

Nonalcoholic fatty liver disease (NAFLD) is increased in offspring exposed to parental obesity. Mechanisms for increased risk for disease are emerging. These mechanisms will guide the development of preventative approaches for NAFLD.
Abstract
The surge of obesity across generations has become an increasingly relevant issue, with consequences for associated comorbidities in offspring. Data from longitudinal birth cohort studies support an association between maternal obesity and offspring nonalcoholic fatty liver disease (NAFLD), suggesting that perinatal obesity or obesogenic diet exposure reprograms offspring liver and increases NAFLD susceptibility. In preclinical models, offspring exposed to maternal obesogenic diet have increased hepatic steatosis after diet‐induced obesity; however, the implications for later NAFLD development and progression are still unclear. Although some models show increased NAFLD incidence and progression in offspring, development of nonalcoholic steatohepatitis with fibrosis may be model dependent. Multigenerational programming of NAFLD phenotypes occurs after maternal obesogenic diet exposure; however, the mechanisms for such programming remain poorly understood. Likewise, emerging data on the role of paternal obesity in offspring NAFLD development reveal incomplete mechanisms. This review will explore the impact of parental obesity and obesogenic diet exposure on offspring NAFLD and areas for further investigation, including the impact of parental diet on disease progression, and consider potential interventions in preclinical models.
Abbreviations
- BMI
body mass index
- DMR
differentially methylated region
- ER
endoplasmic reticulum
- FA
fatty acid
- FGF21
fibroblast growth factor 21
- FMT
fecal microbiome transplantation
- HF/HS
high‐fat/high‐sucrose
- HFD
high‐fat diet
- HFFC
high fat, high fructose, high cholesterol
- IHCL
intrahepatocellular lipid
- miRNA
microRNA
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OR
odds ratio
- Pgc1a
PPAR gamma coactivator 1 alpha
- PPAR
peroxisome proliferator–activated receptor
- PQQ
pyrroloquinoline quinone
- SIRT1
sirtuin 1
- TG
triglyceride
The burgeoning global impact of obesity( 1 ) has increased attention on the cardiometabolic consequences for children born to obese parents. Approximately 55% of women in the United States are overweight or obese at the time of conception.( 2 ) Several works link obesity and gestational diabetes to later development of metabolic syndrome and associated complications,( 3 , 4 , 5 ) most notably nonalcoholic fatty liver disease (NAFLD). This review will summarize the role of parental obesity in NAFLD pathophysiology in offspring, highlighting areas for future study, and discuss potential directions for preventive intervention.
Developmental Origins of Health and Disease
The Developmental Origins of Health and Disease hypothesis posits that in utero or early‐life exposures influence one’s susceptibility to chronic diseases later in life. Epidemiologic and animal models demonstrate the impact of the perinatal environment on programming long‐term risk for chronic disease in offspring. Early studies associated low birthweight with increased cardiovascular disease mortality and hypertension in adulthood.( 6 , 7 ) This work, reported on a male UK cohort from almost 100 years ago, was the first to suggest that perinatal stress has both immediate implications for offspring and long‐term impacts on adult health. Subsequent studies of the Dutch Hunger Winter and the Chinese Famine confirmed the association of gestational malnutrition with risk of chronic disease.( 8 , 9 )
Clinical Evidence for an Association Between Maternal Obesity and Increased Risk for NAFLD in Offspring
With a growing interest in how maternal obesity affects offspring, several clinical studies evaluated potential associations between maternal prepregnancy body mass index (BMI) and offspring risk for NAFLD. The seminal Raine cohort study examined a large Australian longitudinal birth cohort of approximately 2,900 offspring and found that maternal prepregnancy BMI greater than or equal to 30 increased the risk of NAFLD in offspring. This effect was independent of diet and other risk factors, with an odds ratio (OR) of 2.29 on multivariable logistic regression.( 10 ) Subsequent analysis showed that maternal prepregnancy obesity had a significant effect on NAFLD in female offspring.( 11 ) Further evaluation in male offspring will be essential to confirm this sex difference or to account for other variables. Other large studies in the United Kingdom and United States have shown a similar association between maternal prepregnancy BMI and offspring NAFLD, independent of offspring adiposity.( 12 , 13 ) The UK cohort evaluated 1,215 children with liver ultrasound and found an adjusted odds ratio (aOR) of 2.72 for steatosis if there was an elevated maternal prepregnancy BMI.( 12 ) The aOR increased to 6.74 if maternal diabetes or glycosuria was present. In the US cohort, 254 offspring were evaluated for steatosis by measurement of hepatic fat fraction (HFF) with magnetic resonance imaging. Logistic regression models identified a significant association between maternal prepregnancy obesity (BMI ≥ 30) and increased HFF.( 13 )
Although these studies show a clear association between maternal obesity and longitudinal risk for NAFLD in offspring, they also indicate an immediate impact on the developing fetus. Modi et al. measured intrahepatocellular lipid (IHCL) in newborns and found a significant positive correlation between IHCL and maternal prepregnancy BMI, with an 8.6% increase in IHCL for each 1‐point increase in BMI.( 14 ) A similar positive correlation between IHCL and maternal BMI appeared in a separate mother/infant cohort.( 15 ) Infants born to obese mothers averaged a 68% increase in IHCL compared with infants born to mothers with a normal BMI.( 15 ) Both studies highlight the importance of the liver as a storage site for increased lipids in utero. Longitudinal follow‐up will be essential to characterize IHCL resolution or progression and to identify factors affecting disease severity.
Preclinical Models of Maternal Obesogenic Diet and NAFLD in Offspring
Because maternal prepregnancy BMI is associated with offspring NAFLD, animal models have been developed using nonhuman primates and rodents to study the mechanisms linking increased offspring steatosis with maternal obesity (Fig. 1). Most models involve feeding an obesogenic diet before mating and continuing during pregnancy and lactation. This experimental design tests the combined impact of prepregnancy obesogenic diet and exposure during fetal development.
