

Abstract
Despite major advances in cancer treatment, pancreatic cancer is still incurable and the treatment outcomes are limited. The aggressive and therapy-resistant nature of pancreatic cancer warrants the need for novel treatment options for pancreatic cancer management. Drug repurposing is emerging as an effectual strategy in the treatment of various diseases, including cancer. In the present study, we evaluated the anticancer effects of pimavanserin tartrate (PVT), an antipsychotic drug used for the treatment of Parkinson disease psychosis. PVT significantly suppressed the proliferation and induced apoptosis in various pancreatic cancer cells and gemcitabine-resistant cells with minimal effects on normal pancreatic epithelial cells and lung fibroblasts. Growth-suppressive and apoptotic effects of PVT were mediated by the inhibition of the Akt/Gli1 signaling axis. The oral administration of PVT suppressed subcutaneous and orthotopic pancreatic tumor xenografts by 51%–77%. The chronic administration of PVT did not demonstrate any general signs of toxicity or change in behavioral activity of mice. Our results indicate that pancreatic tumor growth suppression by PVT was orchestrated by the inhibition of Akt/Gli1 signaling. Since PVT is already available in the clinic with an established safety profile, our results will accelerate its clinical development for the treatment of patients with pancreatic cancer.
Keywords: pimavanserin, pancreatic cancer, Akt, stem cells, drug repurposing, orthotopic tumors, Gli1, c-Myc, apoptosis
Graphical Abstract
Drug repurposing, a unique drug developmental strategy, can overcome the pitfalls for pancreatic cancer treatment. In this study, we discover the anticancer effects of an antipsychotic drug, pimavanserin, for pancreatic cancer. Our pre-clinical findings suggest that pimavanserin could be translated into the clinic to improve the treatment outcome of pancreatic cancer patients.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive and devastating diseases. Despite the modern era in cancer research, pancreatic cancer has proven to be one of the most fatal malignancies, making it the fourth leading cause of cancer-related deaths in developed countries, including the United States.1 Owing to the poor prognosis of patients diagnosed with advanced pancreatic cancer, the median survival of pancreatic cancer patients is estimated to be ∼6 months.2 The multifactorial nature and aberrant high frequencies of mutations make it more fatal and resistant to current therapies. The success of conventional chemotherapies is also dismal, and patients succumb to its debilitating effects. Novel treatment strategies are thus required for the management of this lethal disease.3
The repurposing of existing drugs is considered as an effective drug developmental strategy to counteract the high attrition rates, exorbitant costs, and time-consuming factors involved in the traditional drug-discovery process.4 Drug repurposing has the potential to surpass several challenges associated with de novo drug discovery and guarantees quick clinical trials due to the already-established pharmacokinetics, tolerability, safety, and toxicity profile of the drug.5 Antipsychotic drugs such as pimozide, penfluridol, fluspirilene, haloperidol, olanzapine, chlorpromazine, and reserpine have been demonstrated to exert antineoplastic effects in various cancer models.6, 7, 8, 9, 10, 11 In this study, we have repurposed pimavanserin tartrate (PVT) as a novel therapeutic option for pancreatic cancer. PVT was approved by the US Food and Drug Administration (FDA) in 2016 for the treatment of Parkinson disease psychosis (PDP).
Among several oncogenic signaling pathways, PI3K (phosphatidylinositol 3-kinase)/Akt signaling is a hallmark of pancreatic cancer, and a vital survival pathway used by cancer cells for their survival and proliferation.12,13 PI3K/Akt signaling confers malignant phenotypes and significantly drives pancreatic cancer progression by acting upstream to various oncogenic signaling pathways. The role of PI3K/Akt signaling in invasion, metastasis, resistance to apoptosis, chemotherapy, and radiation therapy has been well defined in several cancer models, including pancreatic cancer.13, 14, 15
The aberrant activation of Sonic Hedgehog (SHH) signaling was observed in 70% of PDAC tisues.3,16 Gli1 is a downstream effector zinc finger transcription factor in the SHH signaling pathway, the overexpression of which contributes to poor prognosis and survival in patients with resected PDAC.17,18 Gli1 also plays a role in promoting invasion, metastasis, chemoresistance, and epithelial-mesenchymal transition in various cancers, including pancreatic cancer.8,18, 19, 20 Moreover, SHH signaling imparts stem cell properties to pancreatic cancer cells by the Gli1 activation of pluripotency-maintaining factors Oct-4, SOX2, NANOG, and c-Myc.8,13,21,22 Non-canonical activation of Gli1 by PI3K/Akt signaling has been recently identified as a potential target for therapy in renal cell carcinoma.23 In addition, Akt /Gli1 signaling axis has been strongly implicated in cancer progression.8 Targeting PI3K/Akt and SHH signaling suppresses tumor growth and inhibits cancer stem cell characteristics.13,24
In the present study, we evaluated the effects of PVT in pancreatic cancer. Our results indicate that PVT treatment suppressed the growth of various pancreatic cancer cells by inhibiting the Akt/Gli1 signaling axis. The oral administration of PVT significantly suppressed the growth of xenograft and orthotopically implanted pancreatic tumors without exhibiting any general signs of toxicity or apparent changes in behavioral activity. To the best of our knowledge, this study is the first to demonstrate the anticancer effects of PVT.
Results
PVT Inhibits the Proliferation of Pancreatic Cancer Cells
The treatment of human AsPC1, BxPC3, MIAPaCa2, PANC1, and murine PO2 pancreatic cancer cells with increasing concentrations of PVT at 24, 48, and 72 h resulted in the significantly reduced survival of cells in a concentration- and time-dependent manner. The half-maximal inhibitory concentration (IC50) of PVT ranged from 3 to 9 μM after 24, 48, and 72 h of treatment (Figures 1A–1E). However, no significant effect was observed in normal human pancreatic ductal epithelial (HPDE-6) cells and the normal lung fibroblast MRC5 with PVT treatment. For example, treatment with 5 μM PVT for 48 h suppressed 62% of MIAPaCa2 cell survival, whereas under similar conditions, the survival of HPDE-6 and MRC5 cells was the least affected (Figure 1F). These results showed that PVT suppresses the proliferation of pancreatic cancer cells and it is relatively non-toxic to normal cells.
Figure 1.

