

Abstract
Acute liver injury (ALI) is associated with multiple cellular events such as necrosis, apoptosis, oxidative stress and inflammation, which can lead to liver failure. In this study, we demonstrate a new role of microRNA (miR)‐208a in ALI. ALI was induced in wild‐type (WT) and miR‐208a knockout (KO) mice by CCl4 administration. Increased alanine aminotransferase and decreased hepatic miR‐208a levels were found in WT mice after acute CCl4 treatment. Histopathological evaluations revealed increased necrosis and decreased inflammation in miR‐208a KO compared with WT mice after CCl4 treatment. CCl4 treatment induced a higher alanine aminotransferase elevation and increased numbers of circulating extracellular vesicles (exosomes and microvesicles) in miR‐208a KO compared with WT mice. We found increased CCl4‐induced nuclear factor kappa B activation and tumor necrosis factor‐α induction and decreased monocyte chemoattractant protein 1 levels in miR‐208a KO compared with WT mice. Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling assay indicated aggravated hepatic apoptosis and necrosis in CCl4 ‐treated miR‐208a KO compared with WT mice. CCl4 treatment induced a greater increase in cleaved caspase‐8, p18, and caspase‐3 in miR‐208a KO compared with WT mice. p53 is involved in various cell death pathways, including necrosis and apoptosis. Our in silico analysis revealed p53 as a predicted miR‐208a target, and we found enhanced p53 and cyclophilin D protein expressions in miR‐208a KO mice after CCl4 treatment. Increased liver injury in miR‐208a KO mice was further associated with increased Bax (B cell lymphoma 2–associated X protein) and p21 expression. Our in vitro results indicated a role of miR‐208a in cell death. We found that CCl4‐induced cytotoxicity was partially rescued by miR‐208a overexpression in RAW macrophages. Altogether, our results revealed a role of miR‐208a in ALI in mice and suggest a role for miR‐208a in regulating cell death.
In this manuscript, we demonstrate a new role of miR‐208a in acute liver injury. Our results revealed a novel role of miR‐208a in acute liver injury in mice and suggest a role for miR‐208a in the regulation of hepatocyte apoptosis and necrosis.

Abbreviations
- ALF
acute liver failure
- ALI
acute liver injury
- ALT
alanine aminotransferase
- Bax
B cell lymphoma 2–associated X protein
- Bcl2
B cell lymphoma 2
- Bid
BH3‐interacting domain death agonist
- Cq
quantification cycle
- CypD
cyclophilin D
- EMSA
electrophoretic mobility shift assay
- EV
extracellular vesicle
- H&E
hematoxylin and eosin
- KO
knockout
- LDH
lactate dehydrogenase
- MCP1
monocyte chemoattractant protein 1
- miRNA
microRNA
- MMP
matrix metalloproteinase
- MPTP
mitochondrial permeability transition pore opening
- mRNA
messenger RNA
- NF‐κB
nuclear factor kappa B
- ns
nonsignificant
- PCNA
proliferating cell nuclear antigen
- PDCD4
programmed cell‐death 4
- RT‐PCR
reverse‐transcription polymerase chain reaction
- α‐SMA
α smooth muscle actin
- TNFα
tumor necrosis factor α
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- UTR
untranslated region
- VEGF
vascular endothelial growth factor
- WT
wild type
Acute liver injury (ALI) is the leading cause of liver failure.( 1 ) Oxidative stress, necrosis, apoptosis, and inflammation are the main events of ALI, which in some cases can lead to the liver failure. Acute liver failure (ALF) can be caused by various etiologies including drugs, viral hepatitis, and ischemia.( 1 ) ALF is a life‐threatening condition and has a high mortality rate.( 1 ) Liver transplantation is a life‐saving therapy in severe ALF cases. The ALI model of CCl4 has been used widely in the hepatology field, as it induces oxidative stress, apoptosis, necrosis, inflammation, and fibrosis in mice.
microRNAs (miRNAs) are a class of regulatory small noncoding RNAs that have revolutionized our understanding of the intricate network of gene regulation. miRNAs have been shown to play pivotal roles in almost every cellular process. Ample studies have demonstrated various roles of miRNAs in liver diseases, including ALI. miR‐208a is one of the most studied heart‐enriched miRNAs and is a key modulator of pivotal heart functions.( 2 , 3 ) miR‐208a belongs to a member of the miRNA family that also contains miR‐208b and is encoded by an intronic region of the Myh6 gene.( 3 ) In recent years, other roles of miR‐208a in different organs/tissue have emerged. For instance, miR‐208a was shown to be involved in various cancers including gastric cancer,( 4 ) breast cancer,( 5 ) lung cancer cells,( 6 ) and hepatocellular carcinoma.( 7 ) However, the role of miR‐208a in ALI is not known. In this report, we identified a protective role of miR‐208a in CCl4‐induced ALI.
Nuclear factor kappa B (NF‐κB) is a master transcription factor that regulates both hepatocyte survival and proliferation. NF‐κB also plays a fundamental role in the inflammatory signaling pathways in the liver.( 8 ) Depending on stimuli, NF‐κB mediates both pro‐inflammatory and anti‐apoptotic responses. The NF‐κB‐regulated gene tumor necrosis factor α (TNFα), through the TNF‐R1 pathway, can lead to hepatocyte apoptosis by recruiting the adapter proteins TRADD (TNFR1‐associated death domain protein and FADD (Fas‐associated death domain protein), which subsequently recruit procaspase‐8.( 9 ) The active caspase‐8 proteolytically activates several effector caspases, and mediates apoptosis.( 9 )
The tumor suppressor p53 plays a key role in cellular stress and is activated by cellular stress. Depending on the type of stress signals, cell type, and cellular context during the stress exposure and co‐activators, activated p53 causes either growth arrest, senescence, or apoptosis.( 10 ) Recent studies have revealed a noncanonical role of p53 in necrosis, necroptosis, and ferroptosis.( 10 , 11 ) p53 regulates necrotic cell death by activating receptor interacting protein (RIP) independent signaling pathways that include induction of mitochondrial permeability transition pore opening (MPTP) either through poly (adenosine diphosphate ribose) polymerase 1 (PARP1) activation or interaction with cyclophilin‐D or regulating Bax (B cell lymphoma 2–associated X protein)/Bak (B cell lymphoma 2 homologous antagonist/killer) expression to elicit necrosis.( 11 )
p53 induces apoptosis using two distinct apoptotic signaling pathways: extrinsic and intrinsic. The extrinsic pathway involves engagement of death receptors that belong to the TNF family, which leads to the activation of caspases including caspase‐8 and caspase‐3.( 12 ) The intrinsic pathway is triggered in response to DNA damage and release of cytochrome c to the cytoplasm.
Cyclin‐dependent kinase inhibitor p21 is a p53‐inducible gene, which plays a key role in the cellular response to DNA damage; it is transcriptionally regulated by both p53‐dependent and p53‐independent mechanisms.( 13 ) Depending on the type of stimuli, cell type and stage, p21 acts as either pro‐apoptotic or anti‐apoptotic or pro‐necrotic.( 14 , 15 )
In the mouse model of ALI, we show that miR‐208a knockout (KO) mice exhibited increased liver injury with a subsequent increase in extracellular vesicles (EVs). A higher induction of the NF‐κB pathway and genes involved in necrotic and apoptotic cell death was observed in miR‐208a KO mice.
