

Abstract
Alagille syndrome (ALGS) and progressive familial intrahepatic cholestasis (PFIC) are inherited cholestatic disorders with risk of developing end‐stage liver disease requiring liver transplantation (LT). We investigated aspartate aminotransferase‐to‐platelet ratio index (APRI), Fibrosis‐4 score (FIB‐4), and conjugated bilirubin as biomarkers to assess fibrosis severity and risk for LT among children with ALGS and PFIC. This multicenter, cross‐sectional study included 64 children with ALGS or PFIC (per genetics or strict clinical criteria) with APRI, FIB‐4, and conjugated bilirubin levels collected within ±90 days of their most recent liver biopsy. A single, blinded pathologist staged all biopsies (metavir; F0‐F2: nonsevere, F3‐F4: severe). Logistic regression and area under the receiver operating characteristic curve analysis (AUC) were used to assess biomarker associations with fibrosis severity and risk for LT. In ALGS, only APRI distinguished F3‐F4 (AUC 0.72, P = 0.012), with a cutoff greater than 2.97 demonstrating a sensitivity of 61.5% (95% confidence interval 0.32, 0.86) and specificity of 81.5% (0.62, 0.94). In ALGS, a 50% increase of APRI increased the odds of F3‐F4 by 1.31‐fold (1.04, 1.65; P = 0.023). In ALGS, APRI (AUC 0.87; P < 0.001) and FIB‐4 (AUC 0.84; P < 0.001) were able to predict risk for LT. In PFIC, only APRI distinguished F3‐4 (AUC 0.74, P = 0.039), with a cutoff greater than 0.99 demonstrating a sensitivity of 80% (0.44, 0.98) and specificity of 64.3% (0.35, 0.87). In PFIC, only FIB‐4 was able predict risk for LT (AUC 0.80; P = 0.002). In ALGS or PFIC, conjugated bilirubin could not distinguish F3‐F4 or predict risk for LT. Conclusion: This liver biopsy–validated study suggests that APRI is able to distinguish F3‐F4 from F0‐F2 in ALGS and PFIC. APRI and FIB‐4 may also serve as predictors of risk for LT in ALGS (APRI and FIB‐4) and PFIC (FIB‐4).
Abbreviations
- ALGS
Alagille syndrome
- ALT
alanine aminotransferase
- APRI
aspartate aminotransferase‐to‐platelet ratio index
- AST
aspartate aminotransferase
- AUC
area under the receiver operator characteristic curve
- BA
biliary atresia
- cBili
conjugated bilirubin
- CFLD
cystic fibrosis liver disease
- CI
confidence interval
- FIB‐4
fibrosis‐4 score
- GGT
gamma‐glutamyl transferase
- GPR
GGT‐to‐platelet ratio
- IQR
interquartile range
- LT
liver transplantation
- PFIC
progressive familial intrahepatic cholestasis
- ROC
receiver operator characteristic
Alagille syndrome (ALGS) and progressive familial intrahepatic cholestasis (PFIC) represent distinct but rare inherited cholestatic disorders that display wide variability in degrees of liver fibrosis, progression of disease, and time to transplant.( 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 ) Although genetic analysis is emphasized for either diagnosis, percutaneous liver biopsy has historically been an essential tool for histologic staging and monitoring progression of disease.( 9 , 10 ) Risks of liver biopsies are relatively low; however, there is still potential for bleeding, patient discomfort, infection, and side effects of anesthesia.( 10 , 11 )
An autosomal dominant disorder, ALGS is characterized by bile duct paucity and often presents with extrahepatic clinical features such as vertebral abnormalities, renal or cardiac disease, posterior embryotoxon, or syndromic facies( 4 , 12 ) with an estimated frequency of 1 in 30,000 to 50,000 live births per year.( 5 ) Although liver disease may improve with age,( 3 ) an estimated 43.5%‐76% of patients with ALGS will undergo liver transplantation (LT) by adulthood.( 1 , 13 , 14 ) PFIC consists of multiple disease subtypes, each characterized by various defects in transport of bile constituents( 15 , 16 ) with autosomal recessive inheritance, and an estimated incidence of 1 in 50,000 to 100,000 live births per year.( 15 , 17 , 18 ) The clinical course of liver disease in PFIC is sometimes rapid, representing up to 10%‐15% of all childhood liver transplants.( 15 , 19 )
Developed initially to monitor the progression of liver fibrosis in adults with hepatitis C,( 20 , 21 ) the simple biomarker indices aspartate aminotransferase (AST)‐to‐platelet ratio (APRI) and Fibrosis‐4 score (FIB‐4) use standard liver biochemistries. These indices have since been biopsy‐validated for use in distinguishing liver fibrosis severity in pediatric hepatobiliary disorders such as biliary atresia (BA)( 22 ) and cystic fibrosis liver disease (CFLD).( 23 ) Total bilirubin and gamma‐glutamyl transferase (GGT)‐to‐platelet ratio (GPR) have also been shown to be useful biomarkers of outcome in children with BA (transplant‐free survival, death)( 24 ) and CFLD (liver fibrosis detection).( 25 )
The primary objective of this study was to investigate the utility of APRI, FIB‐4, and conjugated bilirubin as biomarkers of liver fibrosis validated by liver biopsy in children with ALGS and PFIC. Secondary objectives include investigation of the change in biomarker scores over time and assessment of biomarkers as predictors of risk for LT.