PVT Suppresses the Survival of Pancreatic Cancer Cells
(A–E) Cytotoxic effects of PVT in (A) AsPC1, (B) BxPC3, (C) MIAPaCa2, (D) PANC1, and (E) PO2 cells. (F) Comparison of the cytotoxic effects of PVT between MIAPaCa2, HPDE-6 and MRC5 lung fibroblast cells treated with 5, 7.5, and 10 μM PVT for 48 h. Cell survival was determined by SRB assay to estimate the IC50 values. The experiments were repeated independently 3 times with 4 replicates in each experiment. (G–I) Anticlonogenic effects of PVT in (G) AsPC1, (H) BxPC3, and (I) PANC1 cells. The colony-forming effects of PVT were assessed by clonogenic assay. The experiments contained2 replicates in each experiment. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001.
PVT Inhibits the Colony Formation of Pancreatic Cancer Cells
The proliferative and propagating ability of pancreatic cancer cells after PVT treatment was evaluated by clonogenic assay. We chose sub-toxic concentrations of PVT as determined by sulforhodamine B (SRB) assay. AsPC1, BxPC3, and PANC1 cells were treated with 2.5 and 5 μM PVT for 48 h. Our results indicated that PVT inhibited 40%–65% of the colony-forming ability of pancreatic cancer cells when treated with 5 μM PVT (Figures 1G–1I). These results indicate the anticlonogenic effects of PVT in pancreatic cancer cells.
PVT Induces Apoptosis in Pancreatic Cancer Cells
To determine the mechanism of the antiproliferative effects of PVT, apoptosis induction by PVT treatment was determined in AsPC1, BxPC3, MIAPaCa2, and PANC1 pancreatic cancer cells by annexin V/fluorescein isothiocyanate (FITC) assay. As shown in Figures 2A–2D, the treatment of AsPC1, BxPC3, MIAPaCa2, and PANC1 cells with varying concentrations of PVT (5, 7.5, and 10 μM) for 48 h resulted in significantly increased apoptosis. The treatment of AsPC1 and BxPC3 cells with 7.5 μM PVT caused a 2-fold increase in apoptotic cells (Figures 2A and 2B), whereas in MIAPaCa2 and PANC1 cells, a 7- and a 4-fold increase, respectively, in apoptotic cells was observed (Figures 2C and 2D). The treatment of BxPC3 and PANC1 cells with 10 μM PVT further increased apoptosis (Figures 2B and 2D). However, under similar experimental conditions, PVT did not significantly induce apoptosis in HPDE-6 cells (Figure 2E). The induction of apoptosis by PVT treatment in AsPC1, BxPC3, MIAPaCa2, and PANC1 cells was confirmed by the cleavage of caspase 3 and cleaved poly (ADP-ribose) polymerase (PARP) (Figures 3A–3D).
Figure 2.

PVT Induces Apoptosis in Pancreatic Cancer Cells in a Concentration-Dependent Manner
(A–E) Apoptotic effects of PVT in (A) AsPC1, (B) BxPC3, (C) MIAPaCA2, (D) PANC1, and (E) HPDE-6 cells. The percentage of apoptotic cells was evaluated by annexin V/FITC apoptosis assay using the Accuri C6 flow cytometer. The experiments were repeated independently 2–3 times. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001.
Figure 3.

PVT Inhibits Akt/Gli1 Signaling
(A–D) Western blot analysis of pAkt(Ser473), Akt, Gli1, Oct-4, SOX2, NANOG, c-Myc, cleaved caspase 3, and cleaved PARP in (A) AsPC1, (B) BxPC3, (C) MIAPaCa2, and (D) PANC1 cells treated with 5, 7.5, and 10 μM PVT for 48 h. β-Actin was used as a loading control. The figures shown are the representative blots of at least 3 independent experiments. The blots were developed using the Optimax X-Ray film processor . (E) Immunofluorescence analysis of pAkt(Ser473) (FITC, green), nucleus (DAPI, blue), phalloidin (TRITC, red) in AsPC1 cells treated with 2.5 μM PVT for 48 h, Scale bars, 200μM. (F) Gli1 activity was determined by Gli1 reporter assay in AsPC1 cells. G) Images of tumorspheres formed from PANC1 cells with or without PVT treatment. Scale bars, 1,000 μM. ∗p ≤ 0.05.
Inhibition of Akt/Gli1 Signaling and CSC-like Traits by PVT
Since we observed significant growth-suppressive effects and the induction of apoptosis by PVT treatment, we wanted to elucidate the molecular mechanism by which PVT mediated the above-mentioned effects. We performed a western blot analysis using whole-cell lysates from AsPC1, BxPC3, MIAPaCa2, and PANC1 cells treated with 0, 5, 7.5, and 10 μM PVT for 48 h. Our results showed that PVT treatment significantly inhibited the expression of Akt and its activation by suppressing its phosphorylation at Ser473 in a concentration-dependent manner in all of the cell lines tested (Figures 3A–3D). Interestingly, the expression of Gli1 was also significantly reduced in a concentration-dependent manner with PVT treatment (Figures 3A–3D). Gli1 is a downstream effector and transcription factor in SHH signaling.17 Previous studies have reported that Gli1 activates cancer stem cell (CSC) markers Oct-4, SOX2, NANOG, and c-Myc. Interestingly, we observed remarkable inhibition in the expression of Oct-4, SOX2, NANOG, and c-Myc by PVT treatment (Figures 3A–3D). A significant cleavage of caspase 3 and PARP was observed in a concentration-dependent manner in pancreatic cancer cells after 48 h of PVT treatment (Figures 3A–3D). In addition, immunofluorescence analysis revealed that AsPC1 cells treated with 2.5 μM PVT for 48 h significantly suppressed the levels of pAkt(Ser473) (Figure 3E). Furthermore, the reduced expression of Gli1 was confirmed by performing the Gli1 luciferase reporter assay in AsPC1 cells. Our results indicated that PVT reduced the Gli1 luciferase activity in a concentration-dependent manner. For example, 5 μM PVT suppressed 72% of the Gli1 reporter activity (Figure 3F). CSC traits have been widely observed in tumorspheres formed from cancer cells.25 The treatment of PANC1 cells with 2.5 and 5 μM PVT suppressed the size and number of PANC1 tumorspheres (Figure 3G). These results show that the antiproliferative effects and apoptosis-inducing effects of PVT were mediated by the inhibition of the Akt/Gli1 signaling cascade.
