

Abstract
Nosocomial infections are common in pediatric patients and can be fatal in infants and immunocompromised patients. In September 2018, a high positive rate of human adenovirus HAdV was occurred among hospitalized children in the Children’s Hospital Affiliated to the Capital Institute of Paediatrics in Beijing. To investigate whether this outbreak of HAdV was related to nosocomial infections or the result of community infections, we collected respiratory specimens from patients with acute respiratory infections in a respiratory ward during June to December 2018, and screened for respiratory viruses. Among 1,840 cases included, 95 (5.2%, 95/1840) were positive for HAdV and 81 were genotyped based on phylogenetic analysis, including seven as HAdV-1 (8.6%), 30 HAdV-3 (37.0%), two HAdV-6 (2.5%), and 42 HAdV-7 (51.9%). More HAdV-positive samples were collected in August (4.7%, 12/255), September (15.0%, 41/274) and October (6.9%, 17/247), with a peak in September 2018. By combining the results of HAdV phylogenetic analysis with clinical data of patients, there were 77 cases (4.2%, 77/1840; 81.1%, 77/95) excluded from nosocomial infections, eight cases representing possible infections transmitted by visitors or attending parents, three cases without sequences that might have been due to infection transmitted by roommates positive for HAdV, one case of a roommate without an HAdV sequence, and six cases that shared highly homologous sequences with those of their roommates, for which nosocomial infections might be considered. In conclusion, genotyping of HAdVs based on phylogenetic analysis combined with clinical information provides a powerful method to distinguish nosocomial infections from community acquired infection, especially when tracing the origins of nosocomial infections.
Electronic supplementary material
The online version of this article (10.1007/s12250-020-00299-y) contains supplementary material, which is available to authorized users.
Keywords: Acute respiratory tract infection, Human adenovirus (HAdV), Phylogenetic analysis, Nosocomial infection
Introduction
Hospital-acquired, or nosocomial, infections often represent a sensitive topic, which are the basis of medical disputes, especially with respect to respiratory infections (Wen et al. 2019). However, nosocomial viral respiratory infections, accounting for 10% of healthcare-related infections, are common in pediatric patients and could be fatal for infants with background diseases and immune-compromised patients (Rutledge-Taylor et al. 2012; Quach et al. 2018). To reduce the incidence of nosocomial infection, point of care testing, commercially available for influenza virus, respiratory syncytial virus (RSV), or human adenovirus, is performed for outpatients with acute respiratory tract infections (ARTIs) before their admission (Ding et al. 2017). For patients positive for the same virus, not only standard precaution but also isolation management strategies should be adopted, such as assigning them to a single room or in a cohort, whereas for patients with negative results, general rooms with multiple patients might be chosen (Kobayashi et al. 2019).
Human adenovirus (HAdV) is a common and important viral pathogen causing ARTIs. Some types of HAdV, such as HAdV3 and HAdV7, can cause outbreaks, especially in infants and immune-compromised people, which can result in rapidly progressive consolidation of lung tissues with severe or even fatal respiratory inflammation (Lion 2014), followed by acute respiratory distress syndrome (Hung and Lin 2015) and respiratory failure (Lai et al. 2013). There have been more than 100 subtypes of HAdV (~ 103 types) reported to date, belonging to seven different groups (A–G). Among them, types 1–52 (serotypes) were identified through serum neutralization tests or hemagglutination inhibition tests, whereas types 53–103 (genotypes) were classified by bioinformatics analysis. In general, HAdV group B (HAdV-3, 7, 11, 14, 16, 21, 34, and 35), C (HAdV-1, 2, 5, and 6), and E (HAdV-4) are associated with respiratory tract infections (Sun et al. 2018; Xu et al. 2018; Dhingra et al. 2019). Further, HAdV has an average incubation period of 3–8 days (Lessler et al. 2009).
In previous studies, HAdV infections have been sporadic throughout the year in Beijing, China, and the positive rate of HAdV in hospitalized children is approximately 2.08% annually, as determined by direct immunofluorescence antigen tests (Deng et al. 2013). However, the positive rate of HAdV among hospitalized children from the Children’s Hospital Affiliated to the Capital Institute of Paediatrics in Beijing, China in September 2018 increased rapidly to nearly five-fold higher levels than the previous annual positive rates, with a few severe cases leading to death, suggesting a local outbreak of HAdV or hinting at the occurrence of HAdV nosocomial infections. To confirm whether this outbreak of HAdV was a result of a community outbreak or was related to nosocomial infections, we combined genotyping of HAdVs based on phylogenetic analysis and clinical information to distinguish pediatric nosocomial HAdV infection from community acquired infection.