Materials and Methods
This study was a multicenter, liver biopsy–validated, cross‐sectional study examining the utility of APRI, FIB‐4, and conjugated bilirubin as surrogates of fibrosis in children with genetically or clinically confirmed ALGS or PFIC. We also performed a longitudinal secondary analysis to track the chronological progression of biomarkers and analyzed biomarkers for prediction of risk for future LT. This study was approved by institutional review boards at each center or granted a waiver due to minimal risk. For inclusion, subjects were required to have at least one liver biopsy (from either native liver or explant) and documented genetic or clinical confirmation of ALGS or PFIC subtype 1‐3 diagnosis. The diagnosis of ALGS or PFIC was first confirmed by documented genetic mutation (JAG‐1 or NOTCH‐2 for ALGS; ATP8B1 for PFIC subtype 1; ABCB11 for PFIC subtype 2; and ABCB4 for PFIC subtype 3). If no records of genetic mutation were available, clinical criteria were used to confirm the diagnosis. Clinical criteria for ALGS included any combination of three or more of the following: (1) peripheral pulmonary stenosis (or tetralogy of Fallot), (2) posterior embryotoxon, (3) butterfly vertebrae, (4) renal manifestations, (5) vascular anomalies, (6) facial features historically described in ALGS,( 4 ) or (7) or bile duct paucity (per pathologic reports). Clinical criteria for PFIC subtypes included the following: (1) PFIC subtype 1 = history of low/normal GGT and electron microscopy with Byler’s bile; (2) PFIC subtype 2 = history of low/normal GGT and absent bile salt export pump protein staining or giant cell hepatitis; and (3) PFIC subtype 3 = high GGT with bile duct proliferation and a regular biliary tree (per imaging).
Liver biopsy cores were a minimum of 1 cm in length with 6‐10 portal tracts to ensure sample adequacy (range of 1‐7 cores per sample). Otherwise, tissue samples were from explants. Histological slides (trichrome/hematoxylin and eosin) were digitalized and reviewed by a single pediatric hepatopathologist who was blinded to all participant information. Each biopsy was staged for fibrosis using the metavir classification (F0 = no fibrosis, F1 = portal fibrosis without septa, F2 = portal fibrosis with few septa, F3 = numerous septa without cirrhosis, and F4 = cirrhosis). For this study, metavir stages 0‐2 were classified as “nonsevere” fibrosis (F0‐F2) and metavir stages 3‐4 were classified as “severe” fibrosis (F3‐F4). The most recent liver specimen was designated as the “final biopsy” and used for correlation with APRI, FIB‐4, and conjugated bilirubin in the primary cross‐sectional analysis. For participants with multiple biopsies (n = 16), histology preceding the final biopsy were individually staged and correlated to biomarkers in a secondary longitudinal analysis. Participants who had an increase of at least one metavir stage of fibrosis over multiple biopsies were categorized as “progressors”; participants without progression in fibrosis over multiple biopsies were categorized as “nonprogressors.”
Labs (including conjugated bilirubin and GGT) were collected within ±90 days of the biopsy. For explant biopsies, labs were collected at least 90 days preceding the biopsy. Age in years at the time of lab collection was used for FIB‐4. For purposes of this study, the upper limit of normal AST was 30 U/L and the upper limit of normal GGT was 40 U/L.
Simple Biomarker Indices Calculations
Statistics
Patient and clinical characteristics are summarized using mean with SDs, median with 25th and 75th percentiles, and frequency with percentages. The summary statistics are stratified by fibrosis severity and compared using two‐sample t test, Wilcoxon rank sum test, Fisher’s exact test, or chi‐square test. Logistic regression accounting for the within‐hospital correlation was used to assess the association between fibrosis severity and log‐transformed APRI, FIB‐4, and conjugated bilirubin. For each predictor, a receiver operating characteristic (ROC) analysis with area under the curve (AUC) was performed. The ROC curves were compared. All analyses were performed using Stata v.15 software (College Station, TX).
Results
Overall Findings
This study included 64 individual participants with either ALGS (n = 40) or PFIC subtypes 1‐3 (n = 24, Supporting Table S1) with liver biopsies performed between January 2003 to December 2015. In total, 33 (51.6%) participants were confirmed genetically (JAG1 = 16, NOTCH2 = 1, ATP8B1 = 7, ABCB11 = 5, ABCB4 = 4), and 30 (46.9%) met strict clinical criteria for either ALGS or PFIC. One participant with PFIC subtype 2 was included based on clinical history of intracellular cholestasis with low GGT and ABCB11 heterozygosity. The overall median age at liver biopsy for all participants was 2.0 years (interquartile range [IQR] 0.5, 6.1), and 67% were male. Thirteen (20.3%) of the final biopsies were obtained from explants. Participants with ALGS had higher levels of conjugated bilirubin (P = 0.028), alanine aminotransferase (ALT) (P = 0.003), and AST (P = 0.034) than those with PFIC. Median APRI was also higher in ALGS (2.1 vs. 1.1; P = 0.033). There were no differences in FIB‐4 (0.1 vs. 0.1; P = 0.934) or platelet levels (310.5 vs. 379.5; P = 0.276) between disease groups at time of final biopsy (Supporting Table S2).
When stratified by individual fibrosis stages (F0, F1, F2, F3, or F4) at the final biopsy, there were no differences in median age (P = 0.917) or in levels of AST, ALT, or conjugated bilirubin. Platelet levels were lowest in participants with F4 (P = 0.027). APRI scores were highest in participants with F4 (P < 0.001; Supporting Table S3 and Fig. S1).
Nonsevere (F0‐F2) Versus Severe (F3‐F4) Fibrosis
F0‐F2 Versus F3‐F4 in ALGS
Thirteen of 40 (32.5%) participants with ALGS were staged as F3‐F4 at final biopsy. Among ALGS, there was no difference in age (years) at time of biopsy between F3‐F4 and F0‐F2 (3.5 vs. 1.6; P = 0.319). There were also no differences in AST, ALT, platelets, conjugated bilirubin, or GPR between those with F3‐F4 versus F0‐F2. However, ALGS with F3‐F4 had higher APRI (3 vs. 1.7; P = 0.029) but not FIB‐4 (0.3 vs. 0.1; P = 0.076) (Table 1).
TABLE 1.
