

Abstract
Myeloperoxidase (MPO) activity has been associated with the metabolic syndrome, cardiovascular and liver disease. Here, we evaluate the therapeutic potential of MPO inhibition on nonalcoholic steatohepatitis (NASH) and NASH‐induced fibrosis, the main determinant of outcomes. MPO plasma levels were elevated in patients with nonalcoholic fatty liver disease (NAFLD) compared with healthy controls. In a second cohort, hepatic MPO messenger RNA expression correlated with higher body mass index and hemoglobin A1c, both being risk factors for NAFLD. We could establish by immunohistochemistry that MPO‐positive cells were recruited to the liver in various mouse models of fibrogenic liver injury, including bile duct ligation, carbon tetrachloride (CCl4) treatment, spontaneous liver fibrogenesis in multidrug resistance 2 knockout (MDR2 KO) mice, and NASH‐inducing diet. Comparison of MPO‐deficient mice and their wild‐type littermates exposed to a high‐caloric diet revealed that MPO deficiency protects against NASH‐related liver injury and fibrosis. In line with this, hepatic gene expression analysis demonstrated a MPO‐dependent activation of pathways relevant for wound healing, inflammation, and cell death in NASH. MPO deficiency did not affect NAFLD‐independent liver injury and fibrosis in MDR2 KO or CCl4‐treated mice. Finally, we treated wild‐type mice exposed to NASH‐inducing diet with an oral MPO inhibitor. Pharmacological MPO inhibition not only reduced markers of MPO‐mediated liver damage, serum alanine aminotransferase levels, and hepatic steatosis, but also significantly decreased NASH‐induced liver fibrosis. MPO inhibitor treatment, but not MPO deficiency, significantly altered gut microbiota including a significant expansion of Akkermansia muciniphila. Conclusions: MPO specifically promotes NASH‐induced liver fibrosis. Pharmacological MPO inhibition attenuates NASH progression and NASH‐induced liver fibrosis in mice and is associated with beneficial changes of intestinal microbiota.
Our study shows that in patients, systemic levels of the neutrophil enzyme myeloperoxidase (MPO) correlate with the presence of nonalcoholic steatohepatitis (NASH) and hepatic MPO expression with risk factors of NASH. Using various mouse models, we demonstrate that MPO is essential for NASH progression and consecutive liver fibrosis but not for steatosis‐independent liver fibrogenesis. In mice treated with an MPO inhibitor, AZM198, we show that MPO inhibition has the potential to attenuate NASH‐induced liver injury and fibrosis, and, moreover, is associated with beneficial changes of intestinal microbiota.

Abbreviations
- ALT
alanine aminotransferase
- BDL, bile duct ligation; BMI
body mass index
- DKO
double knockout
- HbA1c
hemoglobin A1c
- HCC
hepatocellular carcinoma
- HFCholC
high‐fat, high‐cholesterol, high‐carbohydrate
- HFHC
high‐caloric, high‐fat, high‐carbohydrate
- HNE
4‐hydroxynonenal
- HOMA‐IR
homeostasis model assessment of insulin resistance
- KO
knockout
- MDR2
multidrug resistance 2
- MPO
myeloperoxidase
- mRNA
messenger RNA
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- ns
not significant
- PMN
polymorphonuclear neutrophil granulocyte
- qPCR
quantitative polymerase chain reaction
- WT
wild type
- α‐SMA
α‐smooth muscle actin
Nonalcoholic fatty liver disease (NAFLD) is a leading cause of chronic liver injury worldwide. The course of disease is highly variable. While mere nonalcoholic fatty liver (NAFL) is often considered a benign condition, nonalcoholic steatohepatitis (NASH) is defined by hepatocellular injury and inflammation and may lead to progressive liver fibrosis and ultimately liver cirrhosis, accounting for a relevant number of patients with terminal liver disease who are threatened by complications of portal hypertension, chronic liver failure, and development of hepatocellular carcinoma (HCC).( 1 ) Thus, progressive NAFLD is responsible for significant liver‐related mortality.( 2 , 3 , 4 ) Regardless of the distinction between NAFL and NASH, liver fibrosis is the most important histological feature associated with liver‐related and all‐cause mortality.( 5 , 6 ) While not directly predictive of mortality itself, NASH is clearly associated with fibrosis severity and likely the underlying pathomechanism of hepatic fibrogenesis in NAFLD.( 1 ) Thus, effective therapeutic strategies should aim at the reduction of steatohepatitis‐induced liver fibrosis. To date, no approved pharmacological therapy for NAFLD/NASH exists.