Materials and Methods
Ward Setting
This study was carried on at the Department of Respiratory Medicine, Children’s Hospital Affiliated to the Capital Institute of Paediatrics, a tertiary hospital in Beijing, China which is situated on the same floor and consists of 13 wards. These 13 wards include 70 beds; the first 12 wards have five beds each (No. 1–60), and the 13th ward has eight beds (No. 61–68) along with two additional beds (No. 69 and 70) outside the door used temporarily in peak season with space limitations. Each ward contains a shared bathroom and a common balcony. Attending parents and visitors are allowed in these wards.
Clinical Specimens
From June to December 2018, 3 months prior to and 3 months after September 2018 (as the intermediate point), throat swabs, nasopharyngeal swabs (NSs), nasopharyngeal aspirates (NPAs), and bronchoalveolar lavage fluids were collected from pediatric patients with ARTI admitted to the Department of Respiratory Medicine, Affiliated Children’s Hospital, Capital Institute of Pediatrics for respiratory virus screening and were subjected to direct immunofluorescence (DFA) tests (Diagnostic Hybrids, Athens, OH, USA) for antigen testing or NxTAG™ RPP assays (Luminex Molecular Diagnostics Inc., Toronto, Canada) for nucleic acid detection. Specimens were immediately stored at 4 °C and sent to the laboratory within 12 h. Upon arrival at the laboratory, each clinical specimen was handled in a Class II bio-safety cabinet and processed immediately using 2.5 mL of viral transport medium (Yocon Biotechnology Co., Ltd, Beijing, China) and then centrifuged (500 × g, 10 min).
For DFA, cell pellets from all NPA and NS specimens were re-suspended and spotted onto an acetone-cleaned slide. Then, individual monoclonal antibody reagents, labeled with fluorescein isothiocyanate (FITC) against RSV, HAdV, influenza virus (Flu) A and B, and parainfluenza virus (PIV) 1–3 were used for virus identification. Supernatants were stored, and some of the specimen was used for nucleic acid extraction.
For NxTAG™ RPP assays, supernatants from all specimens were treated for nucleic acid extraction using MS-2 bacteriophage as an internal control. Extracts were tested for Flu A (with additional subtyping for H1, H3, and H5 variants) and Flu B, RSV A and B, PIV1–4, HAdV, human metapneumovirus, coronaviruses 229E, NL63, OC43, and HKU1, enterovirus/human rhinovirus, and human bocavirus (HBoV) according to the manufacturer’s instructions in a 96-well plate format. The plate was then analyzed using Luminex xPONENT, and the resultant data were analyzed with the Luminex SYNCT Data Analysis Software (Luminex). A mean fluorescence intensity value above the threshold level for a particular target indicated a positive result for that target. For specimens positive for HAdV, confirmed by one of these two respiratory virus screening methods, genotyping and phylogenetic analyses of HAdV were performed.
Viral DNA Extraction
Viral DNA was extracted from 140 µL of each collected HAdV-positive specimen using the QIAamp® Viral RNA Mini Kit (250) (Qiagen, Germany) according to the manufacturer’s instructions. Remaining specimens were stored at − 80 °C for further use.
Polymerase Chain Reaction (PCR) and Phylogeny-Based Classification of HAdV DNA
All DNA positive for HAdV was tested by PCR to amplify a 301-bp fragment from the hexon gene using the primer pair HAdV-HEX1885 (5′-TGCCCCAATGGTC-3′) and HAdV-HEX1913 (5′-ATCCGCGGGGTGCT-3′) (Allard et al. 2001; Allard et al. 1990; Liu et al. 2009). The PCR conditions were as follows: denaturation at 94 for 5 min, followed by 45 cycles at 94 °C for 40 s, 53 °C for 40 s, and 72 °C for 40 s, with a final extension at 72 °C for 7 min. Then, PCR products were purified, and sequences were determined using the Sanger sequencing method by Sino Geno Max Co., Ltd. (Beijing, China).
To supplement the limitation of using only the hexon for subtyping of HAdV in identifing recombinants, three pairs of universal PCR primers that target the variable regions of the three major capsid genes, i.e. hexon, penton base and fiber genes from bioRxiv preprint (Zhang et al. 2020) were also used to amplify and get sequences of hexon, penton base and fiber genes under the permission of Prof. Qiwei Zhang.
The sequences were then subjected to phylogenetic analysis with NCBI Blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&BLAST_PROGRAMS=megaBlast&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome) to identify their HAdV types.