Clinical Characteristics by Fibrosis Severity
| Fibrosis stage (%) | ALGS (n = 40) | P Value* | PFIC (n = 24) | P Value* | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F0 | 9 (22.5) | F0 | 7 (29.2) | |||||||
| F1 | 4 (10.0) | F1 | 3 (12.5) | |||||||
| F2 | 14 (35.0) | F2 | 4 (16.7) | |||||||
| F3 | 9 (22.5) | F3 | 5 (20.8) | |||||||
| F4 | 4 (10.0) | F4 | 5 (20.8) | |||||||
| Variable | n | F0‐F2 (n = 27) | n | F3‐F4 (n = 13) | n | F0‐F2 (n = 14) | n | F3‐F4 (n = 10) | ||
| Male (%) | 27 | 16 (59.3) | 13 | 11 (84.6) | 0.157 | 14 | 9 (64.3) | 10 | 7 (70.0) | 1.000 |
| Genetic | ||||||||||
| confirmation (%) | 27 | 11 (40.7) | 13 | 7 (53.8) | 0.509 | 14 | 8 (57.1) | 10 | 7 (70.0) | 0.678 |
| Age at biopsy (years) | 27 | 16 (0.2, 4.5) | 13 | 3.5 (1.3, 6.4) | 0.319 | 14 | 1.6 (0.6, 8.4) | 10 | 2.2 (1.4, 3.1) | 0.682 |
| Median (IQR) | Median (IQR | Median (IQR) | Median (IQR) | |||||||
| Platelets (109) | 27 | 415 (208, 526) | 13 | 251 (174, 281) | 0.067 | 14 | 416 (351, 512) | 10 | 333 (153, 454) | 0.089 |
| ALT (IU/L) | 27 | 151 (111, 238) | 13 | 203 (126, 215) | 0.908 | 14 | 69.5 (41, 196) | 10 | 95 (73, 154) | 0.482 |
| AST (IU/L) | 27 | 177 (118, 282) | 13 | 294 (155, 326) | 0.059 | 14 | 103 (77, 132) | 10 | 168.5 (96, 298) | 0.266 |
| Conjugated bilirubin (mg/dL) | 21 | 4.8 (3.3, 5.6) | 12 | 7.5 (1.8, 9.3) | 0.294 | 12 | 3.4 (2.3, 4.8) | 9 | 0.8 (0.0, 2.4) | 0.011 |
| GGT (U/L) | 26 | 342 (171, 563) | 13 | 263 (160, 528) | 0.623 | 13 | 25 (20, 39) | 10 | 54 (26, 264) | 0.063 |
| APRI | 27 | 1.7 (1.0, 2.3) | 13 | 3 (1.9, 6.9) | 0.029 | 14 | 0.8 (0.7, 1.7) | 10 | 2 (1.0, 6.8) | 0.053 |
| FIB‐4 | 27 | 0.1 (0.0, 0.3) | 13 | 0.3 (0.1, 0.5) | 0.076 | 14 | 0.0 (0.0, 0.3) | 10 | 0.1 (0.0, 0.6) | 0.219 |
| GPR | 26 | 2.6 (1.1, 4.0) | 13 | 2.8 (1.4, 5.8) | 0.348 | 13 | 25 (20, 39) | 10 | 1 (0.1, 2.0) | 0.035 |
P values for median comparisons using two‐sample Wilcoxon rank‐sum (Mann‐Whitney) test; P values calculated with exact testing for categorical variables when possible; otherwise chi‐square test.
The AUC of log‐transformed APRI and FIB‐4 to distinguish F3‐F4 in ALGS was 0.72 (P = 0.012) and 0.68 (P = 0.039), respectively (Fig. 1A). An APRI greater than 2.97 demonstrated a sensitivity (95% confidence interval [CI]) of 61.54% (0.32, 0.86) and specificity of 81.48% (0.62, 0.94) in ALGS. With a 50% increase in APRI, the odds of F3‐F4 were 1.31‐fold higher (95% CI: 1.04, 1.65; P = 0.023) in ALGS. Log‐transformed conjugated bilirubin (AUC = 0.69; P = 0.121) was unable to distinguish F3‐F4 in participants with ALGS (Fig. 1A).
FIG. 1.
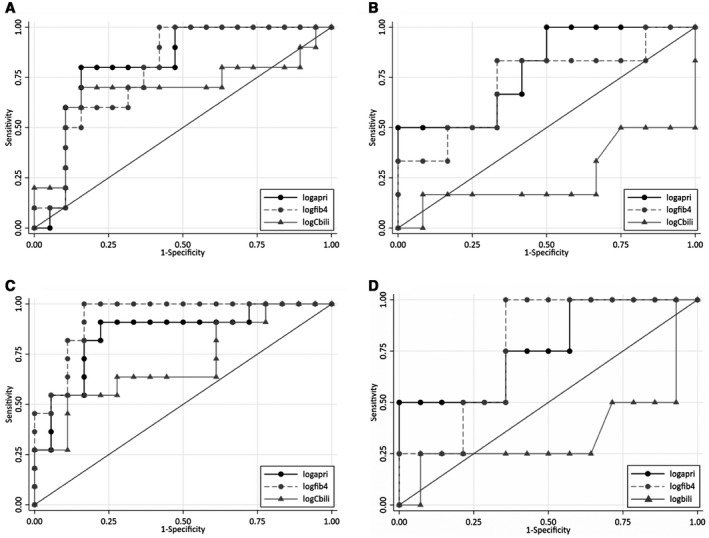
ROC curves for APRI, FIB‐4, and conjugated bilirubin (cBili) to distinguish F3‐F4 from F0‐F2 among ALGS or PFIC (A and B, respectively) and in the prediction of risk for LT in ALGS or PFIC (C and D, respectively). (A) In ALGS, APRI (AUC 0.72) was better at distinguishing F3‐F4 from F0‐F2 than FIB‐4 (AUC 0.68) or cBili (AUC 0.69). (B) In PFIC, APRI (AUC 0.74) was better at distinguishing F3‐F4 from F0‐F2 than FIB‐4 (AUC 0.65) or cBili (AUC 0.26). (C) In ALGS, APRI (AUC 0.87) and FIB‐4 (AUC 0.84) were better at prediction of risk for LT than cBili (AUC 0.71). (D) In PFIC, FIB‐4 (AUC 0.80) was better at the prediction of risk for LT than APRI (AUC 0.66) or cBili (AUC 0.35). Solid line with black dots represents ROC curve of log‐transformed APRI. Hashed line with gray dots represents ROC curve of log‐transformed FIB‐4. Solid line with gray triangles represents ROC curve of log‐transformed cBili.