Hepatic infiltration with inflammatory cells including polymorphonuclear neutrophil granulocytes (PMNs) characterizes NASH.( 7 ) Myeloperoxidase (MPO) is an enzyme predominantly released by PMNs but also by some monocyte populations to generate reactive oxidants such as hypochloric acid in an H2O2‐dependent reaction( 8 ) as a mechanism of host defense. Moreover, MPO has been identified as a main driver of lipid peroxidation in inflamed tissues.( 9 ) Increased numbers of MPO‐positive cells have been described in the livers of patients with NASH compared to patients with NAFL,( 10 ) and MPO‐expressing Kupffer cells have been located in fibrotic septa of cirrhotic livers.( 11 ) Moreover, genetic studies demonstrate an association between gain‐of‐function MPO promoter polymorphisms with unfavorable liver‐related outcomes in patients with hepatitis C, alcoholic liver disease, and haemochromatosis.( 12 , 13 , 14 ) Moreover, systemic MPO levels have been shown to correlate with arterial hypertension, hyperglycemia, and obesity.( 15 , 16 ) These components of the metabolic syndrome are not only established risk factors for NASH progression but also for cardiovascular mortality, which is highly relevant in patients with NASH and even surpasses liver‐related mortality as the leading cause of death.( 5 , 6 )
In summary, a large body of correlative evidence links MPO activity with liver disease progression, as well as with comorbidities of the metabolic syndrome that are associated with NASH. Here, we show that MPO is abundant in patients with NASH and use mouse models to prove that MPO functionally drives NASH. We demonstrate the therapeutic potential of pharmacological MPO inhibition in experimental NASH, focusing on NASH‐related liver fibrosis as the most relevant outcome variable.
Methods
Patients
Plasma MPO levels were measured in patients with biopsy‐proven NAFL (defined by NAFLD activity score < 4), NASH (defined by NAFLD activity score ≥ 4), and liver‐healthy control subjects, who were characterized by normal liver function tests, low liver elastography, and controlled attenuation parameter measurements (FibroScan; Echosens, Paris, France). Hepatic MPO gene expression was analyzed in liver biopsies from a second cohort of patients who had undergone bariatric or other abdominal surgery that has been previously described in detail.( 17 ) The studies were approved by the local ethics committees in Hamburg and Ulm, Germany (PV5036 and 112/2003, respectively) and comply with the 1975 Declaration of Helsinki. No donor organs were obtained from executed prisoners or other institutionalized persons. Written, informed consent was obtained from each subject.
Mice
MPO‐deficient (B6.129X1‐Mpotm1Lus/J) knockout (KO) mice were kindly provided by the group of Stephan Baldus, University Hospital Cologne. Multidrug resistance 2 (MDR2)‐deficient (FVB.129P2‐Abcb4tm1Bor/J) mice and C57BL/6J mice were purchased from Charles River (Sulzfeld, Germany) and kept group‐housed in cages on a 12‐hour light/dark schedule in the animal facility of the Hamburg University Medical Center. Both MPO KO and MDR2 KO mice were back‐crossed with C57BL/6J mice to ensure a homogeneous background. For our experiments, we used age‐ and sex‐matched KO and wild‐type (WT) littermates generated by heterozygous breeding. To evaluate the role of MPO in spontaneous hepatic fibrogenesis and carcinogenesis, we generated animals that were homozygous for the MDR2 deletion and heterozygous for the MPO KO. These mice were then interbred to generate MDR2‐MPO double‐KO (DKO) animals and MDR2 KO–MPO WT littermates that were used for experiments. Different mouse models of liver injury and NASH were analyzed. Further details are outlined in the Supporting Information. Animal care and experiments were in accordance with the ARRIVE guidelines( 18 ) and approved by the review board of the State of Hamburg, Germany (G12/106; G16/50; G10/82).
