is a nutritional requirement for adult humans as well, because they are unable to synthesize it (45). The WHO/FAO requirement for adult men is 10 mg histidine · kg body weight−1 · d−1 (46) so the typical 70-kg individual would require 0.7 g histidine/d in some form in his/her diet.
Histamine
Histidine can be enzymatically decarboxylated to give histamine (Figure 4A). The enzyme involved is histidine decarboxylase (HDC) which requires pyridoxal phosphate as its essential cofactor. HDC has long been known to be localized to mast cells (47) in various tissues and enterochromaffin-like (ECL) cells of the oxyntic mucosa of the stomach (48), but more recently it has also been discovered in the central nervous system (49) and in immune cells (50). Histamine is known as the quintessential inflammatory mediator, giving rise to all aspects of the “triple response” of Lewis (white line, red flare, wheal) in response to injury of the skin (51). Release of histamine from mast cells occurs in response to IgE binding to mast cell membrane receptors as part of the response to allergens. In stomach, histamine has been reported to increase hydrochloric acid secretion by parietal cells (52). HDC activity in ECL cells has been reported to be increased after gastrin treatment, as are histamine synthesis and release from these cells, followed by gastric acid secretion (52). It is not clear whether increased plasma histidine concentrations could lead to increased synthesis of histamine in ECL cells.
FIGURE 4.
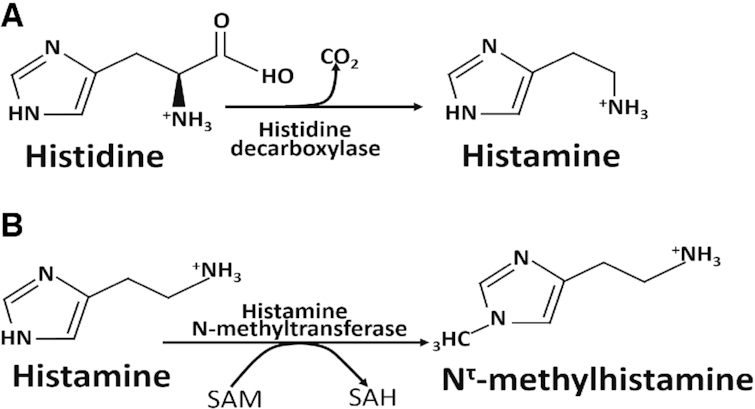
Histidine can be decarboxylated to histamine which may be subsequently methylated to N1-methylhistidine. (A) Conversion of histidine to histamine by histidine decarboxylase. Pyridoxal phosphate is the cofactor. (B) Inactivation of histamine by histamine N-methyltransferase in brain. The methyl donor is SAM, which is converted to SAH. SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
In addition to the role of histamine in gastric acid secretion and the immune response, it also serves as a neurotransmitter in specific regions of the brain (53). Histamine is synthesized in cells of the posterior hypothalamus which have projections in various brain regions (54). In brain of rats, the normal histidine content does not saturate HDC, so an increase in histidine would increase the rate of histamine synthesis (55). Brain histamine controls various functions, such as the sleep–wake cycle, appetite, memory, and stress response (56).
Histamine is removed from synapses by transport into cells and inactivated by histamine N-methyltransferase in the cytoplasm to give N-methylhistidine (Figure 4B). Too little histidine (57), loss-of-function mutation of the HDC gene (58), or too much histamine N-methyltransferase activity (59) would cause low histamine concentrations in brain and neurological symptoms such as anxiety in mice (57) or Tourette syndrome in humans (58). It has recently been suggested that an increase in brain histamine might contribute to the improvement of brain disorders (56).