Materials and Methods
Animal Studies
Ten‐week‐old to 12‐week‐old male C57Bl/6 wild‐type (WT) or miR‐208a‐deficient (KO, total body) mice were used to induce ALI. This study was approved by the University of Massachusetts Medical School (UMMS) Institutional Animal Use and Care Committee (Worcester, MA). The breeding colonies of WT and miR‐208a KO mice were maintained in the animal facility of UMMS. Breeding pairs of miR‐208a KO mice were kindly provided by Dr. Eric Olsen, UT Southwestern Medical Center (Dallas, TX).( 2 ) To induce ALI, male WT or miR‐208a KO mice (n = 6) were injected with either a single dose of corn oil (vehicle control) or CCl4 (1 μL/g intraperitoneally), diluted 1:3 in corn oil as described,( 16 ) and sacrificed after 48 hours of treatment. At the conclusion of the experiment, blood was collected and mice were sacrificed. Liver tissues were collected and snap‐frozen in liquid nitrogen for proteins and in RNA Later (Qiagen, Hilden, Germany) for RNA extraction and analysis. All samples were stored at −80°C.
Blood and Tissue Sample Processing
Blood was collected in serum collection tubes (BD Biosciences, San Jose, CA), kept at 4°C for 30 minutes, and centrifuged at 12,000g for 10 minutes at 4°C. The upper transparent layer was transferred into a new tube and stored at −80°C. A portion of the liver was fixed in 10% phosphate‐buffered formalin, embedded in paraffin blocks, processed histologically, and stained with hematoxylin and eosin (H&E) staining. Histology slides were blindly scored by pathologists for necrosis, inflammation, steatosis, acidophil bodies, regenerative changes, and ballooning degeneration under light microscopy at ×200 and ×400 magnification. Chi‐square tests were used to evaluate the categorical data and studied for histopathological changes using GraphPad Prism 8 software (GraphPad Software, San Diego, CA).
Serum Alanine Aminotransferase Assay
To determine the liver injury at the enzymatic level, serum alanine aminotransferase (ALT) levels were measured using a kinetic method (TECO Diagnostics, Anaheim, CA) according to the manufacturer’s instructions.
Nanoparticle Tracking Analysis
The concentration and diameter of EVs from serum samples were analyzed using the NanoSight NS300 system (NanoSight, Amesbury, United Kingdom) equipped with fast video capture and Nanoparticle Tracking Analysis (NTA) software.( 17 ) Before the measurement, the instrument was calibrated with 100‐nm polystyrene beads (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. The samples were captured for 30 seconds at room temperature. NTA software was used to process the video captures and to quantify the concentration (particles/milliliter) and size distribution (in nanometers) of the measured particles. Measurements were performed in triplicates.
Enzyme‐Linked Immunosorbent Assay
Whole liver cell lysates were used to measure the protein levels of monocyte chemoattractant protein 1 (MCP1), using enzyme‐linked immunosorbent assay as described by the manufacturer (BioLegend Inc., San Diego, CA).
TUNEL Assay
The terminal deoxynucleotidyl transferase‐mediated dUTP‐nick end labeling (TUNEL) assay was used to demonstrate the apoptotic cell death in the liver, which detects 3′ hydroxyl ends in fragmented DNA. The staining was performed on paraffin‐embedded liver sections using the in situ cell death detection kit (TUNEL assay, Abcam, Cambridge, MA) according to the manufacturer’s instructions. DNase I recombinant was used as the positive control. Imaging of TUNEL positive was performed with a light microscope (Zeiss Axiolab, Carl Zeiss Inc., Germany).
Primary Cell Isolation and RNA Extraction
Kupffer cells or hepatocytes were isolated from WT mice using the methods previously described.( 16 , 18 ) Total RNA was extracted from the livers or Kupffer cells or primary hepatocytes using Direct‐zol RNA MiniPrep with on column DNA digestion (Zymo Research, Irvine, CA).
Quantitative Reverse‐Transcription Polymerase Chain Reaction
For messenger RNA (mRNA) transcription analysis, complementary DNA (cDNA) was transcribed with iScript Reverse Transcription Supermix for quantitative reverse‐transcription polymerase chain reaction (RT‐PCR) (Bio‐Rad, Hercules, CA). Quantitative RT‐PCR was performed using the iCycler (Bio‐Rad). All results were normalized to 18S mRNA expression. Primer sequences were the same as described previously.( 17 ) For miRNA quantification, cDNA synthesis and quantitative RT‐PCR were performed using TaqMan miRNA assays (Ambion, Inc., Austin, TX). The quantification cycle (Cq) of target miRNA was normalized to that of snoRNA‐202 Cq as described.( 16 )
Western Blot Analysis
Whole cell lysates were extracted from the liver as described,( 16 ) and equal amounts of protein were used for western blotting. The denatured samples were separated in polyacrylamide gel, transferred onto nitrocellulose membrane, and probed with specific primary antibodies followed by horseradish peroxidase–labeled secondary antibodies. The following primary antibodies were used: caspase 8 (sc‐81662; Santa Cruz Biotechnology, Inc., Dallas, TX), cleaved caspase‐3 (9661; Cell Signaling Technology, Danvers, MA), caspase‐3 (14220; Cell Signaling Technology), truncated BH3‐interacting domain death agonist (BID) (MAB860; R&D Systems, Minneapolis, MN), p53 (sc‐393031; Santa Cruz Biotechnology), proliferating cell nuclear antigen (PCNA) (sc‐56; Santa Cruz Biotechnology), cyclin D1 (SC‐795; Santa Cruz Biotechnology), Bax (L957025; Cell Signaling Technology), cyclophilin D (CypD) (45‐5900; Thermo Fisher Scientific/Invitrogen [Carlsbad, CA]), and B cell lymphoma 2 (Bcl‐2) (SC‐7382; Santa Cruz Biotechnology). Beta actin or glyceraldehyde 3‐phosphate dehydrogenase (Abcam, Cambridge, MA, USA) was used as a loading control.
Electrophoretic Mobility Shift Assay
Nuclear proteins were isolated from liver samples as described.( 16 ) 5 μg nuclear protein was used for conducting electrophoretic mobility shift assay (EMSA) using consensus, double‐stranded oligonucleotide‐specific for NF‐κB (Santa Cruz Biotechnology) as previously described.( 16 )
In Vitro CCl4 Treatment and Cytotoxicity Assay
RAW 264.7 macrophages were cultured and maintained in Dulbecco’s modified high‐glucose medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (HyClone Laboratories, Logan, UT) at 37°C in a 5% CO2 atmosphere as described previously.( 18 ) For in vitro treatments, cells were seeded in 24‐well plates and on the next day were treated either with different concentration of CCl4 (0.1%, 0.2%, 0.3%, and 0.4%) (Sigma Aldrich, St. Louis, MO) dissolved in 0.25% DMSO (Sigma Aldrich) or DMSO (0.25%) for 6 hours. Others have used these doses of CCl4 in HepG2 and Hep3B cells for in vitro treatments.( 19 ) Cell‐free supernatants were used to determine cytotoxicity using lactate dehydrogenase (LDH)–cytotoxicity assay kit II according to the manufacturer’s instructions (Abcam).
Transfection
For miR‐208a overexpression, RAW 264.7 macrophages were either transfected with a negative control miRNA or miR‐208a mimic at 2 pM for 24 hours (Applied Biosystems, Foster City, CA) using Lipofectamine RNAi max reagent (Thermo Fisher Scientific) as described previously.( 18 ) Some cells were treated or not with 0.3% CCl4 for the last 6 hours of the experiment. Cell‐free supernatants were used to determine cytotoxicity using the LDH‐cytotoxicity assay kit II according to the manufacturer’s instructions (Abcam).