F0‐F2 Versus F3‐F4 in PFIC
Ten of 24 (41.7%) participants with PFIC were staged as F3‐F4 at final biopsy. Among PFIC, there was no difference in age at time of biopsy (years) between F3‐F4 and F0‐F2 (2.2 vs. 1.6; P = 0.682). There were also no differences in AST, ALT, platelets, or FIB‐4 between those with F3‐F4 and F0‐F2. However, PFIC with F3‐F4 had lower conjugated bilirubin levels (0.8 vs. 3.4; P = 0.011) and higher APRI (2.0 vs. 0.8; P = 0.053) (Table 1).
The AUC of log‐transformed APRI and FIB‐4 to distinguish F3‐F4 in PFIC was 0.74 (P = 0.039) and 0.65 (P = 0.218), respectively (Fig. 1B). An APRI greater than 0.99 demonstrated a sensitivity (95% CI) of 80% (0.44, 0.98) and specificity of 64.29% (0.35, 0.87) in PFIC. In PFIC, a 50% increase in APRI was not statistically associated with higher odds of F3‐F4. Log‐transformed conjugated bilirubin (AUC = 0.26; P = 0.115) was also unable to distinguish F3‐F4 in participants with PFIC (Fig. 1B).
APRI and FIB‐4: Longitudinal Analysis of Fibrosis in All Participants (ALGS and PFIC)
Sixteen of the 64 (25%) participants (ALGS and PFIC) had two or more liver biopsies performed during the study period. Repeat biopsies were performed due to concern for worsening cholestasis or rising transaminases (n = 7) or were retrieved at time of LT or other procedures (n = 9). Nine participants were progressors (increase of ≥ 1 fibrosis stage between biopsies). The remaining 7 participants were nonprogressors. There were no differences in clinical characteristics (median age of biopsy, labs, APRI, or FIB‐4) between progressors and nonprogressors. Time elapsed (years) between biopsies was also not different between groups (2.8 vs. 1.5, P = 0.368). Progressors demonstrated an increase of APRI (+2.7), FIB‐4 (+0.3), and conjugated bilirubin (+1.3) from time between the first biopsy and final biopsy. In nonprogressors, none of the biomarkers (APRI, FIB‐4, and conjugated bilirubin) demonstrated any change from the time of initial biopsy to final biopsy (Table 2). Overall, 6 (37.5%) participants demonstrated an increase of two or more metavir stages between biopsies. Time elapsed between biopsies was not different in subjects who progressed only one stage versus those who progressed two or more stages (3.28 vs. 3.35 years, P = 0.952).
TABLE 2.
Clinical Characteristics of Progressors Versus Nonprogressors
| Variable | n | Nonprogressors, n = 7 (ALGS = 6, PFIC = 1) | Progressors, n = 9 (ALGS = 7, PFIC = 2) | P Value* |
|---|---|---|---|---|
| Median (IQR) | Median (IQR) | |||
| Age at biopsy (years) | 7 | 2.4 (1.3, 3.4) | 1.7 (1.4, 5.0) | 0.874 |
| Platelets (109) | 7 | 526 (310.0, 537.0) | 279 (194.0, 418.0) | 0.125 |
| ALT (IU/L) | 7 | 129 (93.0, 212.0) | 174 (126.0, 215.0) | 0.427 |
| AST (IU/L) | 7 | 118 (63.0, 301.0) | 326 (120.0, 399.0) | 0.112 |
| Conjugated bilirubin (mg/dL) | 7 | 2.9 (0.0, 4.8) | 6.7 (0.0, 8.5) | 0.453 |
| GGT (U/L) | 7 | 175 (83.0, 563.0) | 160 (52.0, 528.0) | 0.634 |
| APRI | 7 | 0.8 (0.4, 2.7) | 3.9 (1.2, 6.9) | 0.186 |
| FIB‐4 | 7 | 0.1 (0.0, 0.3) | 0.3 (0.0, 0.4) | 0.634 |
| Time between biopsies (years) | 7 | 2.8 (2.0, 6.9) | 1.5 (0.8, 4.7) | 0.368 |
| Δ APRI † | 7 | 0 (0, 1.7) | +2.7 (0.5, 5.9) | 0.082 |
| Δ FIB‐4 † | 7 | 0 (0, 0.2) | +0.3 (0, 0.3) | 0.050 |
| Δ Conjugated † bilirubin (mg/dL) | 5 | 0 (0, 0) | +1.3 (−0.2, 6.8) | 0.651 |
P values for median comparisons using two‐sample Wilcoxon rank‐sum (Mann‐Whitney) test; P values calculated with exact testing for categorical variables when possible; otherwise chi‐square test.
Δ = (APRI, FIB‐4, or cBili at final biopsy) – (APRI, FIB‐4, or cBili at the patient’s first biopsy.
Longitudinal analysis of biomarker trends was performed in 40 participants (ALGS and PFIC; 62.5%) who had multiple sets of labs available (AST, ALT, platelets, and conjugated bilirubin) before final biopsy (Fig. 2). A linear mixed model demonstrated that in 1 year, there was a 10% increase in APRI (95% CI = 3.75, 16.57; P = 0.001) and a 60.2% increase in FIB‐4 (40.79, 82.21; P < 0.001). Using logistic regression, participants who exceeded these percentage changes of APRI and FIB‐4 in 1 year, increased their odds of F3‐F4 by 1.14‐fold (P < 0.001) and 1.01‐fold (P = 0.242), respectively.
FIG. 2.