Phylogenetic Analysis of Viral Homology
Phylogenetic analysis, conducted using MEGA version X software (Molecular Evolutionary Genetics Analysis Version X), was carried on to construct phylogenetic trees based on the sequences collected in the study and those from the GenBank database (numbered with their GenBank number) as controls using the neighbor-joining method and maximum composite-likelihood model. Bootstrap values were estimated with 1000 replications to assess the reliability of the individual nodes in each phylogenetic tree (Tamura and Suzuki 2004; Saitou and Nei 1987; Kumar et al. 2018).
Clinical Data Collection
The medical records from the pediatric patients with confirmed HAdV-positivity were reviewed retrospectively. The following clinical data were extracted from the records: date of admission, date of discharge, bed number (bed No.), ward number (ward No.), date of sample collection.
Results
Genotyping of HAdV
Figure 1 presents a flow chart showing the strategy of virus screening and case inclusion or exclusion. In this study, a total of 2,262 respiratory tract specimens were collected from pediatric patients with ARTI who were admitted to the Department of Respiratory Medicine from June to December 2018, of which 105 specimens were positive for HAdV, as determined by DFA and/or RPP assays. After removing duplicates, a total of 1,840 cases were included for further analysis, including 266 patients admitted to the hospital in June, 246 in July, 255 in August, 274 in September, 247 in October, 262 in November, and 290 in December (Fig. 2A). Among these, 95 were HAdV-positive (5.2%, 95/1840), of which only 81 individual cases, including 56 males (69.1%) and 25 females (30.9%), were genotyped for HAdV by PCR combined with phylogenetic analysis for viral homology of 301-bp fragments of the hexon gene, including seven cases positive for HAdV1 (8.6%, 7/81), 30 for HAdV3 (37.0%, 30/81), two for HAdV6 (2.5%, 2/81), and 42 for HAdV7 (51.9%, 42/81); 14 individual cases were not subjected to phylogenetic analysis due to insufficient PCR products for DNA sequencing.
Fig. 1.

The flow chart of the critical of cases inclusion or exclusion and distinguishing nosocomial HAdV infection from community infection.
Fig. 2.
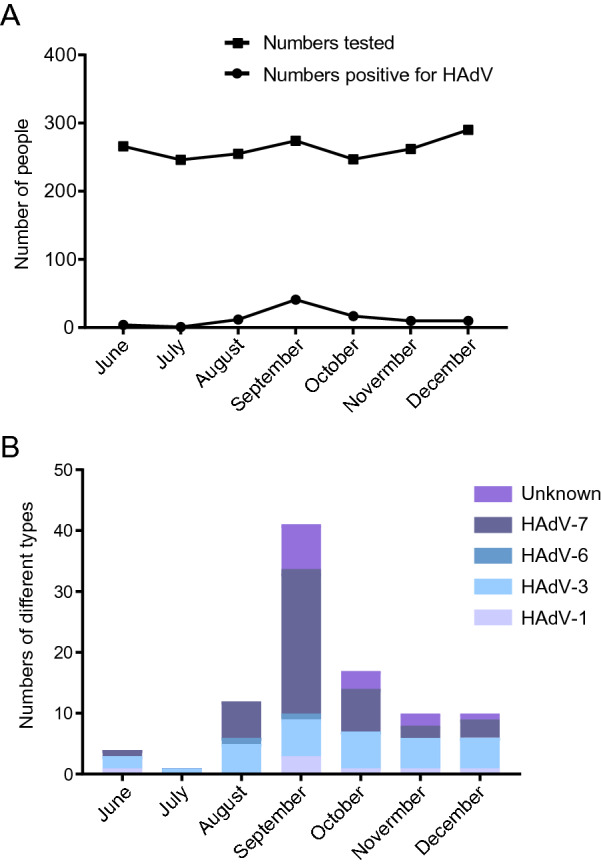
Monthly distributions of HAdV positive cases during June to December, 2018 in the study. A Numbers tested or Numbers positive for HAdV; B Numbers of case positive for different HAdV types.
Using three pairs of universal PCR primers that target the variable regions of the three major capsid genes, we obtained 79 sequences of the penton gene, 75 sequences of the hexon gene, and 66 sequences of fiber gene (Supplementary Figure S1) among those 81 individual cases with 301-bp hexon gene fragments. There were two cases (1W799 and D1476) with three capsid gene sequences belonging to different HAdV types, which might have been due to infection by recombinants or co-infection by different HAdV types, one case (D1909) without any sequence corresponding to those three capsid genes, and 78 cases with sequences in accordance with the genotyping results of the 301-bp hexon gene fragments. Therefore, we selected the genotyping results of the 301-bp fragments of the hexon gene for further analysis.