Statistical Analysis
Statistical significance was determined using either the nonparametric Mann‐Whitney U test or nonparametric one‐way analysis of variance. Data are presented as the mean ± standard error and are considered statistically significant at P < 0.05.
Results
miRNA‐208a KO Mice Display Aggravated CCl4‐Induced Liver Injury
miR‐208a is a highly studied heart miRNA and has recently been shown to be involved in cellular death in gastric and colorectal cancers( 4 , 20 ); however, its role in ALI is yet to be determined. We hypothesized that miR‐208a regulates hepatocyte cell death in ALI. Using a single‐dose CCl4‐induced liver injury, we found decreased hepatic levels of miR‐208a in WT mice compared with oil‐treated control (Fig. 1A). To assess the biological role of miR‐208a in ALI, we tested miR‐208a KO male mice. We found that acute CCl4 treatment induced hepatocyte injury in WT mice, which was further amplified in miR‐208a KO mice. Serum ALT, a liver damage marker, was increased in WT mice, and a greater increase in its levels was found in miR‐208a KO mice after acute CCl4 treatment (Fig. 1B). Cellular death( 21 ) and hypoxic conditions( 22 ) are known to induce EV release in the circulation. Different types of diseases and liver injury models were shown to increase circulating exosomes and larger EVs.( 23 ) Therefore, we evaluated the number of EVs in the circulation and found that CCl4 treatment increased the circulating exosome (particle size = 40‐150 nM) and microvesicle (150‐500 nM) numbers in WT mice, and a further significant increase in exosomes and microvesicles was observed in miR‐208a KO mice (Fig. 1C).
FIG. 1.
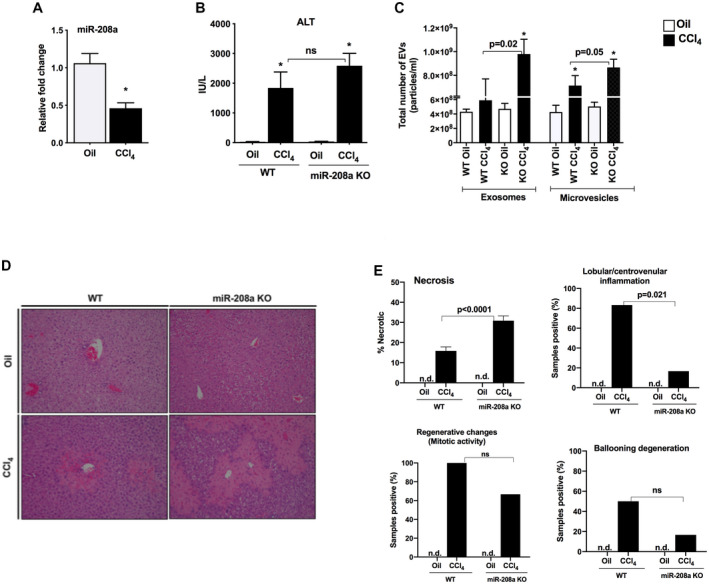
Increased liver injury in miR‐208a KO mice after acute CCl4 treatment. (A) miR‐208a levels were quantified from the livers of WT mice by quantitative PCR after 48 hours of CCl4 treatment, and snoRNA202 was used to normalize the Cq values of each sample. (B) WT or miR‐208a KO mice received a single dose of either oil (n = 4‐5) or CCl4 (n = 6) for 48 hours. Serum ALT levels were determined from serum samples to assess liver damage. (C) Total number of EVs, exosomes, and microvesicles were measured from serum samples using NanoSight. (D) Liver injury was evaluated by H&E staining of the liver sections. (E) Blinded histopathological evaluations of H&E‐stained liver sections were performed, and necrosis, lobular/centrivenular inflammation, regenerative changes (mitotic activity), and ballooning degeneration scoring. Chi‐square tests were used to evaluate the categorical data and studied for histopathological changes using GraphPad Prism 8 software. *P < 0.05 versus oil‐WT or oil‐KO treated mice. Abbreviations: n.d., not detected; ns, nonsignificant.
The H&E staining of liver sections also displayed a higher increase in hepatocyte injury in miR‐208a KO mice compared with WT mice (Fig. 1D). Furthermore, histopathological scoring of H&E‐stained liver sections revealed that CCl4 treatment induced significantly more necrosis (zone 3) and caused decreased inflammation in miR‐208a KO mice compared with WT mice (Fig. 1E, upper panels). Compared with WT CCl4‐treated mice, miR‐208a KO mice exhibited no significant decreases in regenerative changes (mitotic activity) and ballooning degeneration (Fig. 1E, lower panels). Detailed histopathological scores and case images are provided in Supporting Fig. S1. These results suggest that reduction in miR‐208a levels aggravated CCl4‐induced ALI.
Enhanced NF‐κB Activation in Mir‐208a KO Mice After CCl4 Treatment
Acute CCl4 treatment was shown to result in acute inflammation in the liver.( 24 ) Our histopathological analysis revealed a decrease in CCl4‐induced lobular/centrivenular inflammation in miR‐208a KO mice compared with WT mice (Fig. 1E and Supporting Information); therefore, we next checked the inflammation parameters. TNFα expression after CCl4 treatment was increased in WT mice compared with vehicle controls, and the extent of TNFα induction by CCl4 was significantly greater in miR‐208a KO mice compared with the WT (Fig. 2A). TNFα is regulated by different factors including NF‐κB,( 25 ) and we found a significant induction of NF‐κB DNA binding in WT mice after CCl4 treatment, as evaluated by EMSA (Fig. 2B). CCl4 treatment caused a further significant increase in NF‐κB activation in miR‐208a KO compared with WT mice (Fig. 2B). These findings corroborated with enhanced cell death in miR‐208a mice after CCl4 treatment, as both NF‐κB and TNFα were also shown to induce hepatocyte cell death in various models of liver injury.( 9 )
FIG. 2.
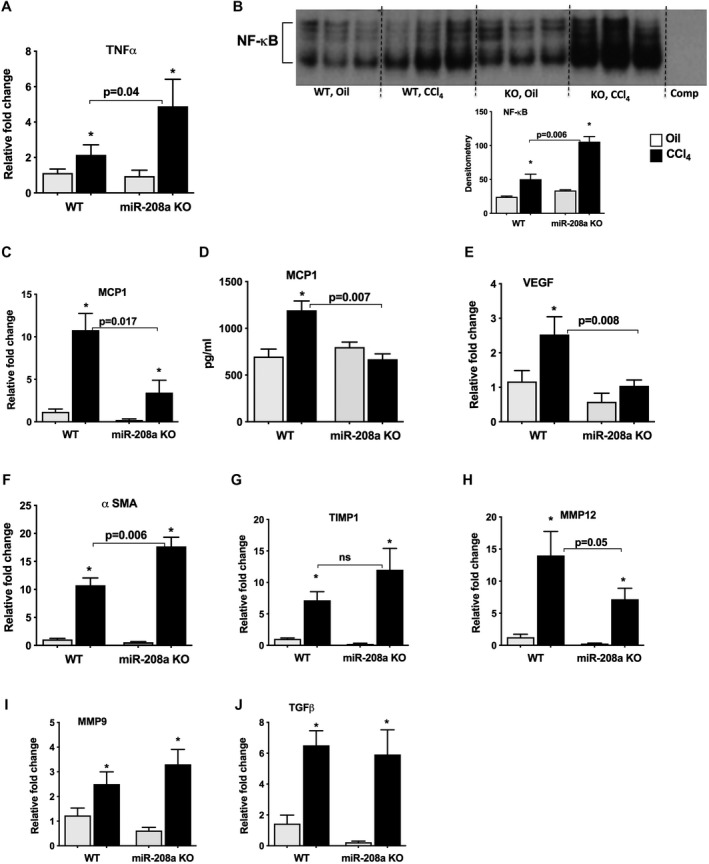
Enhanced NF‐κB activation in miR‐208a KO mice after acute CCl4 treatment. WT or miR‐208a KO mice received oil (n = 4‐5/group) or CCl4 (n = 6/group) treatment for 48 hours. (A) Total RNA was extracted from the liver samples, and the levels of TNFα were determined by quantitative PCR. (B) NF‐κB activation was determined by EMSA from nuclear liver extracts, and representative results are shown along with density units in the bar diagram (B). MCP1 mRNA levels were quantified using quantitative PCR (C), and protein levels of MCP1 were measured from whole cell liver extracts by enzyme‐linked immunosorbent assay (D). (E) VEGF expression was determined by quantitative PCR from RNA samples (E). mRNA levels of α‐SMA (F), TIMP1 (G), MMP12 (H), MMP9 (I), and TGF (J) were determined by quantitative PCR. 18S was used to normalize the Cq values of each sample. *P < 0.05 versus oil‐WT or oil‐KO treated mice. Abbreviation: comp, complementary sequence.