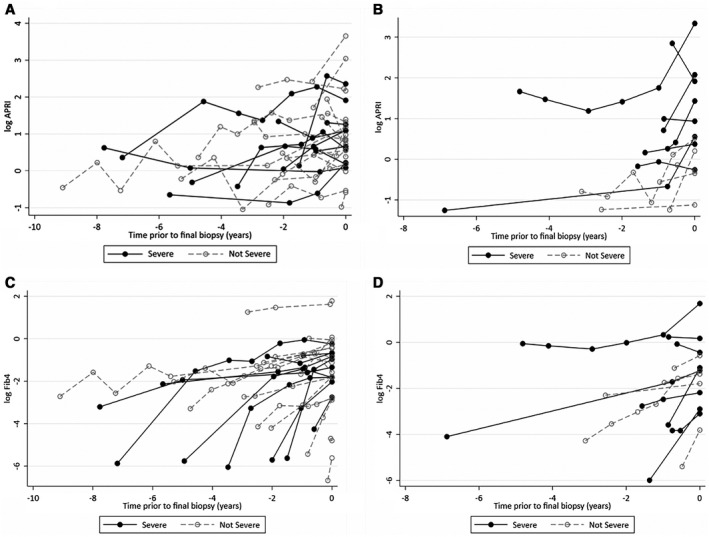
Chronological plot of APRI or FIB‐4 in patients (with two or more sets of labs available) leading up to the time of final biopsy. There is a visual overall upward trend in biomarkers when followed longitudinally. (A) APRI over time in ALGS. (B) APRI over time in PFIC. (C) FIB‐4 over time in ALGS. (D) FIB‐4 over time in PFIC. Individual dots represent log‐transformed APRI or FIB‐4 calculated before the time of final biopsy, which is represented by time point = 0. Filled dots represent APRI or FIB‐4 levels of patients who had F3‐F4 fibrosis at final biopsy. Open dots represent APRI or FIB‐4 levels of patients who had F0‐F2 fibrosis at final biopsy.
LT Versus No LT
LT Versus No LT in ALGS
Thirteen of 40 (32.5%) participants with ALGS eventually underwent LT at a median age of 3.5 years (2.4, 4.3). Of the participants with ALGS who had LT, 6 (46%) demonstrated F3‐F4 fibrosis at final biopsy. The most common LT indications for ALGS were chronic cholestasis (n = 13, 100%), intractable pruritus (n = 10, 76.9%), growth failure/malnutrition requiring either supplemental enteral or parenteral nutrition (n = 9, 69.2%), deforming xanthomas (n = 4, 30.8%), and pathological fractures (n = 3, 23.1%). Median APRI (3.9 vs. 1.7, P < 0.001) and FIB‐4 (0.4 vs. 0.0, P < 0.001) for ALGS LT was higher than ALGS no LT (Table 3).
TABLE 3.
Clinical Characteristics by LT
| Variable | ALGS | P Value* | Variable | PFIC | P Value* | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yes LT, n = 13 (%) | No LT, n = 27 (%) | Yes LT, n = 6 (%) | No LT, n = 18 (%) | ||||||||
| Male (%) | 8 (61.5) | 19 (70.4) | 1.000 | Male (%) | 5 (83.3) | 11 (61.1) | 0.621 | ||||
| F3‐F4 † | 6 (46.2) | 7 (25.9) | 0.284 | F3‐F4 † | 5 (83.3) | 5 (27.8) | 0.050 | ||||
| F0‐F2 † | 7 (53.8) | 20 (74.1) | F0‐F2 † | 1 (16.7) | 13 (72.2) | ||||||
| LT indications | LT indications | ||||||||||
| Chronic cholestasis | 13 (100.0) | Intractable pruritus | 4 (66.7) | ||||||||
| Intractable pruritus | 10 (76.9) | Malnutrition | 3 (50.0) | ||||||||
| Malnutrition | 9 (69.2) | Chronic cholestasis | 2 (33.3) | ||||||||
| Deforming xanthomas | 4 (30.8) | Ascites | 2 (33.3) | ||||||||
| Pathological fractures | 3 (23.1) | ||||||||||
| n | Median (IQR) | n | Median (IQR) | n | Median (IQR) | n | Median (IQR) | ||||
| Age at biopsy (years) | 13 | 2.9 (2.1, 4.3) | 27 | 0.9 (0.2, 5.8) | 0.055 | Age at biopsy (years) | 6 | 6.8 (1.7, 11.7) | 18 | 1.2 (0.6, 3.4) | 0.077 |
| Age at LT (years) | 13 | 3.5 (2.4, 4.3) | 0 | Age at LT (years) | 6 | 6.8 (1.7, 11.7) | 0 | ||||
| Platelets (109) | 13 | 194 (62, 279) | 27 | 420 (267, 528) | <0.001 | Platelets (109) | 6 | 262.5 (153, 512) | 18 | 404 (334, 505) | 0.230 |
| ALT (IU/L) | 13 | 186 (129, 211) | 27 | 151.0 (114.0, 238.0) | 0.697 | ALT (IU/L) | 6 | 114 (42, 154) | 18 | 84.5 (43, 196) | 0.815 |
| AST (IU/L) | 13 | 247 (163, 326) | 27 | 205 (101, 294) | 0.103 | AST (IU/L) | 6 | 175 (79, 298) | 18 | 109.5 (80, 216) | 0.739 |
| Conjugated bilirubin (mg/dL) | 11 | 8.1 (4.3, 18.2) | 22 | 4.2 (0.8, 5.6) | 0.016 | Conjugated bilirubin (mg/dL) | 5 | 0.8 (0.8, 2.4) | 16 | 2.8 (1.9, 4.0) | 0.341 |
| GGT (U/L) | 12 | 254 (143, 422) | 27 | 413.0 (175, 604) | 0.273 | GGT (U/L) | 6 | 51 (20, 345) | 17 | 31.0 (21, 49) | 0.344 |
| APRI | 13 | 3.9 (2.7, 10.6) | 27 | 1.7 (1.0, 2.1) | <0.001 | APRI | 6 | 2.0 (0.8, 8.0) | 18 | 0.9 (0.7, 2.3) | 0.257 |
| FIB‐4 | 13 | 0.4 (0.2, 1.1) | 27 | 0.0 (0.0, 0.3) | 0.001 | FIB‐4 | 6 | 0.2 (0.1, 1.2) | 18 | 0.0 (0.0, 0.3) | 0.033 |
| GPR | 12 | 4.4 (2.0, 5.5) | 27 | 413 (175, 604) | 0.273 | GPR | 6 | 0.5 (0.1, 5.6) | 17 | 0.2 (0.1, 0.4) | 0.257 |
P values for median comparisons using two‐sample Wilcoxon rank‐sum (Mann‐Whitney) test; P values calculated with exact testing for categorical variables when possible; otherwise chi‐square test.