Statistics
All data are expressed as mean ± SEM. Statistical analysis of human data was performed by one‐way analysis of variance with Tukey’s post hoc test and Spearman r two‐tailed correlation, respectively. Murine experiments with two groups were statistically analyzed by Mann‐Whitney U test. For comparison of more than two groups, one‐way analysis of variance with Sidak correlation or with Tukey’s post hoc test was used. Differences were considered significant at P values < 0.05. The statistics for the microarray analysis are described in the Supporting Information. The in vitro experiment was statistically analyzed by Kruskal‐Wallis test with Dunn’s multiple comparisons test.
Additional materials and methods are described in the Supporting Information.
Results
Abundance of MPO in Patients With NAFLD and Mouse Models of Liver Fibrosis
MPO‐positive cells have been characterized as a feature of NASH liver histology.( 10 ) To evaluate our hypothesis that MPO activity is systemically increased in patients with NASH, we measured fasting plasma levels of patients with biopsy‐proven NAFL (n = 9), NASH (n = 10), and liver‐healthy control subjects (n = 17). Indeed, compared with healthy individuals, plasma MPO levels were moderately increased in NAFL and strongly increased in patients with NASH, the latter comparison reaching statistical significance (40.7 ng/mL vs. 16.1 ng/mL; P = 0.0044; Fig. 1A). In a second cohort of patients, we analyzed whether hepatic gene expression of MPO is associated with risk factors for NASH progression. Interestingly, hepatic MPO gene‐expression levels were significantly correlated with body mass index (BMI) and the marker for impaired glucose homeostasis, hemoglobin A1c (HbA1c) (Fig. 1B). Thus, we found that systemic and hepatic MPO levels are increased in the presence of NAFLD and associated comorbidities.
Fig. 1.
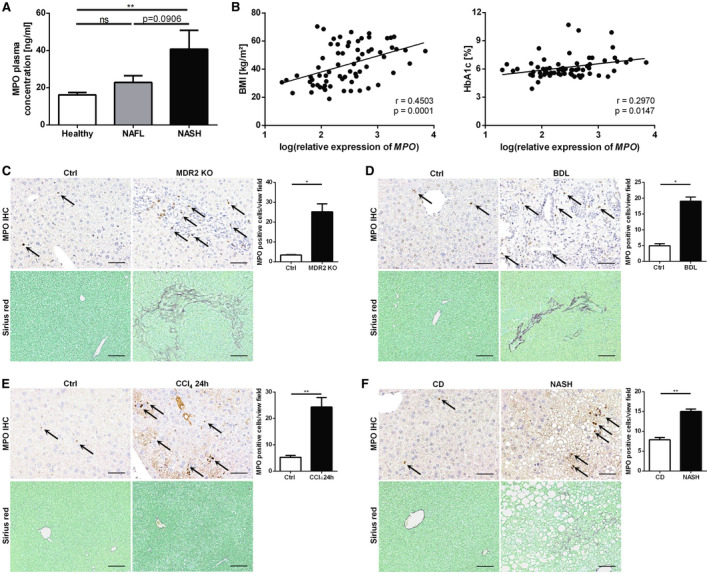
Abundance of MPO in patients with NAFLD and mouse models of liver fibrosis. (A) MPO plasma concentration in liver‐healthy control subjects (“Healthy”; n = 17) and patients with biopsy‐proven NAFL (n = 9) or NASH (n = 10). (B) Correlation of hepatic MPO mRNA expression and BMI (n = 69) and HbA1c (n = 67), respectively, in patients undergoing surgery. (C‐F) Representative images of mouse livers immunostained for MPO (scale bar: 50 µm), quantification of MPO‐positive cells, and hepatic sirius red staining (scale bar: 100 μm) from different profibrogenic liver injury models: 12‐week‐old female WT (“Ctrl”) and MDR2 KO mice (nCtrl = 4; nMDR2 KO = 5) (C); mice untreated (“Ctrl”) and 13 days following BDL (nCtrl = 4; nBDL = 5) (D); and mice untreated (“Ctrl”) or following a single injection of CCl4 (nCtrl = 4; nCCl4 = 6) (E). (F) Mice fed with chow diet (“CD”) or HFHC (“NASH”) for 26 weeks (nCD = 3; nNASH = 12). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01. Abbreviations: Ctrl, control; IHC, immunohistochemistry; ns, not significant.