Conclusion
Histidine metabolism has been studied since histamine was first discovered at the beginning of the previous century. The clinical and biochemical pictures were emphasized in the latter half of the century but there are still many questions to be answered. There is continuing disagreement over how much histidine an adult human needs or can safely ingest. Urinary excretion of 3-methylhistidine has been used as a measure of proteolysis for 50 y (32) but a possible role of the methylated protein has just recently been identified (31) and more work is needed on a possible mechanism. Many protective functions of cis-urocanate in skin have been proposed, but a recent article showed apoptotic cell death due to reactive oxygen species caused by cis-urocanate (12), so is it protective or harmful? Studies on the definitive role of carnosine in muscle (37) are still needed, as are those on a possible role of histamine in providing protection in several brain disorders (56). Thus histidine is a well-known, well-studied amino acid but there is still much to do.
Acknowledgments
The authors’ responsibilities were as follows—both authors: wrote the manuscript, are responsible for its content, and read and approved the final manuscript.
Notes
Supported by Canadian Institutes of Health Research grant MOP-142321 (to MEB).
Author disclosures: The authors report no conflicts of interest.
The 10th Workshop on the Assessment of Adequate and Safe Intake of Dietary Amino Acids was held in Tokyo, Japan, 19-20 November 2019. The Conference was sponsored by the International Council on Amino Acid Science (ICAAS).
This article appears as part of the supplement “10th Amino Acid Assessment Workshop”, sponsored by the International Council on Amino Acid Science (ICAAS). The guest editors of the supplement are D Bier, L Cynober, S Morris, P Stover, M Kadowaki, and R Elango. The travel and accommodation costs of the guest editors and speakers were paid in full by ICAAS. Publication costs for this supplement were defrayed in part by the payment of page charges. The opinions expressed in this publication are those of the authors and are not attributable to the sponsors or the publisher, Editor, or Editorial Board of The Journal of Nutrition.
Abbreviations used: ECL, enterochromaffin-like; FIGLU, N-formiminoglutamate; FTCD, formimidoyltransferase cyclodeaminase; HDC, histidine decarboxylase; PEPT, oligopeptide transporter; THF, tetrahydrofolate; UROC1, urocanate hydratase 1.
Contributor Information
Margaret E Brosnan, Department of Biochemistry, Memorial University of Newfoundland, St John's, Newfoundland, Canada.
John T Brosnan, Department of Biochemistry, Memorial University of Newfoundland, St John's, Newfoundland, Canada.
References
- 1. Jones ME. Albrecht Kossel, a biographical sketch. Yale J Biol Med. 1953;26:80–97. [PMC free article] [PubMed] [Google Scholar]
- 2. Carter P, Wells JA. Dissecting the catalytic triad of serine protease. Nature. 1988;332:564–8. [DOI] [PubMed] [Google Scholar]
- 3. Baldwin J, Chothia C. Haemoglobin: the structural changes related to ligand binding and its allosteric mechanism. J Mol Biol. 1979;129:175–220. [DOI] [PubMed] [Google Scholar]
- 4. Kilejian A. A unique histidine-rich polypeptide from the malaria parasite, Plasmodium lophurae. J Biol Chem. 1974;249:4650–5. [PubMed] [Google Scholar]
- 5. Chalmers GWG, Gosline JM, Lillie MA. The hydrophobicity of vertebrate elastins. J Exp Biol. 1999;202:301–14. [DOI] [PubMed] [Google Scholar]
- 6. Tessari P. Nonessential amino acid usage for protein replenishment in humans: a method of estimation. Am J Clin Nutr. 2019;110:255–64. [DOI] [PubMed] [Google Scholar]