Monthly Distribution of HAdV-Positive Cases
In June, there were four cases (1.5%, 4/266) of HAdV infection, including one with HAdV1, two with HAdV3, and one with HAdV7, whereas there was only one case (0.4%, 1/246) of HAdV3 infection in July. However, cases of HAdV infection increased rapidly in August with 12 cases (4.7%, 12/255), including five HAdV3, one HAdV6, and six HAdV7, as well as in September with 41 cases (15.0%, 41/274), including three HAdV1, six HAdV3, one HAdV6, 23 HAdV7, and eight unknown. Thereafter, HAdV infection cases decreased gradually, with 17 cases (6.9%, 17/247) in October, consisting of one HAdV1, six HAdV3, seven HAdV7, and three unknown, 10 cases (3.8%, 10/262) in November, including one HAdV1, five HAdV3, two HAdV7, and two unknown, and 10 cases (3.4%, 10/290) in December, comprising one HAdV1, five HAdV3, three HAdV7, and one unknown (Fig. 2B).
Phylogenetic Analysis of Viral Homology Combined with Clinical Information
Based on phylogenetic trees of the 301-bp fragment of the hexon gene, sequences of seven cases were typed as HAdV1 in three clusters, 30 as HAdV3 in five clusters, two as HAdV6 in two clusters, and 42 as HAdV7 in eight clusters (Fig. 3). According to clinical information and data of phylogenetic analysis, those 95 cases, including 81 with sequences in phylogenetic trees and 14 without sequences, were grouped step-by-step as shown in Fig. 1. There were 46 cases of patients admitted to the Department of Respiratory Medicine for the first time and confirmed to be HAdV-positive within 48 h after admission, including 40 in phylogenetic trees (in blue, Fig. 3; 1W799, D1476, and D1909 were included) and six without sequences, whereas there were 22 cases with specimens collected within 48 h after admission with more than 8 days isolation from the previous discharging, including 17 in phylogenetic trees (in green, Fig. 3) and five without sequences. All of these 68 (71.6%, 68/95) cases were not considered nosocomial infections.
Fig. 3.

Phylogenetic trees of 301 bp fragments of hexon gene constructed using MEGA X software to identify the genotypes of HAdV-positive specimens. Sequences from the study were named as “No. of specimens–Date of admission–Date of discharging–No. of ward and bed–Typing of HAdV”. Sequences from GenBank were labelled with their GenBank number- typing of HAdV, which were in different clusters (Cluster 1–8) in different HAdV types. Blue: sequences from cases admitted to the Department of Respiratory Medicine for the first time and were confirmed to be HAdV positive within 48 h after admission. Green: sequences from cases with specimens collected in 48 h after the admission with over 8 days isolation from the previous admission. Orange: sequences from cases who were admitted over one time and confirmed with sequences in different sero- or geno-type or in different clusters of one type from their roommates positive for HAdV during their previous admission. Yellow: sequences from cases confirmed to be HAdV after 48 h from admission shared no homology sequence with their roommates. Red: sequences from case owning a roommate positive for HAdV, but without HAdV sequence. Purple: sequences from cases with HAdV sequences shared high homology with that of roommates in the previous or this admission.
Among cases of patients admitted to the Department of Respiratory Medicine more than once, there were nine cases (in orange, Fig. 3), including five cases with 7 days, two cases with 6 days, one case with 4 days, and one case with 3 days of isolation from the previous discharging. These were confirmed to be associated with sequences of different genotypes or different clusters of one type, as compared to those of their HAdV-positive roommates during the previous admissions of these nine patients. Therefore, these nine (9.5%, 9/95) patients might have been infected by the community. Among cases confirmed as HAdV 48 h after admission, there were eight cases (in yellow, Fig. 3) with no homologous sequences shared with those of their roommates, which implied that these patients might have been infected by visitors or attending parents. However, there were three cases from other wards with sequences sharing high homology with those of the eight cases. For these eight patients, most (5/8) might have been infected during Aug 15 to Sep 12.
There were 10 remaining cases, for which nosocomial infections could not be excluded. Among them, there were three groups. In group one, there were three cases without sequences that might have indicated infection by roommates positive for HAdV. In group two, there was one case (in red, Fig. 3) with a roommate positive for HAdV, but without an HAdV sequence. In group three, there were six cases (in purple, Fig. 3) with HAdV sequences that shared high homology with those of roommates during their previous or current admission, which was supported by the high homology of three capsid gene sequences shared with those of their roommates (Supplementary Figure S1). For these 10 cases, most (6/10) might have also been infected during Aug 15 to Sep 12 (Table 1).