Fig. 1.
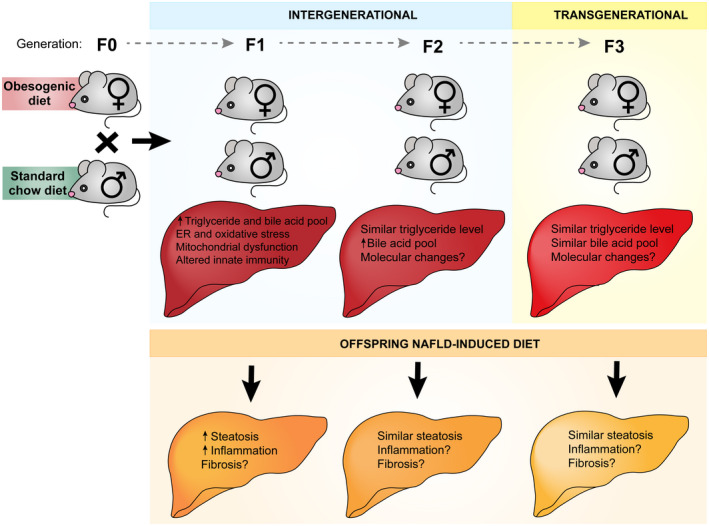
Maternal obesogenic diet exposure contributes to offspring NAFLD in preclinical models. Preclinical models have identified a role for maternal obesogenic diet exposure on offspring NAFLD. F1 generation offspring have increased intrahepatic triglycerides, bile acid pool size, ER stress, oxidative stress, and mitochondrial dysfunction. F1 offspring also exhibit altered innate immunity. Aspects of this phenotype that are passed to subsequent generations remain unclear (F2, F3). After exposure to NAFLD‐inducing diets, F1 offspring exhibit increased steatosis and inflammation. The F2 and F3 generations develop a similar degree of steatosis and inflammation. Use of these preclinical models will be essential in defining the mechanisms that underlie both intergenerational and transgenerational phenotypes. (Illustration by Astrid Rodriguez‐Velez and Anne Robinson in association with InPrint at Washington University in St. Louis. Reprinted with permission.).
A Japanese macaque model of maternal high‐fat diet (HFD) before and during pregnancy showed increased fetal hepatic steatosis early in the third trimester.( 16 ) This observation is consistent with the hypothesis that increased maternal fuel delivery increases fat accumulation in the fetal liver. In addition, offspring from HFD‐fed macaques showed elevated hepatic triglyceride (TG) levels at postnatal days 30 and 180, highlighting persistently increased hepatic fat and long‐term alterations in hepatic lipid metabolism.
Similar findings were observed in rodent models of maternal obesogenic diet. Murine maternal HFD exposure led to persistent steatosis in 15‐week‐old offspring, despite weaning to a standard chow diet. More severe steatosis occurred in offspring exposed to maternal HFD, high‐fat/high‐sucrose (HF/HS) diet, and high‐fat, high‐fructose, high‐cholesterol (HFFC) diet.( 17 , 18 , 19 , 20 , 21 ) Genes involved in TG and fatty acid (FA) synthesis were increased in the liver, including increased sterol regulatory element‐binding protein 1c (SREBP1c).( 17 ) TG and FA synthesis increased, whereas FA oxidation decreased.( 22 , 23 ) This combination of increased lipogenesis and decreased lipid catabolism leads to increased liver lipid accumulation. Two primary regulators of these pathways, peroxisome proliferator–activated receptor alpha (PPARα) and sirtuin 1 (SIRT1), decreased in liver following maternal obesogenic diet exposure.( 24 , 25 ) Notably, activation of both PPARα and SIRT1 decreased hepatic TG levels, supporting the importance of these two targets.( 26 , 27 ) SIRT1 also regulates PPAR gamma coactivator 1 alpha (Pgc1a) expression and ultimately controls targets of PPARγ.( 28 )
Mechanisms of Transmission of Altered Hepatic Metabolism
Multiple animal models show increased hepatic lipid accumulation in offspring exposed to maternal obesogenic diet along with transcription factor and target gene changes consistent with increased hepatic lipogenesis. However, the mechanisms behind this developmental programming of NAFLD are poorly understood. Developmental programming studies mostly focus on epigenetic changes as a primary mechanism for phenotypes observed in offspring. Epigenetic programming may manifest through changes in DNA methylation, histone modifications, and microRNA (miRNA) expression. In addition to epigenetic changes, an altered microbiome may undergo vertical transmission from dams to offspring. Recent studies have begun to evaluate these potential mechanisms.
Epigenetic Modifications
DNA methylation has been the most frequently studied in developmental programming of NAFLD. DNA methylation occurs when methyl groups are added to cytosine or adenine residues, typically repressing gene transcription. Recent studies have focused on identifying differentially methylated regions (DMRs) in rodent models of maternal obesity. Wankhade et al. identified 82 DMRs associated with maternal HFD exposure affecting developmental processes.( 29 ) Notable DMRs included two genes involved in liver metabolism: Pgc1a and fibroblast growth factor 21 (Fgf21). Offspring exhibited Pgc1a hypermethylation and Fgf21 hypomethylation; however, associated gene expression was not measured. PGC1α levels were decreased in a mouse model of hepatic steatosis, consistent with possible hypermethylation.( 30 ) PGC1α promotes FA beta oxidation and mitochondrial biogenesis; thus, decreased levels would support increased hepatic lipid accumulation. Hypomethylation at FGF21 would likely lead to increased expression. Notably, circulating and intrahepatic levels of FGF21 increase in NAFLD.( 31 ) Additional studies identified methylation changes in clock genes following maternal HFD exposure.( 32 ) Hypermethylation occurred in the promoter regions of brain and muscle aryl hydrocarbon receptor nuclear translocator–like protein 1 (Bmal‐1) and period circadian protein 2 (Per2), both of which regulate metabolism.( 32 , 33 , 34 ) These changes were associated with a loss in rhythmic Bmal‐1 and Per2 expression over a 24‐hour period and with increased hepatic TG accumulation. Exposure to maternal HFD also disrupted the rhythmic expression of genes involved in endoplasmic reticulum (ER) stress. For instance, hypermethylation in the glucose‐regulated protein 78 (Grp78) promoter decreases this gene’s expression.( 35 ) Although these changes in hepatic clock genes are expected to increase hepatic TG accumulation, a direct causal link has not been defined.