Inhibiting or Silencing Akt Potentiates the Effects of PVT
Akt, a serine/threonine protein kinase, activates Gli1 in a ligand-independent manner. To establish Akt as a target of PVT in pancreatic cancer, Akt was pharmacologically inhibited by PI3K inhibitor LY294002 in MIAPaCa2 cells. Interestingly, blocking the phosphorylation of Akt at Ser473 by LY294002 resulted in the suppression of Gli1 expression and its downstream targets Oct-4, NANOG, and c-Myc (Figure 4A). These results indicate the regulation of Gli1 by Akt in our model. In addition, Akt inhibition alone increased the cleavage of caspase 3. The effects of PVT in suppressing pAkt(Ser473) and its downstream targets Gli1, Oct-4, NANOG, and c-Myc were substantially enhanced in MIAPaCa2 cells pre-treated with LY294002 (Figure 4A). Similarly, the enhanced cleavage of caspase 3 was observed with PVT treatment in MIAPaCa2 cells pre-treated with LY294002 (Figure 4A). In another experiment, Akt was knocked down using Akt small interfering RNA (siRNA) in AsPC1 cells. Akt siRNA inhibited the expression of Akt and its phosphorylation at Ser473. Silencing Akt induced apoptosis, which was indicated by the cleavage of caspase 3 and PARP. The effects of PVT in reducing the phosphorylation of Akt at Ser473 and the expression of Akt and Gli1 were augmented in cells in which Akt was silenced using Akt siRNA (Figure 4B). Moreover, apoptosis-inducing effects of PVT were significantly increased in Akt-silenced cells, as indicated by the enhanced cleavage of caspase 3 and PARP (Figure 4B). These results indicate that the growth-suppressive effects of PVT in pancreatic cancer cells were mediated by inhibiting Akt/Gli1 signaling.
Figure 4.

Akt Inhibition Enhances the Effects of PVT in Pancreatic Cancer Cells
(A) Western blot analysis of pAkt(Ser473), Akt, Gli1, Oct-4, NANOG, c-Myc and cleaved caspase 3 in MIAPaCa2 cells pre-treated with 50 μM PI3K inhibitor LY294002 and then treated with 7.5 μM PVT for 48 h. (B) Western blot analysis of pAkt(Ser473), Akt, Gli1, cleaved caspase 3, and cleaved PARP in AsPC1 cells transfected with 100 nM Akt siRNA and then treated with 7.5 μM PVT for 48 h. (C) Western blot analysis of Gli1, Oct-4, c-Myc, and cleaved caspase 3 in BxPC3 cells pre-treated with 30 μM Gli1 inhibitor GANT-61 and then treated with 7.5 μM PVT for 48 h. (D) Western blot analysis of Gli1, Oct-4, NANOG, c-Myc, and cleaved caspase 3 in MIAPaCa2 cells transfected with 100 nM Gli1 siRNA and then treated with 7.5 μM PVT for 48 h. (E) Whole-cell lysates of Gli1+/+ and Gli1−/− MEFs were analyzed by western blotting. β-Actin or β-tubulin was used as a loading control.
Gli1 Inhibition Enhances the Effects of PVT
GANT-61, a pharmacological inhibitor of Gli1, significantly inhibited the expression of CSC markers Oct-4 and c-Myc, and enhanced the cleavage of caspase 3 in BxPC3 cells (Figure 4C). Notably, Gli1 and its downstream effector molecules SOX2 and NANOG were found to be reduced in Gli1−/− mouse embryonic fibroblasts (MEFs) as compared to Gli1+/+ MEFs (Figure 4E). These results indicate the potential role of Gli1 in regulating the pancreatic CSC properties. Interestingly, the increased suppression of Gli1, Oct-4, and c-Myc expression was observed with PVT treatment in BxPC3 cells pre-treated with GANT-61 (Figure 4C). A similar trend was observed in the induction of apoptosis as evaluated by the cleavage of caspase 3 (Figure 4C). In another experiment, Gli1 was genetically knocked down by Gli1 siRNA in MIAPaCa2 cells. PVT suppressed Gli1 and its downstream targets Oct-4, NANOG, and c-Myc, and the enhanced cleavage of caspase 3 in Gli1 knocked down MIAPaCa2 cells (Figure 4D). These results prove the role of Gli1 signaling in PVT-mediated growth suppression of pancreatic cancer cells.
PVT Suppresses the Growth of Subcutaneously Implanted Pancreatic Tumors
To examine the in vivo efficacy of PVT and to establish the mechanism of tumor growth inhibition in pancreatic cancer, BxPC3 cells were implanted subcutaneously in female athymic nude mice. Once the tumor volume reached 70 mm3, mice were randomized and an experimental group of mice were treated every day with 10 mg/kg PVT by oral gavage. Our results showed that at the day of sacrifice, the average tumor volume of control group was 873 mm3, whereas in the treatment group, it was 429 mm3, indicating a 51% tumor growth suppression by PVT (Figure 5A). Tumor lysates were analyzed by western blotting. Our results showed that the phosphorylation of Akt at Ser473 and the expression of Gli1 were reduced in the tumors obtained from PVT-treated mice. However, the protein level of Akt remained unchanged. In addition, PVT treatment increased the cleavage of PARP in pancreatic tumors (Figure 5B). Tumor sections were analyzed by immunohistochemistry (IHC) and TUNEL assay. The IHC analysis indicated that PVT treatment decreased pAkt(Ser473), Gli1, Oct-4, and NANOG, and increased the cleavage of PARP (Figure 5C). As evaluated by TUNEL assay, a significant induction of apoptosis was observed in PVT-treated tumors (Figure 5D). The levels of PVT in tumors and pancreas were determined by liquid chromatography-tandem mass spectrometry (LC/MS-MS). After oral administration of 10 mg/kg PVT for 28 days, 0.35 μg PVT per gram tumor, 0.48 μg PVT per gram pancreas, and 0.31 μg PVT per gram brain were observed (Tables S2–S4). The plasma concentration of PVT was estimated to be 0.0618 μg/ml (Table S1). Moreover, in a National Institute of Mental Health Data Archive (NDA) pharmacology and toxicology study (NDA no. 207318), the concentration of PVT in various organs was estimated 72 h post-administration of a single oral dose of 30 mg/kg PVT in male Long Evans rats.26 Changes in mice weight and organ weight are considered parameters for assessing the general signs of toxicity after treatment with any drug. Mice weight during the course of the experiment and the weight of organs such as pancreas, liver, brain, and kidney were recorded after being removed at the day of sacrifice. Our results indicated that mice weight and organ weight remain unchanged with the chronic administration of PVT (Figure S1).
Figure 5.