Most recent biopsy of native liver or at explant.
To distinguish participants with ALGS with LT, the AUC of log‐transformed APRI, FIB‐4, and conjugated bilirubin was 0.87 (P < 0.001), 0.84 (P < 0.001), and 0.71 (P = 0.051), respectively (Fig. 1C). APRI greater than 2.17 had a sensitivity (95% CI) of 92.3% (0.64, 0.99) and specificity of 81.5% (0.62, 0.94). In ALGS, a 50% increase in APRI was associated with 1.97‐fold higher odds for LT (95% CI: 1.20, 3.24; P = 0.007).
LT Versus No LT in PFIC
Six of 24 (25%) participants with PFIC eventually underwent LT at a median age of 6.8 years. Of the participants with PFIC who had LT, 5 (83%) demonstrated F3‐F4 fibrosis at final biopsy. The most common LT indications for PFIC were intractable pruritus (n = 4, 66.7%), malnutrition requiring supplemental enteral nutrition (n = 3, 50%), chronic cholestasis (n = 2, 33%), and portal hypertension with ascites (n = 2, 33%). Median APRI (2.0 vs. 0.9, P = 0.257) and FIB‐4 (0.2 vs. 0.0, P = 0.033) for PFIC LT was higher than PFIC with no LT (Table 3).
To distinguish participants with PFIC with LT, the AUC of log‐transformed APRI, FIB‐4, and conjugated bilirubin was 0.66 (P = 0.315), 0.80 (P = P = 0.002), and 0.35 (P = 0.473), respectively (Fig. 1D). FIB‐4 greater than 0.11 had a sensitivity (95% CI) of 83.3% (0.36, 0.99) and specificity of 66.7% (0.41,0.87). In PFIC, a 50% increase in FIB‐4 was associated with 1.36‐fold higher odds for LT (95% CI: 1.28, 1.44; P = < 0.001).
Discussion
The heterogeneity of liver disease in ALGS and PFIC can present prognosticating challenges to pediatric hepatologists. Over the past two decades, efforts have been made to validate biomarker indices such as APRI and FIB‐4 in pediatric liver disorders.( 22 , 23 , 26 , 27 , 28 , 29 , 30 , 31 ) We evaluated the utility of APRI, FIB‐4, and conjugated bilirubin as biomarkers of liver fibrosis and as predictors of LT in children with ALGS and PFIC. Importantly, our findings are reported from a very young and cholestatic cohort, with close to 35% of our participants demonstrating greater than F3 fibrosis and 10% with cirrhosis at the time of final biopsy. This suggests the potential for rapid and early progression of liver disease in ALGS and PFIC and emphasizes the importance of affordable and reproducible noninvasive alternatives to liver biopsy for reliable assessment of fibrosis and monitoring of disease progression.
In this study, APRI demonstrated fair discriminative ability to distinguish severe fibrosis (F3‐F4) among participants with ALGS, exhibiting high specificity with a cutoff of over 2.97. Similarly, when applied to participants with PFIC, APRI exhibited high sensitivity to distinguish F3‐F4 with a cutoff of over 0.99. APRI was not as adept at distinguishing between individual metavir stages, although there was a statistical increase as fibrosis worsened in both disease groups. Furthermore, APRI was two times higher in participants with ALGS and PFIC with F3‐F4. These findings support that elevated or rising APRI may be associated with hepatic disease progression in ALGS and PFIC with suggested biopsy‐validated cutoffs for clinical prediction of severity of fibrosis. Although clinical decision making in children with ALGS or PFIC is often based on the development of extrahepatic manifestations such as intractable pruritus, disfiguring xanthomas, and severe malnutrition from persistent cholestasis,( 2 , 4 , 32 ) identification of worsening fibrosis severity in these children remains an important prognostic variable, as advanced stages of fibrosis not only lead to complications from cirrhosis but contribute to and parallel the severity of these extrahepatic features. Therefore, despite a somewhat limited sensitivity, APRI appears to be a readily available, noninvasive tool that may complement the overall decision‐making process in children with rare cholestatic disorders.
Multiple pediatric studies, including ours, have demonstrated superior utility of APRI in fibrosis detection when compared with FIB‐4 and other various biomarkers. In a previous study of 67 children with CFLD, APRI outperformed FIB‐4 in its ability to distinguish F3‐F4 versus F0‐F2 and was superior in differentiating patients with CFLD versus those without CFLD.( 23 ) In a separate investigation of 77 children with NAFLD, APRI was superior to FIB‐4 in distinguishing F0‐F1 from F2‐F3.( 28 ) A potential disadvantage of FIB‐4 in pediatrics may stem from the inclusion of age, as this variable may lead to a diminished effect when applied to particularly young and narrow age ranges. In our study, we also evaluated conjugated bilirubin as a potential biomarker of severe fibrosis. Our results demonstrated that conjugated bilirubin may not be a reliable biomarker of liver fibrosis severity in PFIC or ALGS, as it was unable to distinguish F3‐F4 from F0‐F2 among participants in either disease group. Various reports have demonstrated the utility of GPR in distinguishing fibrosis levels in hepatocellular diseases,( 33 , 34 , 35 ) and most recently in pediatric CFLD.( 25 ) However, there were no differences in GPR when compared between F3‐F4 versus F0‐F2 in participants with ALGS. Furthermore, GPR is confounded by the low to normal levels of GGT found in PFIC subtypes 1 and 2, as evidenced by lower GPR levels among participants with PFIC with F3‐F4 (Table 1).