MCP1 is another NF‐κB‐regulated chemokine that recruits immune cells to the inflammation site, and recent studies suggest that MCP1 can play a dual role as an inflammatory chemokine and in the liver repair.( 24 ) We found a significant increase in MCP1, both at the mRNA (Fig. 2C) and protein (Fig. 2D) levels, in WT mice after CCl4 treatment. Consistent with our histopathological findings of reduced inflammatory cell presence in miR‐208a‐deficient livers, miR‐208a KO mice showed significant attenuation in MCP1 expression compared with WT mice (Fig. 2C,D). It has been reported that MCP‐1 plus Siglec‐9 treatment induces transcription of vascular endothelial growth factor (Vegf) and hepatocyte growth factor trophic factors for liver regeneration in a rat model of ALF.( 26 ) Consistent with this report, we found a significant induction in the mRNA levels of VEGF in WT mice; however, miR‐208a KO mice showed significant attenuation in VEGF induction after CCl4 treatment (Fig. 2E).
CCl4 also induces the expression of profibrotic genes( 27 ); therefore, we evaluated the expression of α smooth muscle actin (α‐SMA) and found increased levels in WT mice. The increase in α‐SMA was further augmented in miR‐208a KO mice at the mRNA level (Fig. 2F). No significant differences in the protein levels of α‐SMA were found between the genotypes at 48 hours after CCl4 administration. Activated hepatic stellate cells induce the expression of inhibitor of metalloproteinase 1 (TIMP‐1), which causes the inhibition of matrix metalloproteinase (MMP) activity and subsequent accumulation of matrix proteins in the extracellular space.( 28 ) Our results indicate that mRNA levels of TIMP1 were increased in WT mice, and a trend toward enhanced TIMP1 expression was found in miR‐208a KO mice (Fig. 2G). Next, we checked the expression of MMP and found a significant induction of MMP12 mRNA levels in WT mice and to a lesser extent in miR‐208a KO mice (Fig. 2H), suggesting a possible transient accumulation of matrix proteins in KO mice. CCl4 treatment induced MMP9 and TGFβ mRNA expression in both WT and miR‐208a KO mice to a similar extent (Fig. 2I,J).
Induction of Cellular Death Pathway in miR‐208a KO Mice After CCl4 Treatment
Since we found increased hepatocyte cell damage in miR‐208a KO mice, we postulated a role of miR‐208a in cellular death. TUNEL assay was performed to determine the type of cell death, and we found an increase in apoptotic cell death in miR‐208a KO mice compared with WT mice after CCl4 treatment (Fig. 3A). An increase in the cytosolic TUNEL staining was also observed in the livers of miR‐208a KO mice (Fig. 3A), which is characteristic of necrosis.( 29 ) Caspase‐8 is involved in apoptotic cell death,( 30 ) and miR‐208a was shown to modulate the expression of caspase‐8.( 31 ) We found an increase in p18, the cleaved form of caspase‐8, in WT mice, and a further augmentation in p18 form was detected in miR‐208a KO mice (Fig. 3B). CCl4 treatment also induced the levels of p43, another cleaved form of caspase‐8 in miR‐208a KO mice (Fig. 3B). We found baseline increases in procaspase‐8, caspase p18, and p43 levels in oil‐treated miR‐208a KO mice (Fig. 3B). Next, we checked the caspase‐3 levels and found a significant increase in cleaved caspase‐3 in miR‐208a KO mice compared with WT mice after CCl4 treatment (Fig. 3C). A baseline increase in cleaved caspase‐3 was also observed in oil‐treated miR‐208a KO mice, and a further significant increase in its levels was found after CCl4 treatment in KO mice (Fig. 3C). These results suggest increased activation of apoptotic cell death pathways in miR‐208a KO mice after oil treatment and even more extensively after CCl4 treatment.
FIG. 3.
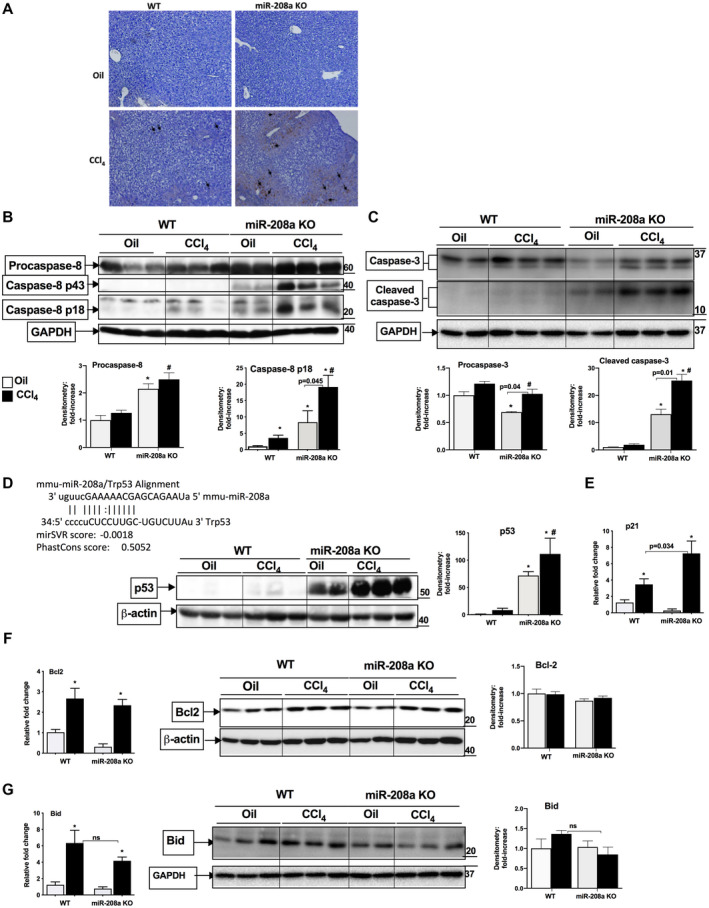
Induction of cellular death in miR‐208a KO mice after acute CCl4 treatment. WT or miR‐208a KO mice received oil (n = 4‐5/group) or CCl4 (n = 6/group) treatment for 48 hours. (A) TUNEL assay was performed on paraffin‐embedded liver sections. The arrows indicate dark brown nuclei for TUNEL‐positive cells. The cytoplasmic staining in miR‐208a KO represents necrosis. Western blot analysis was used to determine the protein levels of procaspase‐8, caspase‐8 (B) and caspase‐3 (C), and density units for procaspase‐8 and p18, active form of caspase‐8 (B), and procaspase‐3 and caspase‐3 (C) are shown in the bar diagram (n = 4 [oil], n = 6 [CCl4]). (D, upper panel) Predicted miR‐208a binding site at the 3′UTR region of p53. (D, lower panel) Western blot analysis for p53 protein and density units are shown in the bar diagram (n = 4 [oil], n = 6 [CCl4]). (E) p21 mRNA levels were determined by quantitative PCR. Western blot analysis for Bcl2 (F) and Bid (G) protein levels and density units are shown in the bar diagram (n = 3 [oil], n = 3 [CCl4]). Glyceraldehyde 3‐phosphate dehydrogenase (B,C,G) or β‐actin was used as a loading control. The mRNA levels of Bcl2 (F) and Bid (G) were determined by quantitative PCR. 18S was used to normalize the Cq values of each sample. *P < 0.05 versus oil‐treated WT mice, # P < 0.05 versus CCl4‐treated WT mice. Abbreviation: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