PVT Suppresses the Growth of Pancreatic Tumors in BxPC3 Subcutaneous Xenograft Model
(A) Oral administration of 10mg/kg PVT everyday suppressed the growth of subcuateous BxPC3 tumor xenografts. (B) Western blot analysis of pAkt(Ser473), Akt, Gli1, and cleaved PARP in BxPC3 tumor lysates. β-Actin was used as a loading control. The protein expression in the control and treatment groups was normalized with the respective β-actin of each group. Each blot or FFPE represents a tumor from an individual mouse (B, C and D). Individual FFPE tumor tissues were sectioned and analyzed for (C) IHC and (D) TUNEL assays. Scale bars, 200 µm . The statistical significance level was considered to be p ≤ 0.05.
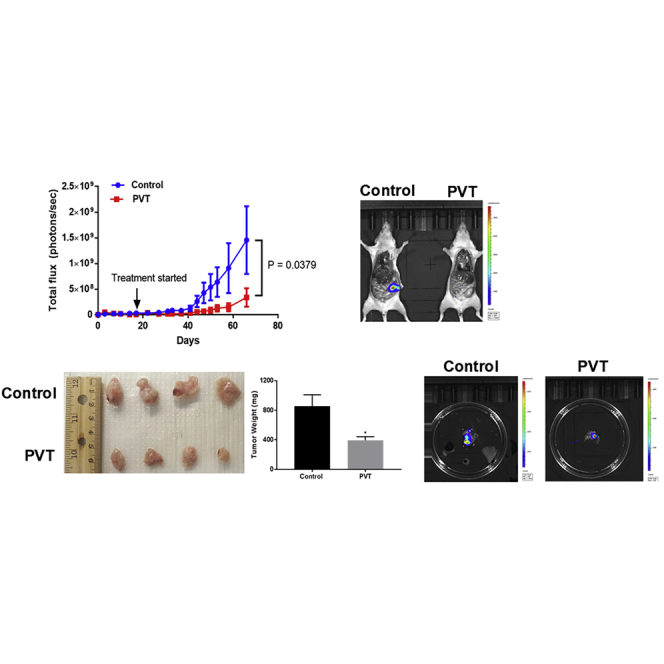
PVT Inhibits the Growth of Orthotopic Pancreatic Tumors
To mimic the clinically relevant condition and further validate the in vivo efficacy of PVT, an orthotopic pancreatic xenograft experiment was performed. PANC-1 luc cells were orthotopically implanted in the pancreas of athymic nude mice. After 17 days of tumor implantation, mice were randomized, and the treatment group of mice received 10 mg/kg PVT by oral gavage every day. Our results showed a constant increase in luminescence in the pancreas of control mice, whereas no significant increase in luminescence was observed in the pancreas of PVT-treated mice. At the end of the experiment, the total flux in control mice was 1.46 × 109 photons per second, while in the treated group, it was observed to be 3.38 × 108 photons per second, indicating a 77% decrease in luminescence with PVT treatment when compared to controls. These results suggest that PVT significantly suppressed orthotopic pancreatic tumor growth (Figure 6A). After terminating the experiment, tumors were removed and weighed. The average weight of PVT-treated tumors was 54% less when compared to the tumors excised from the control group (Figure 6B). Furthermore, we compared the luminescence in the pancreas isolated from both groups. Our results clearly indicated that PVT suppressed 80% of the luminescence in the pancreas, which further validated our observations made in Figure 6A (Figure 6C). PANC1-luc tumor lysates and tumor sections were subjected to western blotting and IHC analysis, respectively. Our results indicated that the level of pAkt(Ser473), Gli1, and the downstream targets SOX2, NANOG, and c-Myc were clearly reduced in the tumors from PVT-treated mice, whereas the cleavage of caspase 3 and PARP was increased with PVT treatment (Figures 6D and 6E). Significant apoptosis was observed in PVT-treated pancreatic tumors as evaluated by TUNEL assay (Figure 6F). Long-term administration of PVT did not change the overall mice weight and organ weight (Figure S2). PVT is an antipsychotic drug and acts as an inverse agonist for 5-HT2A receptors. Owing to its action on the central nervous system, we assessed the behavioral activity of mice after long-term PVT administration. The behavioral activity of mice was monitored using VersaMax (Accuscan Instruments) after 49 days of 10 mg/kg PVT treatment. Our results demonstrated that PVT treatment did not affect behavioral activity determinants such as total distance, horizontal activity, vertical activity, and ambulatory activity when compared to control group mice (Figures S5A–S5D). These results show that the chronic administration of PVT did not alter the behavioral activity of mice.
Figure 6.

PVT Suppresses the Growth of Orthotopically Implanted Pancreatic Tumors
(A) PANC1-luc cells were orthotopically implanted on the pancreas of athymic nude mice, and after 18 days of tumor implantation, mice were treated with 10mg/kg PVT everyday. Tumor luminescence was measured approximately twice per week and plotted against days (n = 5). Representative images of mice from control and PVT-treated groups at the day of sacrifice. (B) Orthotopic pancreatic tumors were aseptically removed, and the tumor weight was compared between control and PVT-treated groups. (C) Individual tumors from control and PVT-treated groups were imaged for bioluminescence. (D) Excised pancreas/tumors were homogenized, lysed, and analyzed for Gli1, SOX2, NANOG, c-Myc, and cleaved caspase 3 by western blotting. β-Actin was used as a loading control. The protein expression in the control and treatment groups was normalized with the respective β-actin of each group. Each lane of blot represents a tumor from an individual mouse. Individual FFPE pancreas/tumors were processed, sectioned, and analyzed for (E) IHC and (F) TUNEL assays. Scale bars, 200 µm. The statistical significance level was considered to be p ≤ 0.05. ∗p ≤ 0.05.