However, little is known about the role of MPO in hepatic fibrogenesis. Therefore, we assessed whether MPO‐positive cells are present in various mouse models of hepatic fibrogenesis. MPO immunohistochemistry revealed that the number of MPO‐positive cells was significantly increased in mouse livers that were profibrogenically injured by carbon tetrachloride (CCl4) or bile duct ligation (BDL) as well as in livers from MDR2 KO mice that develop spontaneous biliary injury and liver fibrosis (Fig. 1C‐E). Notably, mice with NASH, induced by a high‐caloric, high‐fat, high‐carbohydrate (HFHC) diet over 26 weeks, showed an increased number of MPO‐positive cells on liver histology compared with nonsteatotic livers of mice that were fed a control diet (Fig. 1F).
In summary, abundance of MPO is correlated with NAFLD/NASH in patients and mice as well as with murine liver fibrosis.
MPO Deficiency Attenuates Liver Injury and Liver Fibrosis in a Dietary Mouse Model of NASH
Having established that MPO is increased in fibrogenic liver injury in mice and in patients with NAFLD, our next aim was to characterize the functional relevance of MPO in NASH progression. For this purpose, male MPO KO mice and their WT littermates were fed a high‐fat, high‐cholesterol, high‐carbohydrate (HFCholC) diet( 19 ) or a control diet for 24 weeks. We used this cholesterol‐based dietary NASH model as it reliably produces liver fibrosis, the prognostically most relevant histologic feature of NASH. Mice exposed to the HFCholC diet developed significant hepatic steatosis and mild liver fibrosis (Fig. 3A‐E). Furthermore, we could demonstrate an elevated MPO serum concentration and, like in HFHC‐fed mice, an increased hepatic recruitment of MPO‐positive cells in HFCholC‐fed mice compared with control mice (Fig. 2A,B).
Fig. 3.
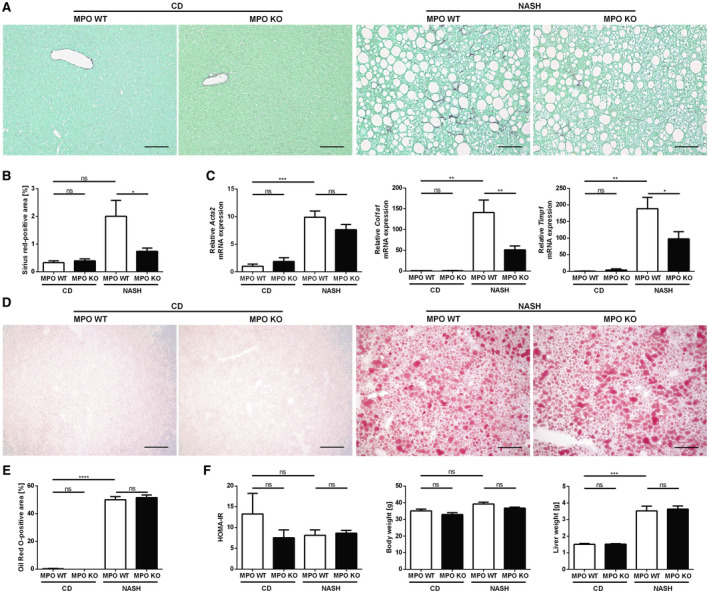
MPO deficiency attenuates liver fibrosis in a dietary mouse model of NASH. Hepatic fibrosis, steatosis, and metabolic parameters were evaluated in MPO KO and WT littermates that were fed chow diet (“CD”) or HFCholC diet (“NASH”) for 24 weeks (nMPO WT CD = 3‐4; nMPO KO CD = 6; nMPO WT NASH = 13‐14; nMPO KO NASH = 16‐17). Liver fibrosis was quantified by sirius red staining (A) (scale bar: 100 µm), quantification of stained area (B), and qPCR (C) of fibrosis‐related genes. (D) Oil Red O staining was used to evaluate hepatic steatosis (scale bar: 200 µm). (E) Quantification of the staining. (F) HOMA‐IR, body weight, and liver weight. Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Abbreviations: Acta2, α‐smooth muscle actin; Col1a1, α1 type I collagen; Timp1, tissue inhibitor of metalloproteinase 1.