- 7. Coltorti M, Di Simone A, Budillon G. Histidase and urocanase activities of liver and plasma. Correlations between tissue enzyme levels and plasmatic increases during human and mouse viral hepatitis. Clin Chim Acta. 1966;13:568–73. [DOI] [PubMed] [Google Scholar]
- 8. Taylor RG, Levy HL, McInnes RR. Histidase and histidemia. Clinical and molecular considerations. Mol Biol Med. 1991;8:101–16. [PubMed] [Google Scholar]
- 9. Dadova J, Galan SRG, Davis BG. Synthesis of modified proteins via functionalization of dehydroalanine. Curr Opin Chem Biol. 2018;46:71–81. [DOI] [PubMed] [Google Scholar]
- 10. Baden HP, Pathak MA. The metabolism and function of urocanic acid in skin. J Invest Dermatol. 1967;48:11–17. [PubMed] [Google Scholar]
- 11. De Fabo EC, Noonan FP. Mechanism of immune suppression by ultraviolet irradiation in vivo. I. Evidence for the existence of a unique photoreceptor in skin and its role in photoimmunology. J Exp Med. 1983;157:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaneko K, Walker SL, Lai-Cheong J, Matsui MS, Norval M, Young AR. cis-Urocanic acid enhances prostaglandin E2 release and apoptotic cell death via reactive oxygen species in human keratinocytes. J Invest Dermatol. 2011;131:1262–71. [DOI] [PubMed] [Google Scholar]
- 13. Levy HL, Madigan PM, Peneva P. Evidence for delayed histidine transamination in neonates with histidinemia. Pediatrics. 1971;47:128–32. [PubMed] [Google Scholar]
- 14. Noguchi T, Okuno E, Kido R. Identity of rat kidney histidine-pyruvate aminotransferase with glutamine-oxo acid aminotransferase. Biochem J. 1977;161:177–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Noguchi T, Okuno E, Kido R. Identity of isoenzyme 1 of histidine-pyruvate aminotransferase with serine-pyruvate aminotransferase. Biochem J. 1976;159;607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brosnan ME, MacMillan L, Stevens JR, Brosnan JT. Division of labour: how does folate metabolism partition between one-carbon metabolism and amino acid oxidation?. Biochem J. 2015;472:135–46. [DOI] [PubMed] [Google Scholar]
- 17. Luhby AL, Cooperman JM, Teller DN. Urinary excretion of formiminoglutamic acid: application in diagnosis of clinical folic acid deficiency. Am J Clin Nutr. 1954;7:397–406. [DOI] [PubMed] [Google Scholar]
- 18. Lyons RT, Pitot HC. The regulation of ornithine aminotransferase synthesis by glucagon in the rat. Arch Biochem Biophys. 1976;174:262–72. [DOI] [PubMed] [Google Scholar]
- 19. Snodgrass PJ, Lin RC, Müller WA, Aoki TT. Induction of urea cycle enzymes of rat liver by glucagon. J Biol Chem. 1978;253:2748–53. [PubMed] [Google Scholar]
- 20. Aleman G, Ortiz V, Langley E, Tovar AR, Torres N. Regulation by glucagon of the rat histidase gene promoter in cultured rat hepatocytes and human hepatoblastoma cells. Am J Physiol Endocrinol Metab. 2005;289:E172–9. [DOI] [PubMed] [Google Scholar]
- 21. Davis SR, Stacpoole PW, Williamson J, Kick LS, Quinlivan EP, Coats BS, Shane B, Bailey LB, Gregory JF III. Tracer-derived total and folate-dependent homocysteine remethylation and synthesis rates in humans indicate that serine is the main one-carbon donor. Am J Physiol Endocrinol Metab. 2004;286:E272–9. [DOI] [PubMed] [Google Scholar]
- 22. Kawai Y, Moriyama A, Asai K, Coleman-Campbell CM, Sumi S, Morishita H, Suchi M. Molecular characterization of histidemia: identification of four missense mutations in the histidase gene. Hum Genet. 2005;116:340–6. [DOI] [PubMed] [Google Scholar]
- 23. Levy HL, Shih VE, Madigan PM. Routine newborn screening for histidinemia. Clinical and biochemical results. N Engl J Med. 1974;291:1214–9. [DOI] [PubMed] [Google Scholar]