Maternal hepatic insulin resistance also induces changes in offspring hepatic DNA methylation. Liver insulin receptor knockout (LIRKO) mice were used to evaluate the impact of maternal hepatic insulin resistance on offspring hepatic metabolism. Offspring of LIRKO dams exhibited altered DNA methylation and expression of two transforming growth factor β superfamily genes: neuronal regeneration related protein (Nrep) (hypomethylation and increased expression) and growth differentiation factor 15 (hypermethylation and decreased expression).( 36 ) In addition, NREP was shown to control hepatic lipid content by regulating FA synthesis and oxidation through expression of Pparγ and Srebp1c. This observation directly linked an epigenetic modification due to maternal insulin resistance to control of hepatic lipid metabolism in the offspring.
Another mechanism of epigenetic regulation involves histone modifications such as methylation and acetylation of specific residues. In mice, exposure to maternal obesogenic diet significantly increased histone 3 lysine 14 acetylation (H3K14ac) and histone 3 9 trimethylation (H3K9me3) in the offspring liver.( 37 ) These differential modifications were enriched in the promoter regions of genes regulating metabolism, including Pparγ, Pparα, retinoid X receptor alpha, and retinoic acid–related orphan receptor alpha. However, gene expression was only affected for Pparα (decreased). H3K14ac also increased in primate offspring exposed to a maternal HFD.( 38 ) Changes in histone modifications also occurred in phosphoenolpyruvate carboxykinase 1 in response to gestational HFD; these modifications include decreased H3K9me3 and increased H3K 4 dimethylation.( 39 ) Because these histone modifications do not necessarily alter gene expression, the impact on offspring hepatic metabolism remains unclear. One possibility includes histone modifications exerting a greater effect on gene expression in the setting of offspring exposure to a nonalcoholic steatohepatitis (NASH)–inducing diet.
MiRNAs regulate expression in a posttranscriptional manner by driving degradation of mRNAs before translation. In mice, maternal HFD exposure led to altered expression of approximately 6% of 579 miRNAs assayed in liver, with 10 miRNAs increased and 23 decreased.( 40 ) Among those down‐regulated was miR‐122, one of the most abundant miRNAs in the liver. A subsequent analysis in mice showed a similar reduction in miR‐122.( 41 ) Notably, miR‐122 decreased in the mouse liver during diet‐induced development of NASH, whereas circulating miR‐122 levels increased.( 42 ) Together, the existing data support a sustained impact of maternal obesogenic diet and insulin resistance on epigenetic modifications in offspring liver. Further studies should delineate the mechanisms of these changes and determine whether similar changes occur in human tissue.
Transmission of Altered Microbiome
Vertical transmission of an altered microbiome represents another potential method for passing phenotypes across generations. A shift in offspring microbiome with maternal obesity or maternal obesogenic diet occurred in both humans and preclinical models.( 29 , 43 ) Maternal obesogenic diet exposure in mice shifted offspring microbial populations by decreasing alpha diversity and increasing the abundance of Firmicutes compared with Bacteroidetes.( 44 ) Changes in the gut microbiome also occurred in nonhuman primates after maternal HFD exposure.( 45 ) Furthermore, the impact of maternal obesity on the offspring microbiome has been studied in humans. For instance, Soderborg et al. used fecal microbiome transplantation (FMT) from infants of obese or normal BMI mothers to gnotobiotic mice. In mice receiving FMT from infants of obese moms, an increased Firmicutes to Bacteroidetes ratio and hepatic steatosis occurred after 6 weeks of Western diet.( 43 ) The increased steatosis was present despite microbiome differences equilibrating between the two groups. The authors concluded that changes transmitted by an altered microbiome occur before offspring diet exposure. Thus, the initial “pioneering” bacteria present in the gut appear to educate the immune system. Indeed, maternal HFD exposure in mice induced changes in offspring innate immunity and likely increased disease severity, specifically through impaired phagocytic function in Kupffer cells and reduced numbers of natural killer T cells.( 46 ) Gut dysbiosis can also induce changes in intestinal permeability, resulting in delivery of harmful bacterial products to the liver through the portal circulation. Adult mice exposed to maternal HFD exhibited an altered gut barrier with increased permeability and as well as levels of circulating lipopolysaccharide (LPS).( 47 )
Perinatal Environment and Progression to Fibrosis in Offspring NAFLD
Although the association between maternal obesity and offspring steatosis has been defined for infants and adolescents, the impact of perinatal exposure on NAFLD progression is unknown. A retrospective analysis from the NASH Clinical Research Network approached this question by using birthweight to stratify biopsy findings in children with NAFLD. Children with high birthweight had a significantly increased OR for steatosis severity and NASH diagnosis, consistent with the aforementioned longitudinal birth cohorts.( 48 ) Conversely, low‐birthweight patients had a higher OR for advanced fibrosis. Although this study was retrospective and did not account for maternal diet, it highlighted the potential role of the perinatal environment on disease progression. Furthermore, this study suggested a potential divergence in the impact of perinatal risk factors on steatosis and inflammation versus fibrosis. This could be related to differences in zonality seen in children with NAFLD, because zone 3 (pericentral) steatosis was more associated with steatohepatitis, whereas zone 1 (periportal) steatosis was more strongly associated with fibrosis.( 49 ) Disease progression in relation to maternal obesogenic diet exposure has also been evaluated in mouse models. Offspring of mice exposed to maternal obesogenic diet developed worse inflammation and fibrosis when they were themselves fed a HFD.( 46 ) Exposure to maternal HFD activated hepatic stellate cells, increased expression of profibrotic genes, and led to liver fibrosis in offspring weaned to a low‐fat diet.( 20 ) Wankhade et al. also demonstrated increased hepatic fibrogenesis in offspring of mothers fed HFD when the offspring were subsequently placed on a methionine/choline‐deficient diet.( 29 ) A separate model using a maternal HFFC diet confirmed increased fibrogenesis in offspring.( 18 ) Notably, disease pathogenesis was accelerated in this model: Significant fibrosis occurred after only 7 weeks of HFFC diet feeding, making this an ideal model for studying rapid disease progression.