PVT Inhibits the Growth of Gemcitabine-Resistant Tumor Cells
Gemcitabine-resistant MIAPaCa2 (MIAPaCa2-GR) cells were evaluated for resistance to gemcitabine using the SRB assay. MIAPaCa2-GR cells exhibited a 50-fold higher resistance to gemcitabine when compared to sensitive MIAPaCa2 cells (Figure 7A). Notably, MIAPaCa2-GR cells, when treated with increasing concentrations of PVT, showed a decrease in cell proliferation in a concentration- and time-dependent manner. The IC50 was 4.5–5 μM after 24, 48, and 72 h of PVT treatment (Figure 7B). Our mechanistic analysis indicated that PVT suppressed the levels of pAkt(Ser473), Akt, Gli1, and the downstream effector molecules SOX2 and c-Myc. The increase in the cleavage of caspase 3 and PARP with PVT treatment indicated apoptosis (Figure 7C). We evaluated the efficacy of PVT in vivo by implanting MIAPaCa2-GR cells in athymic nude mice. Our results revealed that the oral administration of 10 mg/kg PVT suppressed 54% of the tumor growth when compared to the control group (Figure 7D). The weight of the excised tumors at the end of the experiment revealed a 55% reduction in tumor weight, which further validated our observations (Figure 7E). Our western blotting and IHC analysis indicated that PVT treatment decreased the levels of pAkt(Ser473), Akt, and Gli1, and increased the cleavage of caspase 3 and PARP as an indicator of apoptosis in gemcitabine-resistant tumors. Overall mice weight and weight of organs such as pancreas and brain remained unchanged (Figure S3). In addition, PVT did not alter the clinical chemistry parameters such as alanine transaminase (ALT), aspartate transaminase (AST), albumin, calcium and total serum protein, phosphorous, glucose, blood urea nitrogen (BUN), and creatinine (Figure S4). These results indicated that PVT was equally effective in suppressing the growth of gemcitabine-resistant cells in vitro and in vivo as compared to gemcitabine-sensitive tumors and did not exhibit any general signs of toxicity.
Figure 7.

PVT Inhibits the Growth of Gemcitabine-Resistant Cells In Vitro and In Vivo
(A) Cytotoxic effects of gemcitabine in MIAPaCa2 and MIAPaCa2-GR cells. (B) Cytotoxicity of PVT in MIAPaCa2-GR cells. (C) Whole-cell lysates of MIAPaCa2-GR cells treated with 5, 7.5, and 10 μM PVT for 48 h were subjected to western blotting . (D) Oral administration of 10 mg/kg PVT everyday suppressed the growth of subcutaneously implanted MIAPaCa2-GR tumors. The tumor volume was measured once every 3 days using a vernier caliper. (E) Aseptically excised tumors were weighed at the day of sacrifice. (F) Snap-frozen tumors were lysed and subjected to western blotting analysis. The protein expression in the control and treatment groups was normalized with the respective β-actin of each group. Each lane of blot represents a tumor from an individual mouse. Blots were developed by the Chemidoc Touch Imaging System (BioRad). (G) FFPE tissues were analyzed by IHC. Scale bars, 10 mm. The statistical significance level was considered to be p ≤ 0.05. ∗∗p ≤ 0.01.
Discussion
Pancreatic cancer management has become more challenging due to its aggressive nature and ineffective treatment options. As a result, there is a wide scope to develop novel treatment strategies for pancreatic cancer. In our present study, we have demonstrated the in vitro and in vivo antineoplastic effects of an anti-Parkinson drug, PVT, in pancreatic cancer. Considering the heterogenic nature of pancreatic cancer, we investigated the effects of PVT in a series of five different pancreatic cancer cell lines with different phenotype and genotype profile. In fact, BxPC3 cells with wild-type K-ras, and AsPC1, MIAPaCa2, and PANC1 cells with mutant K-Ras were used in this study.27 To rule out the sex and species differences, we used two male PDAC lines (MIAPaCa2 and PANC-1), two female PDAC cell lines (AsPC1 and BxPC3), and one murine pancreatic cancer cell line (PO2).27 In addition, we evaluated the toxic effects of PVT in normal HPDE-6 cells and human lung fibroblasts, and in a mice model. Our results indicated that PVT displayed remarkable antiproliferative and apoptotic effects in all of the pancreatic cancer cell lines tested, whereas it was the least toxic to normal epithelial and fibroblast cells.
A plethora of oncogenic signaling pathways have been established in pancreatic cancer. Akt/PKB, a serine-threonine kinase, has been widely shown to be the central node for pancreatic cancer progression. The hyperactivation of Akt has been reported in 59% of PDAC tumors.28 Akt is activated by phosphorylation at serine or threonine residues.29,30 Activated Akt acts as a regulatory intermediate for various tumor promoting signaling pathways and promotes tumorigenic phenotypes such as apoptosis resistance, CSC characteristics, and cell proliferation and survival.28 Owing to its major role in pancreatic cancer, several PI3K and Akt inhibitors are in clinical trials for pancreatic cancer treatment and providing the rationale to develop more.31 Our results indicate that PVT not only suppressed the phosphorylation of Akt but also reduced the expression of Akt in various pancreatic cancer cells.
CSCs are a subpopulation of tumor-initiating cells that confer phenotypic and functional variations in cancer cells. Their role has been established in resistance to the current treatment options.17,22,32 Gli1, a downstream transcription factor in SHH signaling, has been shown to regulate CSC markers Oct-4, SOX2, NANOG, and c-Myc in pancreatic cancer.17 The ligand-dependent and ligand-independent activation of Gli1 promotes tumorigenic progression in various cancer models. Non-canonically, Gli1 is positively regulated by K-ras, transforming growth factor β (TGF-β), PI3K-Akt, and protein kinase C α (PKC-α).33 Akt/Gli1 signaling has been shown as a potential pathway to target renal cell carcinoma and glioblastoma.8,23 Gli1 is shown to be overexpressed in 70% of human pancreatic tumors. Our results indicate that PVT inhibited Gli1 and stem cell markers Oct-4, SOX2, NANOG, and c-Myc in various pancreatic cancer cells. In addition, PVT treatment increased the cleavage of caspase 3 and PARP, confirming apoptosis. Suppression of pAkt(Ser473) was validated by immunofluorescence analysis. Moreover, PVT reduced the formation of pancreatic cancer cell tumorspheres, indicating the effects of PVT in inhibiting CSC characteristics. Similar to PVT, the inhibition of Akt by PI3K inhibitor LY294002 reduced the expression of Gli1 and its downstream targets Oct-4, NANOG, and c-Myc, and induced apoptosis. These effects were significantly increased in response to combination treatment (LY294002 and PVT). The inhibition of Gli1 by GANT-61 inhibited CSC markers Oct-4 and c-Myc and increased cleaved caspase 3. Interestingly, Gli1 siRNA alone did not have much effect on the expression of Oct-4, NANOG, and c-Myc, but it did enhance the cleavage of caspase 3. This can be attributed to the fact that perhaps the level of Gli1 knockdown achieved in our studies was not sufficient to inhibit its downstream effectors. Nonetheless, the inhibition or silencing of Akt or Gli1 further potentiated the effects of PVT in inhibiting the Akt/Gli1 signaling axis. These results showed that PVT mediated antiproliferative and apoptotic effects by inhibiting Akt/Gli1 signaling in pancreatic cancer cells. Three different in vivo pancreatic tumor models were used to evaluate the antitumor efficacy of PVT. Notably, PVT suppressed 50%–77% of pancreatic tumor growth in subcutaneous and orthotopic xenograft models in mice. Further mechanistic analysis of tumors revealed that pancreatic tumor growth suppression by PVT was associated with the inhibition of Akt/Gli1 signaling, confirming our in vitro observations. Pro-apoptotic effects of PVT in vivo were also confirmed by western blotting, IHC, and TUNEL analysis.