Understanding the degree of injury and progression of liver fibrosis over time in children with ALGS and PFIC is important in the complex decision making regarding novel medical therapies,( 36 ) as well as for the timing of liver transplant evaluation.( 9 , 10 ) Although imaging modalities using elastography have emerged within pediatrics,( 37 ) they remain expensive and are not yet widely available in all pediatric institutions. In contrast, APRI and FIB‐4 require only standard‐of‐care laboratories to calculate and can be easily tracked over time. In our study, we uniquely describe the trajectory of APRI and FIB‐4 in a longitudinal manner among participants with ALGS and PFIC. Our results suggest that participants with ALGS or PFIC who exceed specific percentage changes in APRI and FIB‐4 over a 1‐year period are at greater risk for development of F3‐F4. Furthermore, our findings demonstrate that fibrosis progressors had clinically more abnormal liver biochemistries and platelet counts than nonprogressors at time of final biopsy. Although there was no difference in the time elapsed between biopsies, progressors were generally younger and demonstrated clinically notable changes in APRI, FIB‐4, and conjugated bilirubin from time of initial to final biopsy. In comparison, nonprogressors did not demonstrate any change in APRI or FIB‐4 from biopsy to biopsy. Unfortunately, while these findings are unique, our small subcohort sample size provided insufficient power for statistical significance.
Few pediatric studies have investigated the utility of APRI or FIB‐4 as a biomarker of clinical outcome. In a Finnish study of 29 patients with biliary atresia, APRI was evaluated as a predictor of native liver survival when collected at the time of Kasai portoenterostomy (KPE). Although this study showed no correlation of APRI with metavir (or Ishak) fibrosis, APRI at the time of KPE was significantly higher in the 10 patients who eventually underwent LT.( 31 ) In our analysis, APRI, FIB‐4, and conjugated bilirubin at the time of final biopsy were all significantly higher in the participants who eventually underwent LT versus those who did not. Among participants with ALGS, APRI and FIB‐4 demonstrated good prediction of those who eventually underwent LT. While conjugated bilirubin demonstrated fair ability (AUC 0.71) to predict risk for LT in ALGS, statistical significance was not achieved (P = 0.051). Among participants with PFIC, only FIB‐4 demonstrated good prediction of those who eventually underwent LT. We acknowledge that the processes of listing and prioritization for LT are subjective, and that children with ALGS or PFIC may be transplanted for quality‐of‐life indications such as medically refractory pruritus and malnutrition, although worsening fibrosis often contributes to and corresponds with progression of these symptoms. As such, the potential use of APRI and FIB‐4 as an additional complementary tool may further guide decision making and timing of LT evaluation for children with these rare diseases.
Limitations of this pediatric study include a moderate sample size and generalization of PFIC subtypes 1‐3, although strict inclusion standards requiring either genetic confirmation or a priori clinical criteria and the validation of metavir fibrosis staging by a single, blinded pediatric‐trained hepatopathologist were implemented. Fewer subjects had data available for conjugated bilirubin, longitudinal, and LT analysis. For study inclusion, all participants were required to have at least one liver biopsy. Hence, our cohort may be skewed toward more severe liver disease, as it is possible that participants with milder disease did not undergo liver biopsy during this study period. However, it should be acknowledged that it is often the stable patient without significant liver dysfunction or coagulopathy who can safely undergo liver biopsy. Furthermore, participants with more than one biopsy were subject to potential bias, as biopsies were retrieved primarily at the time of explant or for concern for worsening liver disease. We did not analyze bile acids as a biomarker, as they were not readily available in all participants. Although 3 participants had labs collected outside the 90‐day window (100, 134, and 162 days) for APRI and FIB‐4 calculations, we felt their inclusion was important for the overall analysis of these rare orphan disorders. Prospective, large‐scale studies involving multicenter cohorts or international database consortiums will be needed in the future for further validation of these findings.
In conclusion, the evaluation of noninvasive alternatives to liver biopsy for fibrosis monitoring continues to be an essential and developing field in pediatric liver disorders. Investigation of our young cohort of cholestatic children appears to validate the use of APRI as a noninvasive biomarker for liver fibrosis severity in both ALGS and PFIC. In addition, APRI or FIB‐4, but not conjugated bilirubin, appear to associate with risk for future LT in children with ALGS or PFIC, providing an alternative method of assessment that may complement other existing processes of evaluation for liver transplant candidacy.
Supporting information
Fig S1
Table S1‐S3
Supported by the Bergeron family.
Potential conflict of interest: Dr. Leung received grants from Mirum, Gilead, and AbbVie. Dr. Loomes consults for Mirum, Albireo, and Retrophin. Dr. Molleston received grants from AbbVie, Gilead, Mirum/Shire, and Albireo.
References
- 1. Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, et al. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl 2012;18:940‐948. [DOI] [PubMed] [Google Scholar]
- 2. Liu Y, Sun LY, Zhu ZJ, Wei L, Qu W, Zeng ZG. Liver transplantation for progressive familial intrahepatic cholestasis. Ann Transplant 2018;23:666‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mouzaki M, Bass LM, Sokol RJ, Piccoli DA, Quammie C, Loomes KM, et al. Early life predictive markers of liver disease outcome in an International, Multicentre Cohort of children with Alagille syndrome. Liver Int 2016;36:755‐760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999;29:822‐829. [DOI] [PubMed] [Google Scholar]
- 5. Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner NB. Consequences of JAG1 mutations. J Med Genet 2003;40:891‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davit‐Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma‐glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010;51:1645‐1655. [DOI] [PubMed] [Google Scholar]