Although multiple mouse models clearly demonstrate worse disease progression with maternal obesogenic diet exposure, this phenotype may be nutrient specific. For example, maternal HF/HS‐exposed offspring fed a NASH‐inducing diet were protected from fibrosis despite higher levels of hepatic TGs.( 21 ) Confirming this finding in other models will be essential for comparing epigenetic changes and identifying specific alterations leading to disease progression versus protection. There is precedent for transmitting protection from fibrosis across generations. Inducing fibrosis in male rats led to suppression of fibrosis development in filial 1 (F1) and F2 offspring through epigenetic changes that increased PPARγ, a potential protective factor.( 50 )
Paternal Obesity/Obesogenic Diet and Offspring NAFLD
Fewer studies have focused on the impact of paternal obesogenic diet exposure on offspring liver metabolism. Follow‐up analysis of the Raine cohort addressed paternal obesity in relation to adolescent NAFLD in both sexes.( 11 ) Paternal obesity and NAFLD were positively associated in male offspring; however, multivariate analysis did not confirm this association.
In mice, paternal obesogenic diet exposure led to increased intrahepatic TG levels in the offspring.( 51 ) This was exacerbated if both the dam and stud were fed an HFD. A similar increase in offspring steatosis occurred in a rat model of paternal HF/HS and high‐salt diet.( 52 ) The offspring also had changes in hepatic expression and methylation of glucose and lipid metabolism genes, suggesting that paternal obesity induced epigenetic changes to the sperm. Indeed, HFD changed the histone distribution and methylation in the sperm genome.( 53 , 54 ) Although the number of studies is currently limited, paternal obesity appears to affect offspring hepatic lipid metabolism.
Multigenerational Transmission of Phenotypes
Another important aspect of developmental NAFLD programming is whether these events are transmitted across multiple generations. An outcome following maternal exposure is considered intergenerational if passed from the F0 to the F2 generation, and transgenerational if passed to the F3 generation or beyond (Fig. 1). This designation distinguishes between alterations in the F2 generation, which could directly result from an F0 event or exposure, and findings in the F3 generation, which must be passed epigenetically. Following paternal exposure, transmission to F2 and beyond is considered transgenerational, because the sperm of the F1 offspring are not present in utero. Few studies have evaluated multigenerational passage of parental obesogenic diet exposures in relation to offspring NAFLD. In the maternal HF/HS diet model, a similar degree of steatosis occurs in F2 and F3 offspring, suggesting that the maternal HF/HS diet does not have a transgenerational impact on steatosis.( 21 ) Barbosa et al. showed no increase in intrahepatic TG levels in F2 offspring from a paternal HFD lineage. In fact, after offspring HFD feeding, intrahepatic TG levels increased to a lesser degree than control mice.( 55 ) This study did show transgenerational changes in hepatic immunometabolism.
Offspring from a maternal obesogenic lineage were protected from fibrosis across all three generations.( 21 ) The mechanism for this transgenerational effect has not been defined. However, one male lineage transmitted fibrosis protection across multiple generations in association with epigenetic changes in Pparγ.( 50 ) This same group identified plasma DNA methylation in Pparγ as an independent predictor of fibrosis severity in patients with NAFLD.( 56 )
Li et al. evaluated the impact of repeat HFD from the F0 to the F2 generation.( 57 ) Repetitive generational HFD resulted in more severe steatosis in subsequent generations, with reduced histone methylation in liver X receptor alpha and ER oxidoreductase 1 alpha promoters and decreased expression of the histone methyltransferase responsible for H3K9me2.( 57 ) These findings highlight a feed‐forward cycle of repetitive HFD across generations.
Theoretical Link Between Maternal Obesogenic Diet and Periportal Disease in Offspring
In pediatric NAFLD, children tend to develop steatosis and inflammation in the periportal region rather than the pericentral distribution observed in adult NAFLD.( 58 ) Periportal disease distribution is associated with increased fibrosis and more advanced liver disease.( 59 ) Animal models with periportal disease also demonstrate bile ductular proliferation, or ductular reaction, which has been reported in biopsies from children and adults with NASH and fibrosis.( 60 , 61 ) Furthermore, periportal inflammatory infiltrates strongly predict fibrosis severity in both adult and pediatric NAFLD.( 62 , 63 ) All of these factors likely contribute to increased progressive disease; however, the molecular drivers for this pathologic distinction between periportal and pericentral are unclear (Fig. 2). One possible contributor is altered bile acid homeostasis, as observed in offspring exposed to maternal HF/HS diet. The bile acid pool increased, and the abundance of specific intrahepatic bile acid species shifted, including increased muricholic acid and decreased deoxycholic acid.( 21 ) These shifts occurred partly due to changes in bile acid synthesis, including increased cytochrome P450 7A1 (Cyp7a1) expression and activity. Resident gut bacteria also modulate enterohepatic bile acid metabolism through enzymatic modification of intestinal bile acids. Changes in the microbiome of offspring exposed to maternal obesogenic diet have been reported in humans, nonhuman primates, and rodents. In fact, this phenomenon could play a primary role in bile acid metabolism. Indeed, FMT from infants with obese mothers to gnotobiotic mice showed increased intrahepatic bile acid levels and increased Cyp7a1 expression.( 43 ) In this study, recipient mice also developed periportal inflammation similar to the maternal HF/HS diet model. The presence of periportal inflammation often suggests an altered gut barrier leading to translocation of proinflammatory factors (i.e., LPS) into the portal circulation. However, this inflammation could also indicate underlying cholestatic liver disease. Although only a hypothetical link currently exists, perinatal events, including exposure to maternal obesity, may program a propensity for the development of periportal disease in children. Other factors, such as genetics and environmental exposures, also likely play a role, although these have not yet been studied.