Resistance to gemcitabine is a major problem in the treatment of patients with pancreatic cancer. It is thus vital to find options to inhibit resistance to therapy and enhance the antitumorigenic effects of gemcitabine. Our results demonstrated that PVT inhibited the growth of gemcitabine-resistant MIAPaCa2-GR cells in vitro and in vivo by inducing apoptosis. Since PVT is an inverse agonist of the 5-HT2A receptor, we compared the anticancer effects of PVT with other known inverse agonists and antagonists of 5-HT2A receptors. Surprisingly, nelotanserin (inverse agonist of the 5-HT2A receptor) and volinanserin (antagonist of the 5-HT2A receptor) exhibited the least or no antineoplastic effects in pancreatic cancer cells as compared to PVT. For instance, the IC50 of nelotanserin was observed to be 60 μM, whereas the IC50 of PVT was 6.3 μM after 48 h of treatment in BxPC3 cells, which is 10 times less than the concentration of nelotanserin. Similarly, volinanserin did not significantly reduce the proliferation of BxPC3 cells. We further performed molecular studies in BxPC3 cells treated with 7.5 μM PVT and 75 μM nelotanserin or volinanserin (10× higher concentration when compared with PVT). Our observations indicated that nelotanserin and volinanserin did not inhibit the expression of pAkt(Ser473), Akt, Gli1, and c-Myc, whereas there was significant inhibition of these proteins in PVT treatment. Moreover, nelotanserin and volinanserin did not induce any apoptosis as compared to PVT (Figure S6). However, the underlying mechanism behind this phenomenon needs to be explored and will be the focus of our future studies. At the antitumor dose used in the present study, PVT did not exhibit any general signs of toxicity or changed behavioral activity in mice. However, the side effects associated with the long-term administration of PVT cannot be ruled out and require further in-depth studies.
Overall, our study provides convincing results that PVT could be established as a potential drug for the treatment of patients with pancreatic cancer in the near future. Ours is the first study to report the anticancer effects of PVT.
Materials and Methods
Ethics Statement
All of the animal experiments were carried out in accordance with the ethical standards and according to approved protocol by the Institutional Animal Care and Use Committee (IACUC).
Cell Culture
Human pancreatic cancer cell lines AsPC1, BxPC3, MIAPaCa2, and PANC1 were purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA). Murine pancreatic cancer cell line PO2 was a kind gift from Dr. Guang-Yu Yang (Northwestern University, Chicago, IL, USA). The PANC1 luc cell line used for the orthotopic in vivo study was a kind gift from Dr. Erica K. Sloan (Monash University, Melbourne, VIC, Australia). The MIAPaCa2 gemcitabine-resistant cell line was a benevolent gift from Dr. Terry H. Landowski (University of Arizona Cancer Center, Tucson, AZ, USA). The normal human pancreatic ductal epithelial cell line HPDE-6 was a generous gift from Dr. Ming-Sound Tsao (University of Toronto, Toronto, ON, Canada). Normal human lung fibroblast cell line MRC5 was a kind gift from Dr. Dipankar Ray (University of Michigan, Ann Arbor, MI, USA). Gli1+/+ and Gli1−/− MEF cell lines were a kind gift from Dr. Bushman (University of Wisconsin, Madison, WI, USA). MEFs were maintained as previously described by us.25 All of the cell lines were authenticated by short tandem repeat (STR) analysis at Texas Tech University Health Sciences Center (TTUHSC) core facilities (Lubbock, TX, USA) before starting the experiments. Monolayer cultures of AsPC1, BxPC3, and MIAPaCa2 gemcitabine-resistant cells were maintained in RPMI medium, whereas MRC5, MIAPaCa2, PO2, PANC1, and PANC1-luc cells were cultured in DMEM medium, supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin antibiotic mixture, 2 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 2.2 g/L sodium bicarbonate, and 10 mL/L glucose. All of the cells were cultured at 37°C in an incubator with 5% CO2. HPDE-6 cells were maintained as previously described by us.14,26
Cell Survival Assay
Cells were plated at a density of 3,000–4,000 cells per well in 96-well plates and allowed to attach overnight. After overnight incubation, cells were treated with different concentrations of PVT (Selleck Chemicals) for 24, 48, and 72 h. Cells were then analyzed by SRB assay as described by us previously.6,9 All of the experiments were repeated independently at least 3 times.
Clonogenic Assay
Briefly, 500 cells per well were seeded in a 6-well plate and incubated overnight. The cells were then treated with various concentrations (0–5 μM) of PVT for 48 h. After 48 h, the media was replaced with 2 mL fresh medium and incubated at 37°C with 5% CO2 for 13 days or until the cells in the control group formed large clones. The cells were then fixed with 10% trichloroacetic acid (TCA) overnight and stained with SRB for 2 h. The colonies from the control and treatment groups (n = 2) were quantified using ImageJ (NIH) software.
AnnexinV/FITC Apoptosis Assay
The assay was performed with an annexin V/FITC apoptosis detection kit (BD Biosciences). Briefly, 0.2 × 106 cells were treated with varying concentrations of PVT (0–10 μM) for 48 h. Cells were then assayed for apoptosis according to the manufacturer’s instructions and analyzed by flow cytometer (Accuri C6, Ann Arbor, MI, USA) as previously described by us.8,9 All of the experiments were repeated independently at least 2 times.
Western Blot Analysis
AsPC1, BxPC3, MIAPaCa2, and PANC1 cells were treated with varying concentrations of PVT (0–10 μM) for 48 h. After 48 h, the whole-cell lysates were subjected to SDS-PAGE, and the resolved proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane. The membranes were probed for primary antibodies against pAkt(Ser473), Akt, Gli1, Oct-4, SOX2, NANOG, and c-Myc, cleaved caspase 3, cleaved PARP, β-actin, and β-tubulin (Cell Signaling Technologies, Danvers, MA, USA). The membranes were developed as previously described by us.15 All of the experiments were repeated independently at least 3 times.