- 7. Bull LN, Carlton VE, Stricker NL, Baharloo S, DeYoung JA, Freimer NB, et al. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC‐1] and Byler syndrome): evidence for heterogeneity. Hepatology 1997;26:155‐164. [DOI] [PubMed] [Google Scholar]
- 8. Frider B, Castillo A, Gordo‐Gilart R, Bruno A, Amante M, Alvarez L, et al. Reversal of advanced fibrosis after long‐term ursodeoxycholic acid therapy in a patient with residual expression of MDR3. Ann Hepatol 2015;14:745‐751. [PubMed] [Google Scholar]
- 9. Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD, American Association for the Study of Liver Diseases . Liver biopsy. Hepatology 2009;49:1017‐1044. [DOI] [PubMed] [Google Scholar]
- 10. Dezsőfi A, Baumann U, Dhawan A, Durmaz O, Fischler B, Hadzic N, et al. Liver biopsy in children: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2015;60:408‐420. [DOI] [PubMed] [Google Scholar]
- 11. Chi H, Hansen BE, Tang WY, Schouten JN, Sprengers D, Taimr P, et al. Multiple biopsy passes and the risk of complications of percutaneous liver biopsy. Eur J Gastroenterol Hepatol 2017;29:36‐41. [DOI] [PubMed] [Google Scholar]
- 12. Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet 2016;9:75‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut 2001;49:431‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vandriel S, Wang J‐S, Li L, Piccoli DA, Loomes KM, Sokal EM, et al. Clinical features and outcomes in an international cohort of 731 Alagille Syndrome patients from 19 countries. Hepatology 2019;70(Suppl.):55A‐56A. 10.1002/hep.30940. [DOI] [Google Scholar]
- 15. Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012;36(Suppl. 1):S26‐S35. [DOI] [PubMed] [Google Scholar]
- 16. Bull LN, Thompson RJ. Progressive familial intrahepatic cholestasis. Clin Liver Dis 2018;22:657‐669. [DOI] [PubMed] [Google Scholar]
- 17. Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol 2014;4:25‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baker A, Kerkar N, Todorova L, Kamath BM, Houwen RHJ. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2019;43:20‐36. [DOI] [PubMed] [Google Scholar]
- 19. Mehl A, Bohorquez H, Serrano MS, Galliano G, Reichman TW. Liver transplantation and the management of progressive familial intrahepatic cholestasis in children. World J Transplant 2016;6:278‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003;38:518‐526. [DOI] [PubMed] [Google Scholar]
- 21. Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006;43:1317‐1325. [DOI] [PubMed] [Google Scholar]
- 22. Grieve A, Makin E, Davenport M. Aspartate Aminotransferase‐to‐Platelet ratio index (APRi) in infants with biliary atresia: prognostic value at presentation. J Pediatr Surg 2013;48:789‐795. [DOI] [PubMed] [Google Scholar]
- 23. Leung DH, Khan M, Minard CG, Guffey D, Ramm LE, Clouston AD, et al. Aspartate aminotransferase to platelet ratio and fibrosis‐4 as biomarkers in biopsy‐validated pediatric cystic fibrosis liver disease. Hepatology 2015;62:1576‐1583. [DOI] [PubMed] [Google Scholar]
- 24. Shneider BL, Magee JC, Karpen SJ, Rand EB, Narkewicz MR, Bass LM, et al. Total serum bilirubin within 3 months of hepatoportoenterostomy predicts short‐term outcomes in biliary atresia. J Pediatr 2016;170:211‐217.e211‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sellers ZM, Lee LW, Barth RA, Milla C. New algorithm for the integration of ultrasound into cystic fibrosis liver disease screening. J Pediatr Gastroenterol Nutr 2019;69:404‐410. [DOI] [PubMed] [Google Scholar]
- 26. Díaz JJ, Gura KM, Roda J, Perez‐Atayde AR, Duggan C, Jaksic T, et al. Aspartate aminotransferase to platelet ratio index correlates with hepatic cirrhosis but not with fibrosis in pediatric patients with intestinal failure. J Pediatr Gastroenterol Nutr 2013;57:367‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McGoogan KE, Smith PB, Choi SS, Berman W, Jhaveri R. Performance of the AST‐to‐platelet ratio index as a noninvasive marker of fibrosis in pediatric patients with chronic viral hepatitis. J Pediatr Gastroenterol Nutr 2010;50:344‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang HR, Kim HR, Kim MJ, Ko JS, Seo JK. Noninvasive parameters and hepatic fibrosis scores in children with nonalcoholic fatty liver disease. World J Gastroenterol 2012;18:1525‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mansoor S, Yerian L, Kohli R, Xanthakos S, Angulo P, Ling S, et al. The evaluation of hepatic fibrosis scores in children with nonalcoholic fatty liver disease. Dig Dis Sci 2015;60:1440‐1447. [DOI] [PubMed] [Google Scholar]
- 30. Jackson JA, Konomi JV, Mendoza MV, Krasinskas A, Jin R, Caltharp S, et al. Performance of fibrosis prediction scores in paediatric non‐alcoholic fatty liver disease. J Paediatr Child Health 2018;54:172‐176. [DOI] [PubMed] [Google Scholar]
- 31. Suominen JS, Lampela H, Heikkilä P, Lohi J, Jalanko H, Pakarinen MP. APRi predicts native liver survival by reflecting portal fibrogenesis and hepatic neovascularization at the time of portoenterostomy in biliary atresia. J Pediatr Surg 2015;50:1528‐1531. [DOI] [PubMed] [Google Scholar]
- 32. Englert C, Grabhorn E, Richter A, Rogiers X, Burdelski M, Ganschow R. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation 2007;84:1361‐1363. [DOI] [PubMed] [Google Scholar]
- 33. Lemoine M, Shimakawa Y, Nayagam S, Khalil M, Suso P, Lloyd J, et al. The gamma‐glutamyl transpeptidase to platelet ratio (GPR) predicts significant liver fibrosis and cirrhosis in patients with chronic HBV infection in West Africa. Gut 2016;65:1369‐1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lemoine M, Thursz M, Mallet V, Shimakawa Y. Diagnostic accuracy of the gamma‐glutamyl transpeptidase to platelet ratio (GPR) using transient elastography as a reference. Gut 2017;66:195‐196. [DOI] [PubMed] [Google Scholar]
- 35. Lee J, Kim MY, Kang SH, Kim J, Uh Y, Yoon KJ, et al. The gamma‐glutamyl transferase to platelet ratio and the FIB‐4 score are noninvasive markers to determine the severity of liver fibrosis in chronic hepatitis B infection. Br J Biomed Sci 2018;75:128‐132. [DOI] [PubMed] [Google Scholar]
- 36. Rockey DC. Current and future anti‐fibrotic therapies for chronic liver disease. Clin Liver Dis 2008;12:939‐962, xi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fitzpatrick E, Quaglia A, Vimalesvaran S, Basso MS, Dhawan A. Transient elastography is a useful noninvasive tool for the evaluation of fibrosis in paediatric chronic liver disease. J Pediatr Gastroenterol Nutr 2013;56:72‐76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1‐S3