Fig. 2.
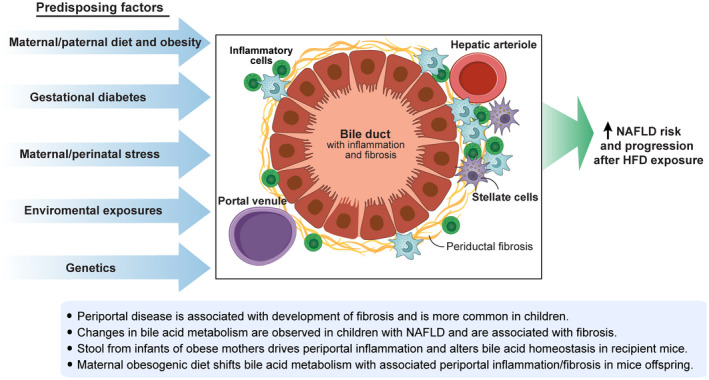
Theoretical model for factors contributing to periportal inflammation/fibrosis, which may contribute to NAFLD progression. Multiple perinatal and environmental factors have been linked to the development of periportal disease (inflammation/fibrosis). In relation to NAFLD, periportal disease is more common in children and associated with the development of fibrosis. Studies in mice have shown that maternal obesogenic diet exposure is associated with baseline periportal inflammation/fibrosis in offspring. Although the mechanisms are not defined, vertical transmission of an altered gut microbiome likely plays a role. A developing hypothesis is that predisposition for periportal disease due to these factors contributes to worse disease progression in NAFLD. (Illustration by Astrid Rodriguez‐Velez and Anne Robinson in association with InPrint at Washington University in St. Louis. Reprinted with permission.).
Potential Interventions in Preclinical Models
Once a role for developmental programming has been determined, preventive methods targeting its mechanism may be effective. One logical approach to eliminating the impact of maternal obesity on offspring would include dietary changes before conception. In the Japanese macaque model of maternal obesity, dietary reversal led to a partial but incomplete resolution of increased fetal hepatic TGs in subsequent pregnancies.( 16 ) Zhou et al. recently determined that the timing of dietary reversal is crucial in a mouse model of maternal HFD exposure.( 64 ) Female mice fed HFD for 12 weeks prior to breeding underwent one of two potential interventions: dietary reversal for 1 week or for 9 weeks before breeding. Reversing maternal diet for 9 weeks abrogated the increased steatosis seen in control offspring (those exposed to maternal HFD through weaning). Conversely, dietary reversal for 1 week before breeding resulted in more severe offspring steatosis than control. These findings suggest that developmental programming related to maternal diet is complex, and some interventional approaches may cause more harm. Thus, understanding the appropriate timing for dietary adjustment is necessary before providing clinical advice. Promotion of breastfeeding also likely benefits offspring, as duration of breastfeeding greater than 6 months was an independent protective factor from steatosis in the Raine cohort.( 10 )
Although prenatal or postnatal lifestyle modifications are the most benign approach, other preventive therapies with a low side‐effect profile could be beneficial. Oxidative stress and mitochondrial dysfunction occurred in livers of offspring exposed to maternal HFD.( 17 ) One study evaluated pyrroloquinoline quinone (PQQ), an antioxidant found in soil and plants, in a mouse model of maternal obesogenic diet exposure. Supplementation with PQQ reduced accumulation of hepatic TGs and ceramides in offspring fed a Western diet.( 65 ) PQQ supplementation during gestation and lactation also reduced hepatic TG accumulation and increased expression of lipid‐catabolism genes in the offspring. PQQ supplementation also reduced expression of proinflammatory genes, including interleukin‐6, nucleotide‐binding oligomerization domain‐like receptor family pyrin domain containing 3, and prostaglandin‐endoperoxide synthase 2, suggesting this compound may prevent disease progression. PQQ reduced thioacetamide‐induced liver fibrosis in mice but has not been studied in NAFLD‐associated fibrosis.( 66 )
Given the role of vertical microbiome transmission in the setting of maternal obesity, microbiome‐directed approaches may be feasible. Microbiome‐targeted therapies include prebiotics (nutrient delivery affecting existing gut bacteria), probiotics (delivery of live bacteria), and synbiotics (both). Administration of the prebiotic oligofructose to dams fed HF/HS diet resulted in reduced steatosis, improved glucose tolerance, and enhanced insulin sensitivity in offspring.( 67 ) Synbiotic treatment of offspring with psyllium seed, Enterococcus, and Lactobacillus in Japanese macaques only resulted in minor, short‐term changes to the gut microbiome and did not protect from later HFD‐induced gut dysbiosis.( 68 ) Therefore, if the altered gut microbiome acts by impaired immune system development early in life, treatments focused on the offspring microbiome may be too late to have a meaningful effect. Given this observation, microbiome‐targeted approaches may be more successful if instituted prenatally to pass on a healthy microbiome to offspring at the time of birth.