Fig. 2.
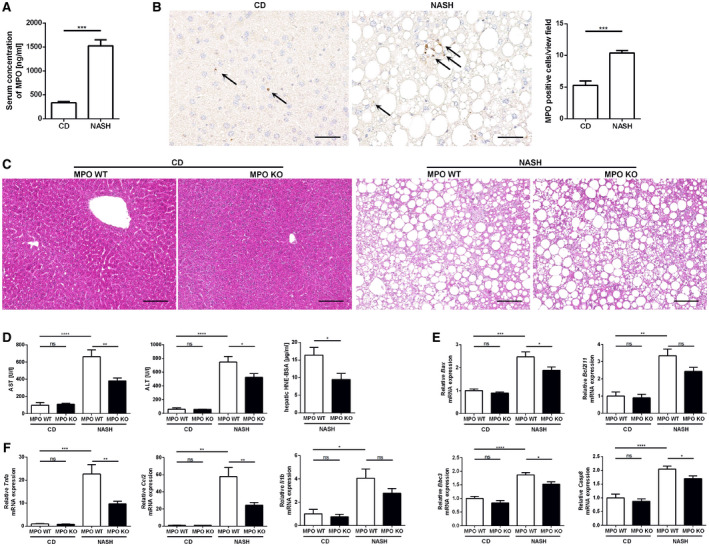
MPO deficiency attenuates liver injury in a dietary mouse model of NASH. Male MPO KO and WT littermates were fed with a chow diet (“CD”) or the HFCholC (“NASH”) diet for 24 weeks (nMPO WT CD = 3‐4; nMPO KO CD = 5‐6; nMPO WT NASH = 12‐14; nMPO KO NASH = 15‐17). (A,B) Comparison of chow‐fed and HFCholC‐fed WT mice, respectively. (A) Serum MPO concentrations. (B) Representative hepatic MPO immunohistochemistries (scale bar: 50 μm) and quantification of MPO‐positive cells per view field. (C‐G) Comparison of chow‐fed and HFCholC‐fed MPO KO mice and their WT littermates. (C) Representative hepatic hematoxylin and eosin stainings (scale bar: 100 μm). (D) Serum aspartate aminotransferase and ALT levels and hepatic content of HNE bovine serum albumin. (E) qPCR of apoptosis‐related genes. (F) qPCR of inflammation‐associated genes. Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Abbreviations: AST, aspartate aminotransferase; BSA, bovine serum albumin; Bax, Bcl‐2‐associated X protein; Bcl2l11, Bcl‐2‐like protein 11; Bbc3, p53 up‐regulated modulator of apoptosis; Casp8, caspase 8; Ccl2, monocyte chemoattractant protein 1; Il1b, interleukin 1 β; Tnfa; tumor necrosis factor α.