To gain insights into the mechanism of increased necrosis and apoptosis in miR‐208a KO mice, we carried out in silico analysis for miR‐208a target genes. Our in silico analysis revealed a miR‐208a‐predicted site at the 3′ untranslated region (UTR) region of the p53 gene (Fig. 3D, upper panel). In recent years, noncanonical functions of p53 have been discovered in various types of cell death, including necrosis and necroptosis.( 10 , 11 ) Evaluation of p53 protein levels in our CCl4 acute injury model showed a slight increase in p53 levels in WT mice at this time. However, in miR‐208a KO mice, the hepatic protein levels of p53 were increased in oil‐treated controls and the p53 increase was more robust in miR‐208a KO mice after CCl4 treatment compared with WT mice (Fig. 3D, lower panel). The p53 target gene, p21, is involved in both necrotic and apoptotic cell death,( 15 ) and down‐regulation of miR‐208a has been shown to cause increased apoptosis by targeting p21 in human lung cancer cells.( 6 ) We found increased p21 transcription in WT mice, which was further amplified in miR‐208a KO mice after CCl4 treatment (Fig. 3E). Next, we checked the expression of Bcl2 (anti‐apoptotic) and Bid (pro‐apoptotic) genes and found no changes in Bcl2 protein; however, its mRNA levels were increased to a similar extent between genotypes after CCl4 treatment (Fig. 3F). Compared with WT mice, Bid mRNA levels showed no significant decrease in miR‐208a KO mice after CCl4 treatment, and no significant change in the levels of total Bid protein was observed in either genotype (Fig. 3G).
Induction of Bax and CypD in miR‐208a KO Mice After CCl4 Treatment
p53 can induce necrosis through its effects on different genes involved in this process,( 11 ) and recently miR‐208a was shown to modulate Bax expression.( 31 ) Bax, a pro‐apoptotic gene, is also regulated by p53. Recent studies suggest a role of Bax in the MPTP formation, a key process in necrosis.( 11 ) We found that CCl4 treatment increased Bax protein levels in the livers of WT mice, and to a greater extent in miR‐208a KO mice (Fig. 4A). p53 can also elicit necrosis through its interaction with CypD and induces the opening of MPTP.( 11 ) Therefore, we checked the protein levels of CypD and found a slight increase in its expression in WT mice after CCl4 treatment (Fig. 4B). Compared with WT mice, CypD levels were increased in oil‐treated miR‐208a KO mice. CCl4 treatment caused a significant higher increase in CypD levels in miR‐208a KO compared with WT mice (Fig. 4B). Altogether, our results suggest that miR‐208a modulates cell death (necrosis and apoptosis) directly or indirectly involving p53‐CypD‐caspase8 axis in CCl4 ‐induced ALI.
FIG. 4.
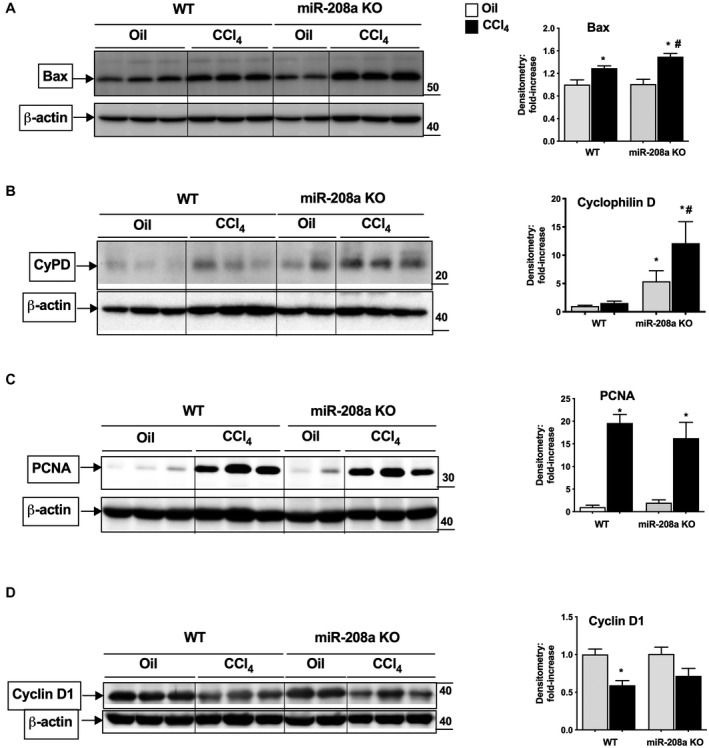
Increased expression of genes involved in necrosis in miR‐208a KO mice after acute CCl4 treatment. WT or miR‐208a KO mice received oil (n = 4‐5/group) or CCl4 (n = 6/group) treatment for 48 hours. Whole‐liver cell lysates were used to determine the protein levels of Bax (A), CypD (B), PCNA (C), and cyclin D1 (D) by western blot analysis. β‐actin was used as a loading control. Density units are shown in the bar diagram from mice, Bax (n = 4 [oil], n = 6 [CCl4]), CypD (n = 4 [oil]; n = 6 [CCl4]), PCNA (n = 3 [oil], n = 3 [CCl4]), and cyclin D1 (n = 3 [oil], n = 3 [CCl4]). *P < 0.05 versus oil‐treated WT mice, # P < 0.05 versus CCl4‐treated WT mice.
To determine whether there is any change in cell proliferation rate in miR‐208a KO mice compared with WT, we evaluated the proliferation parameters. We found induction in the protein levels of nuclear antigen marker associated with cell proliferation, PCNA, in WT mice, whereas miR‐208 KO mice showed a slight insignificant decrease in its levels after CCl4 treatment (Fig. 4C). The protein levels of cell cycle regulator, cyclinD1, were found to be decreased in both WT and miR‐208a KO mice after CCl4 treatment (Fig. 4D). Compared with WT mice, histopathological analysis also showed insignificant decreases in the regenerative changes in miR‐208a KO mice after CCl4 treatment (Fig. 1E). These results suggest that proliferation is not affected in miR‐208a KO mice, at least after 48 hours of the induction of acute liver injury.
miR‐208a Overexpression Attenuates CCl4‐Induced Cytotoxicity in RAW Macrophages
To further explore the role of miR‐208a in cellular death, we performed in vitro experiments. We analyzed miR‐208a levels in Kupffer cells and primary hepatocytes and found a higher expression of miR‐208a in Kupffer cells (average Cq of about 33.86) compared with hepatocytes (average Cq of about 38.87) (Fig. 5A). Because we found a higher level of miR‐208a in Kupffer cells, we optimized in vitro CCl4 treatment in RAW 264.7 macrophages using the same concentrations as previously described for hepatocyte cell lines.( 19 ) RAW macrophages contain cytochrome p450 genes for metabolism of drugs and organic compounds.( 32 )
FIG. 5.