Another potential intervention may be targeted molecular therapies. Several studies of maternal obesogenic diet exposure in mice have noted a decrease in PPARα and PPARγ signaling in offspring liver. Magliano et al. used bezafibrate, a dual PPAR agonist, in offspring exposed to maternal obesogenic diet to reduce hepatic TG levels.( 26 ) In this study, treatment was performed in offspring fed a standard chow diet. Testing the performance of PPAR‐directed therapies in the setting of offspring HFD will be an important next step. Future studies should also explore other molecular therapies. However, such targeted treatments will likely have side‐effect profiles, precluding preventive use during pregnancy or childhood.
Conclusions and Future Directions
Both human and animal data support maternal obesity or obesogenic diet exposure leading to increased hepatic steatosis in newborns and enhanced future risk for NAFLD/NASH. This finding holds across several models and different types of maternal dietary exposures. Recent studies point to epigenetic modifications and vertical transmission of an altered microbiome as key mechanistic factors in transmission of this phenotype. However, there are still important topics that need further study and clarification in this field. Based on the existing literature, the following key areas require further investigation:
The impact of maternal obesity on risk of disease progression is not clear. Although most preclinical studies support that inflammation and fibrosis are worse in offspring exposed to maternal obesogenic diet, this observation may depend on the maternal and/or offspring diet. With maternal HF/HS diet in mice, less inflammation and fibrosis occur in offspring after Western diet exposure. To present some conflicting evidence, high birthweight associates with increased steatosis grade and NASH, whereas low birthweight associates with worse fibrosis in children with NAFLD.( 48 ) Reconciling these discrepancies will be essential to fully understand how maternal obesity impacts disease progression in offspring. Future studies should include pathologic analysis in existing longitudinal birth cohorts.
Current studies evaluating paternal obesity and offspring are insufficient and primarily limited to mouse models. Including paternal data in longitudinal birth cohorts will be crucial to identify whether paternal weight, diet, or both affect offspring in a clinically relevant way. Early indications from mouse models suggest an impact on hepatic lipid metabolism that could be additive if there is maternal obesity. However, further analysis of preclinical models with mechanistic studies will be key.
The most powerful outcome from studies of perinatal determinants of NAFLD could be biomarker discovery. Identifying circulating biomarkers that predict long‐term risk in existing preclinical models will be an important step. Ultimately, these findings can be translated to circulating biomarker identification in humans. These predictive factors will be essential in recognizing which patients will achieve maximum benefit from preventive therapies.
Given that epigenetic changes occur in the setting of parental obesity, their possible transmission across multiple generations remains an important question. Although preclinical rodent models are beginning to address this question, studying changes in multiple generations of mice is costly and time‐intensive. Models with a shorter generation time (i.e., Caenorhabditis elegans, Drosophila, or zebrafish) could provide rapid, cost‐effective, and large‐scale analysis of the transgenerational impact of obesity on NAFLD.
Significant progress has been made in establishing the role of maternal diet in offspring NAFLD risk. However, studies of interventions to prevent developmental programming of offspring liver disease require expansion. Although the best approach still needs identification, the recent study from Zhou et al. also highlights the importance of correct timing for each intervention.( 64 ) Depending on the treatment, administration may occur before pregnancy, perinatally, or postnatally, with consideration for the intervention’s maternal impact. Targeting these important questions will illuminate the mechanisms underlying parental diet programming of developmental liver phenotypes. Understanding these mechanisms will ultimately drive the development of preventive approaches to attenuating NAFLD in a rapidly growing population of patients at risk.
Acknowledgments
The author thanks Dr. Nicholas Davidson for his guidance and assistance in editing the manuscript, Astrid Rodriguez‐Velez and Anne Dolan at InPrint for their assistance with illustrations, and InPrint for their editing assistance.
Supported by the American Gastroenterological Association Research Foundation (P19‐04361) and the National Institute of Diabetes and Digestive and Kidney Diseases (DK‐020579, DK‐052574, DK‐056342, and DK‐122018).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Spieker EA, Pyzocha N. Economic impact of obesity. Prim Care 2016;43:83‐95, viii‐ix. [DOI] [PubMed] [Google Scholar]
- 2. Hinkle SN, Sharma AJ, Kim SY, Park S, Dalenius K, Brindley PL, et al. Prepregnancy obesity trends among low‐income women, United States, 1999‐2008. Matern Child Health J 2012;16:1339‐1348. [DOI] [PubMed] [Google Scholar]
- 3. Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005;115:e290‐e296. [DOI] [PubMed] [Google Scholar]
- 4. Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G. Glucose tolerance and insulin secretion in children of mothers with pregestational IDDM or gestational diabetes. Diabetologia 1997;40:1094‐1100. [DOI] [PubMed] [Google Scholar]
- 5. Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G. Overweight and obesity in infants of mothers with long‐term insulin‐dependent diabetes or gestational diabetes. Int J Obes Relat Metab Disord 1997;21:451‐456. [DOI] [PubMed] [Google Scholar]
- 6. Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 1989;2:577‐580. [DOI] [PubMed] [Google Scholar]
- 7. Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. BMJ 1990;301:259‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lumey LH, Van Poppel FW. The Dutch famine of 1944‐45: mortality and morbidity in past and present generations. Soc Hist Med 1994;7:229‐246. [DOI] [PubMed] [Google Scholar]
- 9. Wang N, Wang X, Li Q, Han B, Chen Y, Zhu C, et al. The famine exposure in early life and metabolic syndrome in adulthood. Clin Nutr 2017;36:253‐259. [DOI] [PubMed] [Google Scholar]