Table 1.
Information of the ten cases for whom nosocomial infection were considered.
| Groups | No. | The previous admission | The admission | Date confirmed as HAdV positive | Sero- or Geno-type of HAdV | Cluster | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Date of admission | Date of discharge | No. of bed | Date of admission | Date of discharge | No. of bed | |||||
| 1 | D1334 | – | – | – | 8/18 | 9/7 | 66, 23, 21 | 9/3 | Unknown | Unknown |
| D1834 | – | – | – | 11/27 | 12/5 | 46 | 12/2 | Unknown | Unknown | |
| D1380 | 8/26 | 9/8 | 42 | 9/11 | 10/22 | 59 | 9/11 | Unknown | Unknown | |
| 2 | D1873 | – | – | – | 11/30 | 12/18 | 44 | 12/10 | HAdV7 | 1 |
| 3 | 1w300 | – | – | – | 9/26 | 10/19 | 69, 9 | 10/11 | HAdV7 | 1 |
| D1376 | – | – | – | 9/4 | 9/19 | 48 | 9/11 | HAdV7 | 1 | |
| 99533 | 9/3 | 9/7 | 30, 8 | 9/11 | 9/27 | 63 | 9/11 | HAdV3 | 1 | |
| D1399 | 8/28 | 9/7 | 34 | 9/13 | 9/22 | 69 | 9/13 | HAdV7 | 1 | |
| D1419 | 9/1 | 9/12 | 31 | 9/16 | 10/9 | 8 | 9/16 | HAdV7 | 1 | |
| D1656 | 10/13 | 10/19 | 31 | 10/26 | 11/7 | 70 | 10/26 | HAdV7 | 1 | |
| Groups | Possible date of infection | Nosocomial infection was considered | Possible sources of infection | |||||
|---|---|---|---|---|---|---|---|---|
| No. | Sero- or Geno-type of HAdV | Cluster | Date of admission | Date of discharge | No. of bed | |||
| 1 | 8/18–9/3 | Yes | 99180 | HAdV-7 | 1 | 8/15 | 9/7 | 61 |
| 98638 | HAdV-6 | 1 | 8/10 | 8/23 | 68 | |||
| 11/27–/12/2 | Yes | D1806 | Unknown | Unknown | 11/27 | 12/11 | 50 | |
| 8/26–/9/8 | Yes | D1302 | HAdV-7 | 5 | 8/16 | 9/4 | 44 | |
| 2 | 11/30–12/10 | Yes | D1806 | Unknown | Unknown | 11/27 | 12/11 | 50, 40 |
| 3 | 9/26–10/10 | Yes | 99943 | HAdV-7 | 1 | 9/26 | 10/8 | 70 |
| 9/4–9/11 | Yes | D1341 | HAdV-7 | 1 | 9/1 | 9/8 | 49 | |
| 9/3–9/7 | Yes | 99416 | HAdV-3 | 1 | 9/5 | 9/8 | 32, 8 | |
| 8/28–9/6 | Yes | 99058 | HAdV-7 | 1 | 8/27 | 9/8 | 34, 9 | |
| 9/1–9/12 | Yes | 99547 | HAdV-7 | 1 | 9/11 | 9/10 | 33, 6 | |
| 10/13–10/19 | Yes | 1w671 | HAdV-7 | 1 | 10/11 | 10/24 | 34 | |
Discussion
HAdV infections and outbreaks usually occur worldwide year-round and are mainly sporadic, although higher rates of HAdV infections have sometimes been reported in the winter and spring in northern China and in the spring and summer in southern China (Li et al. 2015). However, in the present study, an outbreak of HAdV was observed among children from a respiratory ward of a children’s hospital in northern China in August (4.7%, 12/255), September (15.0%, 41/274), and October (6.9%, 17/247), with a peak in September 2018. Among over 100 types of HAdVs, some have tropism for the respiratory tract, especially HAdV types 3, 4, 7, 14, 21, and 55. Moreover, HAdV-7, and in particular the genomic variant HAdV-7d, has recently emerged in the US and is more commonly associated with severe respiratory disease with a higher mortality rate than that with other HAdV types (Potter et al. 2012). In this report, among those types identified, such as HAdV1, HAdV3, HAdV6, and HAdV7, HAdV 3 (37.0%, 30/81) and 7 (51.9%, 42/81) were the major pathogens, which was consistent with the results of previous studies indicating that HAdV7 and HAdV3 were the most prevalent (Chen et al. 2015; Lu et al. 2019).