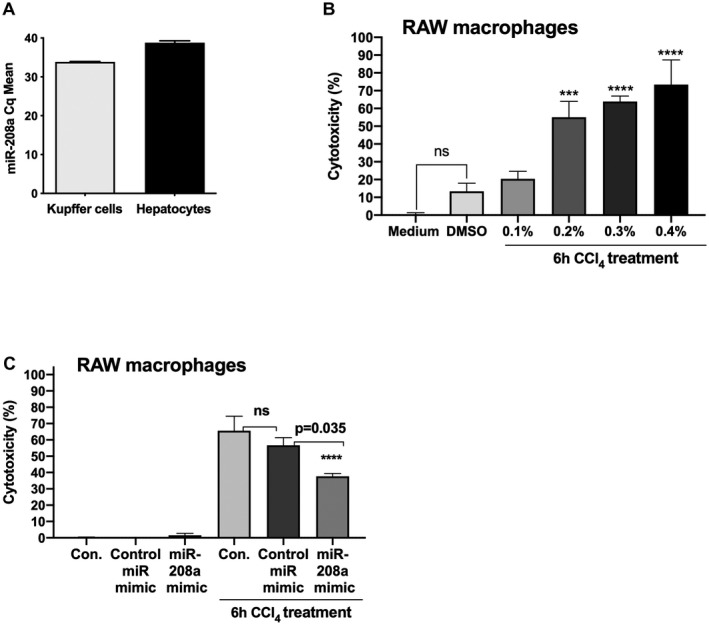
Overexpression of miR‐208a attenuates CCl4‐induced cytotoxicity in RAW macrophages. miR‐208a expression was determined from Kupffer cells (n = 4) and primary hepatocytes (n = 4) isolated from WT mice using quantitative PCR. (A) SnoRNA‐202 was used as an endogenous control to normalize Cq values. (B) RAW 264.7 macrophages were treated either with DMSO control (0.25%) or 0.1%, 0.2%, 0.3%, and 0.4% concentration of CCl4 for 6 hours. Cell‐free supernatants were used to test cytotoxicity using an LDH‐cytotoxicity assay kit II according to the manufacturer’s instructions (n = 7). (C) Raw macrophages were either transfected with negative control miR mimic or miR‐208a mimic at the concentration of 2 pM for 24 hours. Cells were either treated or not with 0.3% CCl4 for the last 6 hours of experiment. Cell‐free supernatants were used to test cytotoxicity using an LDH‐cytotoxicity assay kit II (n = 7). In vitro experiments were repeated three times, and results are shown as an average of two independent experiments. ***P < 0.001, ****P < 0.0001.
In vitro CCl4 treatment for 6 hours at the concentration of 0.2%, 0.3%, and 0.4% caused significant cytotoxicity in RAW macrophages (Fig. 5B). Cytotoxicity of approximately 55%, 63%, and 73% was found in RAW macrophages treated with 0.2%, 0.3%, and 0.4% CCl4, respectively, as measured by LDH assay (Fig. 5B). Therefore, for testing the potential protection of miR‐208a overexpression on cell death, 0.3% CCl4 concentrations were chosen in vitro. Transfection of miR‐208 mimic and not‐negative control caused an approximate 100‐fold increase in miR‐208a levels in RAW macrophages. Our results indicated that CCl4‐induced cytotoxicity was partially rescued by miR‐208a overexpression in RAW macrophages. CCl4 treatment induced about 65% cytotoxicity in control cells and about 56% in negative‐control miRNA‐treated cells, respectively, whereas miR‐208a‐overexpressed macrophages exhibited approximately 37% cytotoxicity 6 hours after 0.3% CCl4 treatment (Fig. 5C). These data suggest a role of miR‐208a in modulating cell death.
Discussion
ALI is characterized by excessive oxidative stress, cell death and inflammation, which in some cases can be fatal. In this study, we revealed a previously unknown role of miR‐208a in an ALI mouse model of CCl4. Earlier studies have demonstrated an imperative role of miR‐208a in heart diseases.( 2 , 3 ) Here, we discovered that miR‐208a KO mice were more susceptible to ALI compared with WT mice. We found that oil‐treated miR‐208a KO mice showed no baseline increases in ALT and in cell death (necrosis and apoptosis) after histopathological evaluations; however, caspase‐8, caspase‐3, p53, and CypD protein levels were increased even without induction of liver injury compared with WT mice. Furthermore, miR‐208a KO mice had exacerbated liver injury, apoptosis, and necrosis in the liver after CCl4 challenge compared with WT mice.
Our results indicated a decrease in hepatic miR‐208a levels after the induction of ALI. The baseline expression of miR‐208a in the liver is low (Cq ~35) compared with its high abundance in the heart. miR‐208a is co‐expressed from the Myh6 gene encoding fast myosin and is essential for the up‐regulation of slow myosin Myh7 and miR‐208b in the adult heart.( 2 ) Not much is reported about the transcriptional regulation of miR‐208a gene. Thyroid hormone signaling is shown to modulate miR‐208 levels.( 33 ) Our database search revealed Atf3, Nrf1, Arnt, and PPARg as some of the predicted transcriptional factor binding sites at the enhancer region of miR‐208a gene (genecards.org).
The liver injury in miR‐208a KO mice was associated with increased levels of circulating EVs after CCl4 treatment. Previous studies, including ours, have shown an increase in EV secretion in response to cellular stress.( 34 , 35 ) Interestingly, miR‐208a KO mice showed the greatest increase in circulating exosomes (40‐150 nm) that are smaller than the usual apoptotic bodies (>800 nm). Exosomes are typically released by stressed cells, and their release involves active cell‐sorting mechanisms through the endosome–multivesicular body intracellular pathways.( 35 ) Consistent with increased CCl4‐induced liver injury/cell death in miR‐208a KO mice, our results indicated enhanced activation of NF‐κB and increased TNFα levels in these mice. During liver injury, NF‐κB induces transcription of various target genes, including those of anti‐apoptotic functions, Bcl2 family, and inflammatory functions.( 36 ) NF‐κB has been implicated in hepatocyte apoptosis, necroptosis, and proliferation.( 36 ) TNFα is a pleiotropic cytokine and is involved in various liver processes such as inflammation, cell death and proliferation,( 37 ) and overproduction of TNFα leads to cell death in hepatocytes by TNF‐R pathway.( 36 )
The histopathological evaluations revealed less lobular/centrivenular inflammation associated with necrosis in miR‐208 KO mice, and consistent with this observation we found significant attenuation in CCl4‐induced MCP1 levels in miR‐208a KO mice. Recently, a role of MCP1 in liver repair has also been demonstrated in a rat model of ALF, in which it promoted the liver regeneration by inhibiting apoptosis and inducing trophic factors like VGEF.( 26 ) In line with this, induction of VEGF found in WT mice was attenuated in KO mice, suggesting that attenuated MCP1 levels might affect both liver inflammation and repair in miR‐208a KO mice. Tang et al. showed that ultraviolet‐induced p53 increase in mouse macrophages caused a decrease in LPS‐induced MCP1 production.( 38 ) p53 negatively regulates MCP1 through its binding to the 5′UTR region of the MCP1 gene,( 38 ) and we found a significant increase in p53 levels in miR‐208a KO mice after CCl4 treatment, suggesting a potential role of p53 in MCP1 regulation.