- 10. Ayonrinde OT, Oddy WH, Adams LA, Mori TA, Beilin LJ, de Klerk N, et al. Infant nutrition and maternal obesity influence the risk of non‐alcoholic fatty liver disease in adolescents. J Hepatol 2017;67:568‐576. [DOI] [PubMed] [Google Scholar]
- 11. Ayonrinde OT, Adams LA, Mori TA, Beilin LJ, de Klerk N, Pennell CE, et al. Sex differences between parental pregnancy characteristics and nonalcoholic fatty liver disease in adolescents. Hepatology 2018;67:108‐122. [DOI] [PubMed] [Google Scholar]
- 12. Patel S, Lawlor DA, Callaway M, Macdonald‐Wallis C, Sattar N, Fraser A. Association of maternal diabetes/glycosuria and pre‐pregnancy body mass index with offspring indicators of non‐alcoholic fatty liver disease. BMC Pediatr 2016;16:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bellatorre A, Scherzinger A, Stamm E, Martinez M, Ringham B, Dabelea D. Fetal overnutrition and adolescent hepatic fat fraction: the exploring perinatal outcomes in children study. J Pediatr 2018;192:165‐170.e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Modi N, Murgasova D, Ruager‐Martin R, Thomas EL, Hyde MJ, Gale C, et al. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr Res 2011;70:287‐291. [DOI] [PubMed] [Google Scholar]
- 15. Brumbaugh DE, Tearse P, Cree‐Green M, Fenton LZ, Brown M, Scherzinger A, et al. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J Pediatr 2013;162:930‐936.e931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, et al. Maternal high‐fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009;119:323‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, et al. Maternal high‐fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009;50:1796‐1808. [DOI] [PubMed] [Google Scholar]
- 18. Gutierrez Sanchez LH, Tomita K, Guo Q, Furuta K, Alhuwaish H, Hirsova P, et al. Perinatal nutritional reprogramming of the epigenome promotes subsequent development of nonalcoholic steatohepatitis. Hepatol Commun 2018;2:1493‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oben JA, Mouralidarane A, Samuelsson AM, Matthews PJ, Morgan ML, McKee C, et al. Maternal obesity during pregnancy and lactation programs the development of offspring non‐alcoholic fatty liver disease in mice. J Hepatol 2010;52:913‐920. [DOI] [PubMed] [Google Scholar]
- 20. Thompson MD, Cismowski MJ, Trask AJ, Lallier SW, Graf AE, Rogers LK, et al. Enhanced steatosis and fibrosis in liver of adult offspring exposed to maternal high‐fat diet. Gene Expr 2016;17:47‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson MD, Derse A, Ferey J, Reid M, Xie Y, Christ M, et al. Transgenerational impact of maternal obesogenic diet on offspring bile acid homeostasis and nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab 2019;316:E674‐E686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bayol SA, Simbi BH, Fowkes RC, Stickland NC. A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic fatty liver disease in rat offspring. Endocrinology 2010;151:1451‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shankar K, Kang P, Harrell A, Zhong Y, Marecki JC, Ronis MJ, et al. Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology 2010;151:2577‐2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamaguchi R, Nakagawa Y, Liu YJ, Fujisawa Y, Sai S, Nagata E, et al. Effects of maternal high‐fat diet on serum lipid concentration and expression of peroxisomal proliferator‐activated receptors in the early life of rat offspring. Horm Metab Res 2010;42:821‐825. [DOI] [PubMed] [Google Scholar]
- 25. Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, et al. A maternal high‐fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J 2012;26:5106‐5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magliano DC, Bargut TC, de Carvalho SN, Aguila MB, Mandarim‐de‐Lacerda CA, Souza‐Mello V. Peroxisome proliferator‐activated receptors‐alpha and gamma are targets to treat offspring from maternal diet‐induced obesity in mice. PLoS One 2013;8:e64258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nguyen LT, Chen H, Zaky A, Pollock C, Saad S. SIRT1 overexpression attenuates offspring metabolic and liver disorders as a result of maternal high‐fat feeding. J Physiol 2019;597:467‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC‐1{alpha}. J Biol Chem 2005;280:16456‐16460. [DOI] [PubMed] [Google Scholar]
- 29. Wankhade UD, Zhong Y, Kang P, Alfaro M, Chintapalli SV, Thakali KM, et al. Enhanced offspring predisposition to steatohepatitis with maternal high‐fat diet is associated with epigenetic and microbiome alterations. PLoS One 2017;12:e0175675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aharoni‐Simon M, Hann‐Obercyger M, Pen S, Madar Z, Tirosh O. Fatty liver is associated with impaired activity of PPARγ‐coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab Invest 2011;91:1018‐1028. [DOI] [PubMed] [Google Scholar]
- 31. Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol 2010;53:934‐940. [DOI] [PubMed] [Google Scholar]
- 32. Mouralidarane A, Soeda J, Sugden D, Bocianowska A, Carter R, Ray S, et al. Maternal obesity programs offspring non‐alcoholic fatty liver disease through disruption of 24‐h rhythms in mice. Int J Obes (Lond) 2015;39:1339‐1348. [DOI] [PubMed] [Google Scholar]
- 33. Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2004;2:e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grimaldi B, Bellet MM, Katada S, Astarita G, Hirayama J, Amin RH, et al. PER2 controls lipid metabolism by direct regulation of PPARγ. Cell Metab 2010;12:509‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soeda J, Cordero P, Li J, Mouralidarane A, Asilmaz E, Ray S, et al. Hepatic rhythmicity of endoplasmic reticulum stress is disrupted in perinatal and adult mice models of high‐fat diet‐induced obesity. Int J Food Sci Nutr 2017;68:455‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Jesus DF, Orime K, Kaminska D, Kimura T, Basile G, Wang CH, et al. Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. J Clin Invest 2020;130:2391‐2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suter MA, Ma J, Vuguin PM, Hartil K, Fiallo A, Harris RA, et al. In utero exposure to a maternal high‐fat diet alters the epigenetic histone code in a murine model. Am J Obstet Gynecol 2014;210:463.e1‐ 463.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aagaard‐Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, et al. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol 2008;41:91‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strakovsky RS, Zhang X, Zhou D, Pan YX. Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J Physiol 2011;589:2707‐2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang J, Zhang F, Didelot X, Bruce KD, Cagampang FR, Vatish M, et al. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor‐2 and key microRNAs in the adult offspring. BMC Genom 2009;10:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benatti RO, Melo AM, Borges FO, Ignacio‐Souza LM, Simino LA, Milanski M, et al. Maternal high‐fat diet consumption modulates hepatic lipid metabolism and microRNA‐122 (miR‐122) and microRNA‐370 (miR‐370) expression in offspring. Br J Nutr 2014;111:2112‐2122. [DOI] [PubMed] [Google Scholar]