- 24. Brosco JP, Sanders LM, Dharia R, Guez G, Feudtner C. The lure of treatment: expanded newborn screening and the curious case of histidinemia. Pediatrics. 2010;125:417–9. [DOI] [PubMed] [Google Scholar]
- 25. Kalafatic Z, Lipovac K, Jezerinac Z, Juretic D, Dumic M, Zurga B, Res L. A liver urocanase deficiency. Metabolism. 1980;29:1013–9. [DOI] [PubMed] [Google Scholar]
- 26. Espinós C, Pineda M, Martinez-Rubio D, Lupo V, Ormazabal A, Vilaseca MA, Spaapen LJM, Palau F, Artuch. Mutations in the urocanase gene URO1 are associated with urocanic aciduria. J Med Genet. 2009;46:407–11. [DOI] [PubMed] [Google Scholar]
- 27. Glinton KE, Levy HL, Kennedy AD, Pappan KL, Elsea SH. Untargeted metabolomics identifies unique though benign biochemical changes in patients with pathogenic variants in UROC1. Mol Genet Metab Rep. 2019;18:14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perry TL, Applegarth DA, Evans ME, Hansen S. Metabolic studies of a family with massive formiminoglutamic aciduria. Pediatr Res. 1975;9:117–22. [DOI] [PubMed] [Google Scholar]
- 29. Arakawa T. Congenital defects in folate utilization. Am J Med. 1970;48:594–8. [DOI] [PubMed] [Google Scholar]
- 30. Hilton JE, Christensen KE, Watkins D, Raby BA, Renaud Y, de la Luna S, Estivill X, MacKenzie RE, Hudson TJ, Rosenblatt DS. The molecular basis of glutamate formiminotransferase deficiency. Hum Mut. 2003;22:67–73. [DOI] [PubMed] [Google Scholar]
- 31. Wilkinson AW, Diep J, Dai S, Liu S, Ooi YS, Song D, Li T-M, Horton JR, Zhang X, Liu C et al. SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature. 2019;565:372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Munro HN, Young VR. Urinary excretion of NT-methylhistidine (3-methylhistidine): a tool to study metabolic responses in relation to nutrient and hormonal status in health and disease of man. Am J Clin Nutr. 1978;31:1608–14. [DOI] [PubMed] [Google Scholar]
- 33. Boldyrev AA, Aldini G, Derave W. Physiology and pathophysiology of carnosine. Physiol Rev. 2013;93:1803–45. [DOI] [PubMed] [Google Scholar]
- 34. Aldini G, Orioli M, Carini M, Facino RM. Profiling histidine-containing dipeptides in rat tissues by liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2004;39:1417–28. [DOI] [PubMed] [Google Scholar]
- 35. Jappar D, Hu Y, Keep RF, Smith DE. Transport mechanisms of carnosine in SKPT cells: contribution of apical and basolateral membrane transporters. Pharm Res. 2009;26:172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bröer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 2008;88:249–86. [DOI] [PubMed] [Google Scholar]
- 37. Sale C, Saunders B, Harris RC. Effect of beta-alanine supplementation on muscle carnosine concentrations and exercise performance. Amino Acids. 2010;39:321–33. [DOI] [PubMed] [Google Scholar]
- 38. Trexler ET, Smith-Ryan AE, Stout JR, Hoffman JR, Wilborn CD, Sale C, Kreider RB, Jäger R, Earnest CP, Bannock L et al. International society of sports nutrition position stand: beta-alanine. J Int Soc Sports Nutr. 2015;12:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Janssen I, Heymsfield SB, Wang Z, Ross R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J Appl Physiol. 2000;89:81–8. [DOI] [PubMed] [Google Scholar]
- 40. Everaert I, De Naeyer H, Taes Y, Derave W. Gene expression of carnosine-related enzymes and transporters in skeletal muscle. Eur J Appl Physiol. 2013;113:1169–79. [DOI] [PubMed] [Google Scholar]
- 41. Baguet A, Reyngoudt H, Pottier A, Everaert I, Callens S, Achten E, Derave W. Carnosine loading and washout in human skeletal muscles. J Appl Physiol. 2008;106:837–42. [DOI] [PubMed] [Google Scholar]