p53 is an essential cellular stress sensor that plays a vast role in various biological functions including apoptosis, necrosis, and necroptosis.( 10 , 11 ) Our bioinformatic in silico analysis revealed a miR‐208a binding site at the 3′UTR region of the p53 gene. p53 regulates various genes involved in both extrinsic and intrinsic apoptotic signaling pathways.( 12 ) p53 can activate the extrinsic pathway through engagement of Fas‐TNF‐R family genes, leading to the activation of various caspases, including caspase‐8 and caspase‐3, which in turn induces apoptosis.( 12 ) miR‐208a was also shown to modulate the expression of caspase‐8.( 31 ) We found increased protein levels of p53 and cleaved active forms of caspase‐8 (p18) and caspase‐3 (a downstream target of cleaved caspase‐8) in miR‐208a KO mice after ALI.
The intrinsic apoptosis pathway is activated in response to DNA damage and involves the Bcl‐2 family members that include various anti‐apoptotic (Bcl2) and pro‐apoptotic (Bax, Bid) genes.( 12 ) We did not find any significant changes in the protein levels of Bcl2 and Bid between both genotypes; however, a higher increase in Bax protein levels was found in miR‐208a KO mice after CCl4 treatment. Overexpression of miR‐208a in cardiomyocytes was shown to have an effect on Bax expression.( 31 ) Although Bax is known as an pro‐apoptotic molecule, recent studies suggest a role of Bax in necrosis.( 11 ) We cannot rule out the dual role of Bax in apoptotic and necrotic cell death in miR‐208a KO mice.
p53 mediates necrotic cell death by activating RIP‐independent signaling pathways and causes the induction of MPTP either through PARP1 activation or interaction with CypD or by regulating Bax/Bak expression.( 11 ) CypD is a key regulator of MPTP, and our results indicated enhanced expression of CypD in miR‐208a KO mice after CCl4 treatment. These results suggest that p53 might be inducing necrosis through its interaction with CypD and possibly by regulating Bax expression in miR‐208a KO mice. However, we cannot rule out the possibility of direct or indirect regulation of Bax by miR‐208a in inducing apoptosis and necrosis.
Interestingly, histopathological evaluations and ALT assay did not show any increase in cell death in oil‐treated miR‐208a KO mice; however, a baseline increase in the protein levels of p53, active forms of caspase‐8 and caspase‐3, and CypD was found in oil‐treated miR‐208a KO mice, suggesting that miR‐208a regulates these genes in general and after the induction of ALI. miR‐208a has been found to modulate the expression of a large cohort of genes (e.g., caspase‐8, Bax, Box, p21, programmed cell‐death 4 [PDCD4]) involved in cell death in other cell types.( 4 , 6 , 31 ) It was shown that miR‐208a overexpression in cardiomyocytes causes a reduction in pro‐caspase‐8 expression.( 31 ) Interestingly, pro‐caspase‐8 can dimerize and activate itself,( 39 ) and our results indicated an increase in pro‐caspase‐8 levels in the livers of miR‐208a KO mice. Adding to the evidence supporting the role of miR‐208a in modulating cell death, miR‐208a overexpression in gastric cancer cells has been shown to suppress apoptosis through PDCD4, with a subsequent decrease in cleaved caspase‐3 (a downstream target of cleaved caspase‐8).( 4 ) Another study demonstrated that overexpression of miR‐208a in the cardiomyocytes of neonatal rats can reduce the incidence of apoptosis.( 40 ) Supporting other findings for a role of miR‐208a in apoptosis, our data suggest that miR‐208a might have an intrinsic role in modulating cell death (apoptosis/necrosis), at least in this liver injury mouse model.
The observation of higher expression of miR‐208a in Kupffer cells suggests that miR‐208a might modulate the function of myeloid cells and indirectly regulate hepatocyte cell death. Consistent with this, we found that overexpression of a miR‐208a mimic in RAW macrophages attenuates CCl4‐induced cytotoxicity. Other studies have also reported the role of miR‐208a in macrophages. Overexpression of miR‐208a in bone marrow–derived macrophages and cardiomyocytes caused a dose‐dependent induction in macrophage inflammatory protein 2 (MIP2) levels.( 41 ) Moreover, miR‐208a was shown to play a role in acute inflammatory response in a murine peritonitis,( 42 ) and overexpression of miR‐208a in human macrophages resulted in a reduction in the mRNA levels of CD14, CD40 ligand, prostacyclin I2 receptor, and PDCD4.( 42 ) These studies support the role of miR‐208a in macrophages.
Finally, it is important to note that we found increased liver damage in miR‐208a KO mice without evidence of compensatory up‐regulation of regenerative markers in the liver. Although our study is limited by investigation of one time point in relation to CCl4 administration, lack of compensatory regenerative pathways may contribute to the higher liver damage in miR‐208a KO mice. In support of this, histopathological evaluations revealed a higher inflammation in WT mice, whereas miR‐208a KO mice exhibited less inflammation in ALI and a higher activation of cell death genes at the biochemical level. This suggests that WT mice are already in the second phase of liver injury, which is the inflammatory phase, whereas KO mice might have either sustained liver injury or lack in liver repair signal.
Because miR‐208a has been found to modulate the expression of a large cohort of genes involved in cell death including caspase‐8, Bax, p21 and PDCD4,( 4 , 6 , 31 ) it is likely that enhanced liver injury in miR‐208a KO mice is not solely through p53 but because of cumulative effects of its target genes involved in the cell death pathway. Moreover, we cannot rule out the possibility of indirect regulation of p53 by miR‐208a. This observation deserves further investigation. Altogether, our results suggest a link between miR‐208a and cell death in CCl4‐induced ALI.
In summary, our findings demonstrate that miR‐208a is an important regulator of ALI and inflammation in the CCl4‐induced acute liver injury mouse model. Our results suggest that miR‐208a might be involved in the regulation of cell death pathways (apoptosis and necrosis) in an ALI.
Supporting information
Fig S1
This work was supported by the National Institute on Alcohol Abuse and Alcoholism (R01AA020744, U01AA026933, and UH2AA026970).
Potential conflict of interest: Dr. Szabo owns stock in and consults for Glympse and Zomagen. She consults for Allergen, Alnylam, Arrow Diagnostics, Durect Corporation, Generon, the Mayday Foundation, Novartis, Pandion Therapeutics, Quest Diagnostics, Surrozen, and Terra Firma. She received grants from Gilead. Dr. Szabo is also Editor‐in‐Chief of Hepatology Communications and author in UpToDate Inc.
Contributor Information
Shashi Bala, Email: shashi.bala@umassmed.edu.
Gyongyi Szabo, Email: Gyongyi.Szabo@umassmed.edu.
References
Author names in bold designate shared co‐first authorship.