Immunofluorescence
AsPC1 cells were plated on a coverslip in a 24-well plate at a concentration of 4 × 104 cells per well and allowed to attach overnight. Cells were then treated with 2.5 μM PVT for 48 h. For the immunofluorescence analysis, cells were washed with 1× PBS twice and then fixed with 4% paraformaldehyde (PFA) for 15 min. PFA-fixed cells were washed 3 times with 1× PBS and permeabilized using 0.1% Triton-X 100 for 5 min. Cells were washed with 1× PBS and then blocked using 6% goat serum in 1% BSA and 0.1% Triton-X 100 for 1 h. Cells were then incubated with antibody against anti-pAkt(Ser473) (1:400) overnight. The next day, cells were washed twice with 1× PBS and incubated with anti-rabbit Alexa Fluor 488 conjugate secondary antibody (1:1,000) in 3% goat serum prepared in 0.5% BSA for 1 h. Cells were washed and incubated with Alexa Fluor 594 phalloidin (1:500) in 3% BSA for 15 min. Cells were washed and coverslips were inversely mounted on the slides using DAPI mounting medium. Images were taken in 512 pixels using a multiphoton confocal microscope (Nikon).
Gli1 Luciferase Reporter Assay
The activity of Gli1 was assessed by performing the Gli1 luciferase reporter assay. The assay was performed using the Cignal Gli reporter assay kit according to the manufacturer’s instructions (QIAGEN). Briefly, 40,000 cells per well in a 96-well plate were transfected with the Cignal Gli reporter. Post-24 h of transfection, cells were treated with 5, 7.5, and 10 μM PVT for 48 h. GANT-61, a known pharmacological inhibitor of Gli1, was used as a positive control, in which the cells were treated with 20 μM GANT-61 for 48 h. After 48 h, the luciferase assay was developed according to the manufacturer’s instructions using Dual-Glo luciferase assay system (Promega).
Tumorsphere Assay
PANC1 cells were seeded at a density of 1,000 cells per well in a 24-well ultra-low attachment plate. Tumorspheres were cultured in DMEM/F12 medium supplemented with 5% FBS, 2% B27 supplement, 20 ng/mL epidermal growth factor (EGF), 20 ng/mL basic fibroblast growth factor (bFGF), and 5 μg/mL insulin. After overnight incubation, cells were treated with 0–5 μM PVT and incubated for 12 days for the formation of tumorspheres. Images were taken using a light microscope (Leica).
Subcutaneous Pancreatic Tumor Implantation
Female athymic nude mice (4–6 weeks old) were purchased from Envigo (Houston, TX, USA) and Charles River Laboratories (Houston, TX, USA). Approximately 1 × 106 BxPC3 or 6.7 × 106 MIAPaCa2-GR cells (1:1 PBS:Matrigel mixture) were implanted in the right flanks of the mice. Once the tumor volume was observed to be 70–100 mm3, mice were randomly divided into 2 groups with 5 mice in each group. Group I was assigned as the control group, which received the vehicle only (PBS). Group II mice received 10 mg/kg PVT by oral gavage every day. Treatment with PVT was started at day 5 (MIAPaCa2-GR) or day 13 (BxPC3). Tumor growth was monitored by measuring the tumor volume twice per week until day 30 (MIAPaCa2-GR) or day 40 (BxPC3) using a vernier caliper. The experiment was terminated due to tumor burden. The mice were euthanized, and the tumors were aseptically removed. A part of the tumor was snap frozen for western blotting and the other part was fixed in formalin for IHC and TUNEL analysis. The weight of the mice was monitored once in 10–14 days.
Orthotopic Pancreatic Tumor Model
Female athymic nude mice (4–6 weeks old) were used for the orthotopic injection of pancreatic cancer cells. Mice were anesthetized by using isoflurane, and a minor incision was made in the left abdomen. Stably transfected luciferase expressing PANC1 (PANC1-luc) cells were orthotopically implanted in the pancreas. Injected into the subcapsular region of the pancreas using a 30-G sterile needle were 1 × 106 exponentially growing PANC1-luc cells in a 20-μL PBS suspension. The peritoneum and skin incisions were closed sequentially with absorbable sutures. Buprenorphine was administered to mice as a pain killer every 8 h for 2 days. The growth of orthotopically implanted tumors was monitored by measuring luminescence via In Vivo Imaging System (IVIS) (Caliper Life Sciences). To determine the basal luminescence value, mice were imaged on the same day after injecting luciferin (3 mg per mouse, intraperitoneal [i.p.]). Mice were randomly divided into 2 groups (control and treatment) on day 17. The control group received vehicle only, whereas the treatment group received 10 mg/kg PVT by oral gavage every day. Mice weight was periodically monitored. The experiment was terminated at day 69 by humanely euthanizing the mice using CO2 overdose. The mice were dissected and representative mice from the control and treatment groups were imaged for luminescence. The pancreas/tumors from the control and treatment groups were aseptically excised out and snap frozen for western blot analysis; few tumors were fixed in formalin for immunohistochemical and TUNEL analysis. Mice weight was monitored once per week. The weight of excised organs such as liver, spleen, kidney, pancreas, lungs, and brain were recorded.
Statistical Analysis
Statistical analysis was performed by Prism 7.0 software (GraphPad Software, San Diego, CA, USA). The results are presented as means ± standard deviations (SDs) for in vitro experiments or standard error of the mean (SEM) for in vivo experiments. Statistical significance was analyzed using the Student’s t test followed by Fisher’s F test and the outcomes were considered statistically significant at p ≤0.05.
Author Contributions
Conception & Design, S.R. and S.K.S. Development of Methodology, S.R. and S.K.S. Data Acquisition, S.R. Analysis and Interpretation of Data (e.g., statistical analysis, biostatistics, computational analysis), S.R. and S.K.S. Writing, Review, and/or Revision of the Manuscript, S.R. and S.K.S. Administrative, Technical, or Material Support (e.g., reporting or organizing data, constructing databases), S.K.S. Study Supervision, S.K.S.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported in part by R01 grant CA129038 (to S.K.S.) awarded by the National Cancer Institute, NIH. The Syngenta Fellowship Award in collaboration with the Society of Toxicology to S.R. is acknowledged. The authors also appreciate the funding from the Dodge Jones Foundation.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.08.019.