As a type of non-enveloped double-stranded DNA virus, HAdVs can persist in the environment for a long time. Moreover, additional factors, such as close living quarters, arduous mental and physical settings, and difficulty maintaining personal hygiene, contribute to the transmission and associated morbidity of HAdVs (Bautista-Gogel et al. 2020). Therefore, an analysis of an unusual increase of HAdV detected in the respiratory ward in this study indicated if nosocomial infection caused by HAdV had occurred. To answer this question, genotyping of HAdV based on a phylogenetic analysis was combined with clinical information for nosocomial infection identification.
Based on clinical data only, 68 among 95 cases were positive for HAdVs, including 46 admitted to the respiratory ward for the first time and confirmed to be HAdV-positive within 48 h after admission, as well as 22 cases with specimens collected 48 h after admission with more than 8 days between isolation and the previous admission, for which HAdV infections were confirmed as community acquired. It has been assumed that if the HAdV infection associated with the second case is transmitted from the first case, the viral nucleic acids of the two samples should naturally fall within the same cluster, whereas if the viral nucleic acids of two samples exhibit evolutionary distance from each other, they would not have originated from the same source and would be grouped into different clusters (Liu et al. 2009). Combined with the genotyping of HAdV based on phylogenetic analysis, there were nine cases confirmed with sequences of different genotypes or in different clusters when compared to sequences from HAdV-positive roommates during a previous admission, for which HAdV infections were also confirmed to be community acquired. Therefore, 77 (4.2%, 77/1840; 81.1%, 77/95) cases were excluded from nosocomial infections, which revealed that the majority of cases in the local significant increase in patients with adenovirus infection were not associated with nosocomial infection.
Combining HAdV genotyping based on phylogenetic analysis with clinical information, there were eight cases of patients that might have been infected by visitors or attending parents, whereas there were 10 cases, including three without sequences, of patients that might have been infected by roommates positive for HAdV, including one with a roommate without an HAdV sequence and six sharing highly homologous sequences with their roommates; for these, nosocomial infections might be considered. For these 18 cases, most (11/18) might have been infected during Aug 15 to Sep 12, which implied a period of careless ward management. As indicated previously herein, more HAdV infections are detected in the winter and spring in northern China (Li et al. 2015), whereas more restricted methods are used to reduce the incidence of nosocomial infection (Ding et al. 2017). However, standard precaution and isolation management for pediatric patients with respiratory symptoms, even if they have negative results in rapid tests, should be adopted. There are many transmission routes for HAdV, including inhaling air from an infected person sneezing or coughing, through direct physical contact with carriers, or touching a surface contaminated by HAdV, among others. Therefore, as the guideline of the Healthcare Infection Control Practices Advisory Committee recommends, isolation from other patients, in addition to standard precaution, is recommended for patients with respiratory infection if possible (Civardi et al. 2013; Kobayashi et al. 2019; Mao et al. 2019). In addition, attending parents were observed to be walking around the corridors without protective masks and clothing, which might increase their chance of becoming a carrier of infection that could be transmitted to other wards (Dare and Talbot 2016; Topoulos et al. 2019). Therefore, strict supervision of the nursing system, regular disinfection of the floor, armrests, chairs, and air in the ward, and preventing viral infections among healthcare workers and caregivers could be helpful to reduce nosocomial infections (Wang et al. 2019). Kobayashi et al. (2019) used a similar strategy to retrospectively investigate two episodes of nosocomial infection by HBoV and human rhinovirus in a pediatric ward in a tertiary hospital in Japan. More data should be accumulated to evaluate the practical value of applying this strategy in clinical treatment.
In conclusion, genotyping of HAdVs based on phylogenetic analysis and that combined with clinical information provides a powerful method to distinguish nosocomial infection from community infection, especially when tracing the origin of the nosocomial infection. In this study, most cases were confirmed as community acquired HAdV infection, which implied a significant increase in patients with adenovirus infection observed. However, there were 18 cases for which nosocomial infection could not be excluded, indicating the requirement of more strict measures to prevent nosocomial infection.
There were several limitations to this study. There were 14 cases that lacked HAdV sequences, which resulted in four cases that could not be excluded from nosocomial infections. HAdV infection results in varied long-term incubation, which means some cases might have been confirmed as HAdV-positive beyond 48 h after admission. Viral transmission caused by visitors also made the analysis more complicated. Moreover, the results indicated that only 301 bp sequence of hexon gene is not enough to accurately determine HAdV type or identify recombinants, which should be supplemented by phylogenetic analysis of sequences of hexon, penton base and fiber genes.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants from Beijing Municipal Commission of Health and Family (No. 2060399 PXM2017_026268_00005_ 00254486) and The Pediatric Medical Coordinated Development Center of the Beijing Hospitals Authority (No. XTZD20180505).