- 1. Blackmore L, Bernal W. Acute liver failure. Clin Med (Lond) 2015;15:468‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress‐dependent cardiac growth and gene expression by a microRNA. Science 2007;316:575‐579. [DOI] [PubMed] [Google Scholar]
- 3. Oliveira‐Carvalho V, Carvalho VO, Bocchi EA. The emerging role of miR‐208a in the heart. DNA Cell Biol 2013;32:8‐12. [DOI] [PubMed] [Google Scholar]
- 4. Yin K, Liu M, Zhang M, Wang F, Fen M, Liu Z, et al. miR‐208a‐3p suppresses cell apoptosis by targeting PDCD4 in gastric cancer. Oncotarget 2016;7:67321‐67332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun X, Jiang S, Liu J, Wang H, Zhang Y, Tang SC, et al. MiR‐208a stimulates the cocktail of SOX2 and beta‐catenin to inhibit the let‐7 induction of self‐renewal repression of breast cancer stem cells and formed miR208a/let‐7 feedback loop via LIN28 and DICER1. Oncotarget 2015;6:32944‐32954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tang Y, Cui Y, Li Z, Jiao Z, Zhang Y, He Y, et al. Radiation‐induced miR‐208a increases the proliferation and radioresistance by targeting p21 in human lung cancer cells. J Exp Clin Cancer Res 2016;35:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu P, Wu D, You Y, Sun J, Lu L, Tan J, et al. miR‐208‐3p promotes hepatocellular carcinoma cell proliferation and invasion through regulating ARID2 expression. Exp Cell Res 2015;336:232‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luedde T, Schwabe RF. NF‐κB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2011;8:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wullaert A, van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor‐kappaB: effects on liver homeostasis and beyond. Endocr Rev 2007;28:365‐386. [DOI] [PubMed] [Google Scholar]
- 10. Moulder DE, Hatoum D, Tay E, Lin Y, McGowan EM. The roles of p53 in mitochondrial dynamics and cancer metabolism: the pendulum between survival and death in breast cancer? Cancers (Basel) 2018;10:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ying Y, Padanilam BJ. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D‐overlapping pathways of regulated necrosis? Cell Mol Life Sci 2016;73:2309‐2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998;281:1305‐1308. [DOI] [PubMed] [Google Scholar]
- 13. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009;9:400‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen A, Huang X, Xue Z, Cao D, Huang K, Chen J, et al. The role of p21 in apoptosis, proliferation, cell cycle arrest, and antioxidant activity in UVB‐irradiated human HaCaT keratinocytes. Med Sci Monit Basic Res 2015;21:86‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kwon YH, Jovanovic A, Serfas MS, Tyner AL. The Cdk inhibitor p21 is required for necrosis, but it inhibits apoptosis following toxin‐induced liver injury. J Biol Chem 2003;278:30348‐30355. [DOI] [PubMed] [Google Scholar]
- 16. Bala S, Csak T, Saha B, Zatsiorsky J, Kodys K, Catalano D, et al. The pro‐inflammatory effects of miR‐155 promote liver fibrosis and alcohol‐induced steatohepatitis. J Hepatol 2016;64:1378‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bala S, Csak T, Momen‐Heravi F, Lippai D, Kodys K, Catalano D, et al. Biodistribution and function of extracellular miRNA‐155 in mice. Sci Rep 2015;5:10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bala S, Csak T, Kodys K, Catalano D, Ambade A, Furi I, et al. Alcohol‐induced miR‐155 and HDAC11 inhibit negative regulators of the TLR4 pathway and lead to increased LPS responsiveness of Kupffer cells in alcoholic liver disease. J Leukoc Biol 2017;102:487‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ozerkan D, Ozsoy N, Yilmaz E. Vitamin D and melatonin protect the cell's viability and ameliorate the CCl4 induced cytotoxicity in HepG2 and Hep3B hepatoma cell lines. Cytotechnology 2015;67:995‐1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu H, Xu L, Chen Y, Xu C. MiR‐208a‐3p functions as an oncogene in colorectal cancer by targeting PDCD4. Biosci Rep 2019;39:BSR20181598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Lehmann BD, Paine MS, Brooks AM, McCubrey JA, Renegar RH, Wang R, et al. Senescence‐associated exosome release from human prostate cancer cells. Cancer Res 2008;68:7864‐7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gupta S, Knowlton AA. HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol 2007;292:H3052‐H3056. [DOI] [PubMed] [Google Scholar]
- 23. Szabo G, Momen‐Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Rev Gastroenterol Hepatol 2017;14:455‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo R, Lin B, Pan JF, Liong EC, Xu AM, Youdim M, et al. Inhibition of caspase‐9 aggravates acute liver injury through suppression of cytoprotective autophagy. Sci Rep 2016;6:32447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayden MS, Ghosh S. Regulation of NF‐kappaB by TNF family cytokines. Semin Immunol 2014;26:253‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ito T, Ishigami M, Matsushita Y, Hirata M, Matsubara K, Ishikawa T, et al. Secreted ectodomain of SIGLEC‐9 and MCP‐1 synergistically improve acute liver failure in rats by altering macrophage polarity. Sci Rep 2017;7:44043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li X, Wang L, Chen C. Effects of exogenous thymosin beta4 on carbon tetrachloride‐induced liver injury and fibrosis. Sci Rep 2017;7:5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol 2015;44‐46:147‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang M, Antoine DJ, Weemhoff JL, Jenkins RE, Farhood A, Park BK, et al. Biomarkers distinguish apoptotic and necrotic cell death during hepatic ischemia/reperfusion injury in mice. Liver Transpl 2014;20:1372‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 2013;5:a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tony H, Meng K, Wu B, Yu A, Zeng Q, Yu K, et al. MicroRNA‐208a dysregulates apoptosis genes expression and promotes cardiomyocyte apoptosis during ischemia and its silencing improves cardiac function after myocardial infarction. Mediators Inflamm 2015;2015:479123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Butzer U, Weidenbach H, Gansauge S, Gansauge F, Beger HG, Nussler AK. Increased oxidative stress in the RAW 264.7 macrophage cell line is partially mediated via the S‐nitrosothiol‐induced inhibition of glutathione reductase. FEBS Lett 1999;445:274‐278. [DOI] [PubMed] [Google Scholar]
- 33. van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell 2009;17:662‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Momen‐Heravi F, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol‐treated hepatocytes horizontally transfer liver specific miRNA‐122 and sensitize monocytes to LPS. Sci Rep 2015;5:9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 2018;75:193‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Faletti L, Peintner L, Neumann S, Sandler S, Grabinger T, Mac Nelly S, et al. TNFα sensitizes hepatocytes to FasL‐induced apoptosis by NFκB‐mediated Fas upregulation. Cell Death Dis 2018;9:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF‐alpha‐induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol 2006;290:G583‐G589. [DOI] [PubMed] [Google Scholar]
- 38. Tang X, Asano M, O'Reilly A, Farquhar A, Yang Y, Amar S. p53 is an important regulator of CCL2 gene expression. Curr Mol Med 2012;12:929‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chang DW, Xing Z, Capacio VL, Peter ME, Yang X. Interdimer processing mechanism of procaspase‐8 activation. EMBO J 2003;22:4132‐4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meng LD, Meng AC, Zhu Q, Jia RY, Kong QZ. Effect of microRNA‐208a on mitochondrial apoptosis of cardiomyocytes of neonatal rats. Asian Pac J Trop Med 2015;8:747‐751. [DOI] [PubMed] [Google Scholar]
- 41. Feng Y, Zou L, Yan D, Chen H, Xu G, Jian W, et al. Extracellular microRNAs induce potent innate immune responses via TLR7/MyD88‐dependent mechanisms. J Immunol 2017;199:2106‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Recchiuti A, Krishnamoorthy S, Fredman G, Chiang N, Serhan CN. MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1‐miRNA circuits. FASEB J 2011;25:544‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1