Supplemental Information
References
- 1.Collisson E.A., Bailey P., Chang D.K., Biankin A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019;16:207–220. doi: 10.1038/s41575-019-0109-y. [DOI] [PubMed] [Google Scholar]
- 2.Tas F., Sen F., Keskin S., Kilic L., Yildiz I. Prognostic factors in metastatic pancreatic cancer: older patients are associated with reduced overall survival. Mol. Clin. Oncol. 2013;1:788–792. doi: 10.3892/mco.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onishi H., Katano M. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J. Gastroenterol. 2014;20:2335–2342. doi: 10.3748/wjg.v20.i9.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 5.Parvathaneni V., Kulkarni N.S., Muth A., Gupta V. Drug repurposing: a promising tool to accelerate the drug discovery process. Drug Discov. Today. 2019;24:2076–2085. doi: 10.1016/j.drudis.2019.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranjan A., Gupta P., Srivastava S.K. Penfluridol: An Antipsychotic Agent Suppresses Metastatic Tumor Growth in Triple-Negative Breast Cancer by Inhibiting Integrin Signaling Axis. Cancer Res. 2016;76:877–890. doi: 10.1158/0008-5472.CAN-15-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiklund E.D., Catts V.S., Catts S.V., Ng T.F., Whitaker N.J., Brown A.J., Lutze-Mann L.H. Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int. J. Cancer. 2010;126:28–40. doi: 10.1002/ijc.24813. [DOI] [PubMed] [Google Scholar]
- 8.Ranjan A., Srivastava S.K. Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget. 2017;8:32960–32976. doi: 10.18632/oncotarget.16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ranjan A., Srivastava S.K. Penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis. Sci. Rep. 2016;6:26165. doi: 10.1038/srep26165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ranjan A., Wright S., Srivastava S.K. Immune consequences of penfluridol treatment associated with inhibition of glioblastoma tumor growth. Oncotarget. 2017;8:47632–47641. doi: 10.18632/oncotarget.17425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw V., Srivastava S., Srivastava S.K. Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin. Cancer Biol. 2019 doi: 10.1016/j.semcancer.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoxhaj G., Manning B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer. 2020;20:74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma N., Nanta R., Sharma J., Gunewardena S., Singh K.P., Shankar S., Srivastava R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget. 2015;6:32039–32060. doi: 10.18632/oncotarget.5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boreddy S.R., Pramanik K.C., Srivastava S.K. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin. Cancer Res. 2011;17:1784–1795. doi: 10.1158/1078-0432.CCR-10-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loganathan S., Kandala P.K., Gupta P., Srivastava S.K. Inhibition of EGFR-AKT axis results in the suppression of ovarian tumors in vitro and in preclinical mouse model. PLOS ONE. 2012;7:e43577. doi: 10.1371/journal.pone.0043577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelleher F.C. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis. 2011;32:445–451. doi: 10.1093/carcin/bgq280. [DOI] [PubMed] [Google Scholar]
- 17.Cochrane C.R., Szczepny A., Watkins D.N., Cain J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers (Basel) 2015;7:1554–1585. doi: 10.3390/cancers7030851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagai S., Nakamura M., Yanai K., Wada J., Akiyoshi T., Nakashima H., Ohuchida K., Sato N., Tanaka M., Katano M. Gli1 contributes to the invasiveness of pancreatic cancer through matrix metalloproteinase-9 activation. Cancer Sci. 2008;99:1377–1384. doi: 10.1111/j.1349-7006.2008.00822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maréchal R., Bachet J.B., Calomme A., Demetter P., Delpero J.R., Svrcek M., Cros J., Bardier-Dupas A., Puleo F., Monges G. Sonic hedgehog and Gli1 expression predict outcome in resected pancreatic adenocarcinoma. Clin. Cancer Res. 2015;21:1215–1224. doi: 10.1158/1078-0432.CCR-14-0667. [DOI] [PubMed] [Google Scholar]
- 20.Inaguma S., Riku M., Hashimoto M., Murakami H., Saga S., Ikeda H., Kasai K. GLI1 interferes with the DNA mismatch repair system in pancreatic cancer through BHLHE41-mediated suppression of MLH1. Cancer Res. 2013;73:7313–7323. doi: 10.1158/0008-5472.CAN-13-2008. [DOI] [PubMed] [Google Scholar]
- 21.Fu J., Rodova M., Roy S.K., Sharma J., Singh K.P., Srivastava R.K., Shankar S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013;330:22–32. doi: 10.1016/j.canlet.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasad S., Ramachandran S., Gupta N., Kaushik I., Srivastava S.K. Cancer cells stemness: A doorstep to targeted therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165424. doi: 10.1016/j.bbadis.2019.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J., Zhu G., Huang J., Li L., Du Y., Gao Y., Wu D., Wang X., Hsieh J.T., He D., Wu K. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: A novel potential therapeutic target. Cancer Lett. 2016;370:313–323. doi: 10.1016/j.canlet.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Liang H., Zheng Q.L., Fang P., Zhang J., Zhang T., Liu W., Guo M., Robinson C.L., Chen S.B., Chen X.P. Targeting the PI3K/AKT pathway via GLI1 inhibition enhanced the drug sensitivity of acute myeloid leukemia cells. Sci. Rep. 2017;7:40361. doi: 10.1038/srep40361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee C.H., Yu C.C., Wang B.Y., Chang W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget. 2016;7:1215–1226. doi: 10.18632/oncotarget.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atrakchi A. 2016. Application Numver: 207318Orig1s000 Pharmacology Review(s)https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/207318Orig1s000PharmR.pdf [Google Scholar]
- 27.Deer E.L., González-Hernández J., Coursen J.D., Shea J.E., Ngatia J., Scaife C.L., Firpo M.A., Mulvihill S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlieman M.G., Fahy B.N., Ramsamooj R., Beckett L., Bold R.J. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br. J. Cancer. 2003;89:2110–2115. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murthy D., Attri K.S., Singh P.K. Phosphoinositide 3-Kinase Signaling Pathway in Pancreatic Ductal Adenocarcinoma Progression, Pathogenesis, and Therapeutics. Front. Physiol. 2018;9:335. doi: 10.3389/fphys.2018.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Massihnia D., Avan A., Funel N., Maftouh M., van Krieken A., Granchi C., Raktoe R., Boggi U., Aicher B., Minutolo F. Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer. J. Hematol. Oncol. 2017;10:9. doi: 10.1186/s13045-016-0371-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuoka T., Yashiro M. Molecular targets for the treatment of pancreatic cancer: Clinical and experimental studies. World J. Gastroenterol. 2016;22:776–789. doi: 10.3748/wjg.v22.i2.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsui W.H. Cancer stem cell signaling pathways. Medicine (Baltimore) 2016;95(1, Suppl 1):S8–S19. doi: 10.1097/MD.0000000000004765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pietrobono S., Gagliardi S., Stecca B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019;10:556. doi: 10.3389/fgene.2019.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.