Author Contributions
LZ and LC conceived and designed the manuscript. CY, JD, RZ, FW and YS performed laboratory work. CZ, LY and BZ analyzed clinical data. CY, LZ, and YQ analyzed the laboratory data. FW, DC, QG and YZ contributed reagents/materials/analysis tools. CY and LZ wrote the paper. YQ, LC and LZ reviewed the manuscript. All authors have read and approved the final manuscript.
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interest.
Animal and Human Rights Statement
This study was approved by the Institutional Review Board of the Capital Institute of Pediatrics (SHERLLM 2015 012). Clinical samples were collected from children with ARTIs for routine respiratory virus tests with consent from parents or legal guardians. The residual samples were used for HAdV sequencing in this study. Additional informed consent was obtained from all patients for which identifying information is included in this article.
Contributor Information
Ling Cao, Email: caoling9919@163.com.
Linqing Zhao, Email: linqingz525@163.com.
References
- Allard A, Girones R, Juto P, Wadell G. Polymerase chain reaction for detection of adenoviruses in stool samples. J Clin Microbiol. 1990;28:2659–2667. doi: 10.1128/jcm.28.12.2659-2667.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard A, Albinsson B, Wadell G. Rapid typing of human adenoviruses by a general PCR combined with restriction endonuclease analysis. J Clin Microbiol. 2001;39:498–505. doi: 10.1128/JCM.39.2.498-505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista-Gogel J, Madsen CM, Lu X, Sakthivel SK, Froh I, Kamau E, Gerber SI, Watson JT, Cooper SS, Schneider E. Outbreak of respiratory illness associated with human adenovirus type 7 among persons attending officer candidates school, Quantico, Virginia, 2017. J Infect Dis. 2020;221:697–700. doi: 10.1093/infdis/jiz060. [DOI] [PubMed] [Google Scholar]
- Chen M, Zhu Z, Huang F, Liu D, Zhang T, Ying D, Wu J, Xu W. Adenoviruses associated with acute respiratory diseases reported in Beijing from 2011 to 2013. PLoS ONE. 2015;10:e0121375. doi: 10.1371/journal.pone.0121375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civardi E, Tzialla C, Baldanti F, Strocchio L, Manzoni P, Stronati M. Viral outbreaks in neonatal intensive care units: what we do not know. Am J Infect Control. 2013;41:854–856. doi: 10.1016/j.ajic.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dare RK, Talbot TR. Health care-acquired viral respiratory diseases. Infect Dis Clin North Am. 2016;30:1053–1070. doi: 10.1016/j.idc.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Qian Y, Zhao L, Zhu R, Sun Y, Tian R. Identification and typing of adenovirus from acute respiratory infections in pediatric patients in Beijing from 2003 to 2012. Bing Du Xue Bao. 2013;29:615–620. [PubMed] [Google Scholar]
- Dhingra A, Hage E, Ganzenmueller T, Böttcher S, Hofmann J, Hamprecht K, Obermeier P, Rath B, Hausmann F, Dobner T, Heim A. Molecular evolution of human adenovirus (HAdV) species C. Sci Rep. 2019;9:1039. doi: 10.1038/s41598-018-37249-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding YX, Tian R, Qian Y, Sun Y, Deng J, Wang F, Zhu RN, Zhao LQ. Clinical value of a rapid respiratory syncytial virus antigen detection in point-of-care testing. ZhongHua Er Ke Za Zhi. 2017;55:139–143. doi: 10.3760/cma.j.issn.0578-1310.2017.02.017. [DOI] [PubMed] [Google Scholar]
- Hung KH, Lin L. Adenovirus pneumonia complicated with acute respiratory distress syndrome: a case report. Medicine. 2015;94:e776. doi: 10.1097/MD.0000000000000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Shinjoh M, Sudo K, Kato S, Morozumi M, Koinuma G, Takahashi T, Takano Y, Tamura Y, Hasegawa N. Nosocomial infection by human bocavirus and human rhinovirus among paediatric patients with respiratory risks. J Hosp Infect. 2019;103:341–348. doi: 10.1016/j.jhin.2019.05.002. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CY, Lee CJ, Lu CY, Lee PI, Shao PL, Wu ET, Wang CC, Tan BF, Chang HY, Hsia SH, Lin JJ, Chang LY, Huang YC, Huang LM , Taiwan Pediatric Infectious Disease Alliance Adenovirus serotype 3 and 7 infection with acute respiratory failure in children in Taiwan, 2010–2011. PLoS ONE. 2013;8:e53614. doi: 10.1371/journal.pone.0053614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessler J, Reich NG, Brookmeyer R, Perl TM, Nelson KE, Cummings DA. Incubation periods of acute respiratory viral infections: a systematic review. Lancet Infect Dis. 2009;9:291–300. doi: 10.1016/S1473-3099(09)70069-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou W, Zhao Y, Wang Y, Xie Z, Loou Y, Tan W. Molecular typing and epidemiology profiles of human adenovirus infection among paediatric patients with severe acute respiratory infection in China. PLoS ONE. 2015;10:e0123234. doi: 10.1371/journal.pone.0123234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lion T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev. 2014;27:441–462. doi: 10.1128/CMR.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LY, Zhang Y, Deng J, Qian Y. Developing of a method for detection and identification of entero-adenovirus in fecal specimens from infantile diarrhea. ZhongHua Jian Yan Yi Xue Za Zhi. 2009;32:857–860. [Google Scholar]
- Lu G, Shi W, Zhang D, Liu Y, Zhang L, Sun Y, Yang P, Wang Q. The epidemiological and etiological features of human adenovirus outbreaks among children in Beijing in 2016. ZhongGou Yu Fang Yi Xue Za Zhi. 2019;20:21–26. [Google Scholar]
- Mao N, Zhu Z, Rivailler P, Yang J, Li Q, Han G, Yin J, Yu D, Sun L, Jiang H, Zhan Z, Xiang X, Mei H, Wang X, Zhang B, Yu P, Li H, Lei Z, Xu W. Multiple divergent human mastadenovirus C co-circulating in mainland of China. Infect Genet Evol. 2019;76:104035. doi: 10.1016/j.meegid.2019.104035. [DOI] [PubMed] [Google Scholar]
- Potter RN, Cantrell JA, Mallak CT, Gaydos J. Adenovirus-associated deaths in US military during postvaccination period, 1999–2010. Emerg Infect Dis. 2012;18:507–509. doi: 10.3201/eid1803.111238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach C, Shah R, Rubin LG. Burden of healthcare-associated viral respiratory infections in children’s hospitals. J Pediatric Infect Dis Soc. 2018;7:18–24. doi: 10.1093/jpids/piw072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge-Taylor K, Matlow A, Gravel D, Embree J, Le Saux N, Johnston L, Suh K, Embil J, Henderson E, John M, Roth V, Wong A, Shurgold J, Taylor G, Program Canadian Nosocomial Infection Surveillance. A point prevalence survey of health care-associated infections in Canadian pediatric inpatients. Am J Infect Control. 2012;40:491–496. doi: 10.1016/j.ajic.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sun J, Xiao Y, Zhang M, Ao T, Lang S, Wang J. Serum inflammatory markers in patients with adenovirus respiratory infection. Med Sci Monit. 2018;24:3848–3855. doi: 10.12659/MSM.910692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Suzuki K. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topoulos S, Giesa C, Gatermann S, Fussen R, Lemmen S, Ewig S. Analysis of acute respiratory infections due to influenza virus A, B and RSV during an influenza epidemic 2018. Infection. 2019;47:425–433. doi: 10.1007/s15010-018-1262-x. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhou KH, Chen W, Yu Y, Feng SF. Epidemiology and risk factors for nosocomial infection in the respiratory intensive care unit of a teaching hospital in China: a prospective surveillance during 2013 and 2015. BMC Infect Dis. 2019;19:145. doi: 10.1186/s12879-019-3772-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen S, Lv F, Chen X, Zhu L, Li H, Lin L, Zhang H. Application of a nucleic acid-based multiplex kit to identify viral and atypical bacterial aetiology of lower respiratory tract infection in hospitalized children. J Med Microbiol. 2019;68:1211–1218. doi: 10.1099/jmm.0.001006. [DOI] [PubMed] [Google Scholar]
- Xu L, Liu J, Liu C, Duan Y, Zhu Y, Xu B, Xie Z. Case-control study of the epidemiological and clinical features of human adenovirus 55 and human adenovirus 7 infection in children with acute lower respiratory tract infections in Beijing, China, 2008–2013. BMC Infect Dis. 2018;18:634. doi: 10.1186/s12879-018-3520-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Sridhar S, Lau Susanna KP, Liu J, Ou J, Yan Y, Zhao S, Lan W, Guan W, Wu J, Woo Patrick CY, Seto D, Zhang Q. Molecular typing of human respiratory adenoviruses with universal PCR and sequencing primers for three major capsid genes: penton base, hexon, and fiber. BioRxiv. 2020 doi: 10.1101/2020.03.17.995373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.