- 42. Tamaki N, Funatsuka A, Fujimoto S, Hama T. The utilization of carnosine in rats fed on a histidine-free diet and its effect on the levels of tissue histidine and carnosine. J Nutr Sci Vitaminol (Tokyo). 1984;30:541–51. [DOI] [PubMed] [Google Scholar]
- 43. Stifel FB, Herman RH. Is histidine an essential amino acid in man?. Am J Clin Nutr. 1972;25:182–5. [DOI] [PubMed] [Google Scholar]
- 44. Holt LE., Jr Some problems in dietary amino acid requirements. Am J Clin Nutr. 1968;21:367–75. [DOI] [PubMed] [Google Scholar]
- 45. Elango R, Ball RO, Pencharz PB. Amino acid requirements in humans: with a special emphasis on the metabolic availability of amino acids. Amino Acids. 2009;37:19–27. [DOI] [PubMed] [Google Scholar]
- 46. FAO. Protein and amino acid requirements in human nutrition. Report of a joint WHO/FAO/UNU expert consultation. WHO technical report series, no. 935 Geneva (Switzerland): WHO; 2007. [Google Scholar]
- 47. Hirasawa N. Expression of histidine decarboxylase and its roles in inflammation. Int J Mol Sci. 2019;20:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aures D, Håkanson R, Schauer A. Histidine decarboxylase and DOPA decarboxylase in the rat stomach. Properties and cellular localization. Eur J Pharmacol. 1968;3:217–34. [DOI] [PubMed] [Google Scholar]
- 49. Schartz JC, Lampart C, Rose C. Histamine formation in rat brain in vivo: effects of histidine loads. J Neurochem. 1972;19:801–10. [DOI] [PubMed] [Google Scholar]
- 50. Best J, Nijhout HF, Samaranayake S, Hashemi P, Reed M. A mathematical model for histamine synthesis, release, and control in varicosities. Theor Biol Med Model. 2017;14:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Greaves MW, Sabroe RA. Histamine: the quintessential mediator. J Dermatol. 1996;23:735–40. [DOI] [PubMed] [Google Scholar]
- 52. Andersson K, Chen D, Mattsson H, Sundler F, Håkanson R. Physiological significance of ECL-cell histamine. Yale J Biol Med. 1998;71:183–93. [PMC free article] [PubMed] [Google Scholar]
- 53. Haas HL, Sergeeva OA, Selbach O. Histamine in the central nervous system. Physiol Rev. 2008;88:1183–241. [DOI] [PubMed] [Google Scholar]
- 54. Watanabe T, Taguchi Y, Shiosaka S, Tanaka J, Kubota H, Terano Y, Tohyama M, Wada H. Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as marker. Brain Res. 1984;295:13–25. [DOI] [PubMed] [Google Scholar]
- 55. Prell GD, Hough LB, Khandelwal J, Green JP. Lack of a precursor-product relationship between histamine and its metabolites in brain after histidine loading. J Neurochem. 1996;67:1938–44. [DOI] [PubMed] [Google Scholar]
- 56. Yoshikawa T, Nakamura T, Yanai K. Histamine N-methyltransferase in the brain. Int J Mol Sci. 2019;20:737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yoshikawa T, Nakamura T, Shibakusa T, Sugita M, Naganuma F, Iida T, Miura Y, Mohsen A, Harada R, Yanai K. Insufficient intake of L-histidine reduces brain histamine and causes anxiety-like behaviors in male mice. J Nutr. 2014;144:1637–41. [DOI] [PubMed] [Google Scholar]
- 58. Baldan LC, Williams KA, Gallezot J-D, Pogorelov V, Rapanelli M, Crowley M, Anderson GM, Loring E, Gorczyca R, Billingslea E et al. Histidine decarboxylase deficiency causes Tourette syndrome; parallel findings in humans and mice. Neuron. 2014;81:77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Naganuma F, Nakamura T, Yoshikawa T, Iida T, Miura Y, Karpati A, Matsuzawa T, Yanai A, Mogi A, Mochizuki T et al. Histamine N-methyltransferase regulates aggression and the sleep-wake cycle. Sci Rep. 2017;7:15899. [DOI] [PMC free article] [PubMed] [Google Scholar]