- 42. Povero D, Eguchi A, Li H, Johnson CD, Papouchado BG, Wree A, et al. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS One 2014;9:e113651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soderborg TK, Clark SE, Mulligan CE, Janssen RC, Babcock L, Ir D, et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat Commun 2018;9:4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Myles IA, Fontecilla NM, Janelsins BM, Vithayathil PJ, Segre JA, Datta SK. Parental dietary fat intake alters offspring microbiome and immunity. J Immunol 2013;191:3200‐3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ma J, Prince AL, Bader D, Hu M, Ganu R, Baquero K, et al. High‐fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model. Nat Commun 2014;5:3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mouralidarane A, Soeda J, Visconti‐Pugmire C, Samuelsson AM, Pombo J, Maragkoudaki X, et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013;58:128‐138. [DOI] [PubMed] [Google Scholar]
- 47. Xue Y, Wang H, Du M, Zhu MJ. Maternal obesity induces gut inflammation and impairs gut epithelial barrier function in nonobese diabetic mice. J Nutr Biochem 2014;25:758‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Newton KP, Feldman HS, Chambers CD, Wilson L, Behling C, Clark JM, et al. Low and high birth weights are risk factors for nonalcoholic fatty liver disease in children. J Pediatr 2017;187:141‐146.e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Africa JA, Behling CA, Brunt EM, Zhang N, Luo Y, Wells A, et al. In Children with nonalcoholic fatty liver disease, zone 1 steatosis is associated with advanced fibrosis. Clin Gastroenterol Hepatol 2018;16:438‐446.e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, et al. Multigenerational epigenetic adaptation of the hepatic wound‐healing response. Nat Med 2012;18:1369‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ornellas F, Souza‐Mello V, Mandarim‐de‐Lacerda CA, Aguila MB. Programming of obesity and comorbidities in the progeny: lessons from a model of diet‐induced obese parents. PLoS One 2015;10:e0124737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li J, Lu YP, Tsuprykov O, Hasan AA, Reichetzeder C, Tian M, et al. Folate treatment of pregnant rat dams abolishes metabolic effects in female offspring induced by a paternal pre‐conception unhealthy diet. Diabetologia 2018;61:1862‐1876. [DOI] [PubMed] [Google Scholar]
- 53. Terashima M, Barbour S, Ren J, Yu W, Han Y, Muegge K. Effect of high fat diet on paternal sperm histone distribution and male offspring liver gene expression. Epigenetics 2015;10:861‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Youngson NA, Lecomte V, Maloney CA, Leung P, Liu J, Hesson LB, et al. Obesity‐induced sperm DNA methylation changes at satellite repeats are reprogrammed in rat offspring. Asian J Androl 2016;18:930‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de Castro BT, Alm PS, Krook A, Barres R, Zierath JR. Paternal high‐fat diet transgenerationally impacts hepatic immunometabolism. FASEB J 2019;33:6269‐6280. [DOI] [PubMed] [Google Scholar]
- 56. Hardy T, Zeybel M, Day CP, Dipper C, Masson S, McPherson S, et al. Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non‐alcoholic fatty liver disease. Gut 2017;66:1321‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol 2012;56:900‐907. [DOI] [PubMed] [Google Scholar]
- 58. Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005;42:641‐649. [DOI] [PubMed] [Google Scholar]
- 59. Brunt EM, Kleiner DE, Wilson LA, Unalp A, Behling CE, Lavine JE, et al. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD‐Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009;49:809‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez‐Pinto H, et al. Accumulation of duct cells with activated YAP parallels fibrosis progression in non‐alcoholic fatty liver disease. J Hepatol 2015;63:962‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nobili V, Carpino G, Alisi A, Franchitto A, Alpini G, De Vito R, et al. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012;56:2142‐2153. [DOI] [PubMed] [Google Scholar]
- 62. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393‐1405. [DOI] [PubMed] [Google Scholar]
- 63. Carpino G, Nobili V, Renzi A, De Stefanis C, Stronati L, Franchitto A, et al. Macrophage activation in pediatric nonalcoholic fatty liver disease (NAFLD) correlates with hepatic progenitor cell response via Wnt3a pathway. PLoS One 2016;11:e0157246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou Y, Peng H, Xu H, Li J, Golovko M, Cheng H, et al. Maternal diet intervention before pregnancy primes offspring lipid metabolism in liver. Lab Invest 2020;100:553‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jonscher KR, Stewart MS, Alfonso‐Garcia A, DeFelice BC, Wang XX, Luo Y, et al. Early PQQ supplementation has persistent long‐term protective effects on developmental programming of hepatic lipotoxicity and inflammation in obese mice. FASEB J 2017;31:1434‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jia D, Duan F, Peng P, Sun L, Ruan Y, Gu J. Pyrroloquinoline‐quinone suppresses liver fibrogenesis in mice. PLoS One 2015;10:e0121939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Paul HA, Collins KH, Nicolucci AC, Urbanski SJ, Hart DA, Vogel HJ, et al. Maternal prebiotic supplementation reduces fatty liver development in offspring through altered microbial and metabolomic profiles in rats. FASEB J 2019;33:5153‐5167. [DOI] [PubMed] [Google Scholar]
- 68. Pace RM, Prince AL, Ma J, Belfort BDW, Harvey AS, Hu M, et al. Modulations in the offspring gut microbiome are refractory to postnatal synbiotic supplementation among juvenile primates. BMC Microbiol 2018;18:28. [DOI] [PMC free article] [PubMed] [Google Scholar]