Liver transaminases were increased in mice on HFCholC diet, but significantly lower in MPO‐deficient mice compared with WT, indicating attenuated hepatocellular injury (Fig. 2D). Moreover, hepatic 4‐hydroxynonenal (HNE), a marker of lipid peroxidation, was significantly lower in MPO KO mice (Fig. 2D). In response to the NASH‐inducing diet, apoptosis‐related genes Bcl2l11, Bax, Bbc3, and Casp8 were up‐regulated in the steatotic livers of WT mice, while in MPO KO mice the gene induction of Bax, Bbc3, and Casp8 was significantly lower (Fig. 2E), suggesting that absence of MPO attenuates the activation of proapoptotic signals in NASH livers. Quantitative polymerase chain reaction (qPCR) of proinflammatory genes showed a strong induction of hepatic expression of Il1b, Tnfα, and Ccl2 in mice with steatohepatitis compared with chow‐fed mice. NASH‐induced up‐regulation of these proinflammatory genes was blunted in MPO‐deficient mice (Fig. 2F). There was no difference, however, in the amount of liver macrophages between MPO KO mice and their WT littermates with NASH, as indicated by F4/80 immunostaining and corresponding Adgre1 qPCR in whole‐liver tissue (Supporting Fig. S1A‐C). Anti‐Ly6G/Ly6C immunostaining revealed a significantly increased hepatic recruitment of infiltrating PMNs in murine NASH compared with control mice, whereas there was no difference between MPO‐deficient or WT mice (Supporting Fig. S1D,E).
Importantly, HFCholC diet–induced liver fibrosis was significantly less pronounced in MPO KO mice compared with WT mice, as shown by sirius red staining for hepatic collagen deposition (Fig. 3A,B). In line with this, NASH‐induced up‐regulation of liver fibrosis–associated genes Col1a1 and Timp1 was significantly attenuated in MPO KO mice (Fig. 3C), and there was a trend to a lower hepatic hydroxyproline content in MPO KO mice compared to WT mice with NASH (Supporting Fig. S2). Oil Red O staining of hepatocellular lipid droplets illustrated that both, WT and MPO KO mice developed similar diet‐induced hepatic steatosis (Fig. 3D,E). Consistent with the observed hepatic steatosis, both groups showed a significant increase in liver weight, hepatic triglycerides, and cholesterol without any difference between the genotypes (Fig. 3F and Supporting Fig. S3). Obesity and type 2 diabetes have been identified as independent risk factors of NASH progression. However, the HFCholC diet did not induce weight gain and insulin resistance as indicated by homeostasis model assessment of insulin resistance (HOMA‐IR) (Fig. 3F).
In summary, our data demonstrate that MPO deficiency decreases steatohepatitis‐related hepatic lipid peroxidation, hepatocellular damage, inflammation, and liver fibrosis independent of body weight gain, hepatic lipid accumulation, and insulin resistance.
MPO‐Dependent Gene Expression in NASH Involves Pathways Relevant for Wound Healing, Inflammation, and Cell Death
MPO‐dependent regulation of NASH‐relevant hepatic gene expression was analyzed by microarray following a stepwise strategy. First, 2,902 genes that were significantly up‐regulated or down‐regulated in WT mice with HFCholC‐induced NASH compared with chow‐fed WT mice were identified out of 20,201 analyzed genes (Supporting Fig. S4B). Next, these NASH‐relevant genes in WT mice were compared with hepatic gene expression patterns in MPO KO littermates with HFCholC‐induced NASH (Supporting Fig. S4C), to identify MPO‐specific gene regulation in NASH, followed by pathway analysis of differentially regulated genes. The most significant MPO‐induced pathways in NASH included gene sets related to neutrophil‐mediated inflammation, extracellular matrix organization, and wound healing (Fig. 4A). Selected MPO‐induced pathway‐related gene‐expression patterns are displayed as heat maps in Fig. 4B. There was less evidence of pathways up‐regulated in the absence of MPO—all of which were related to metabolism and showed only weak induction (Supporting Fig. S4D).
Fig. 4.
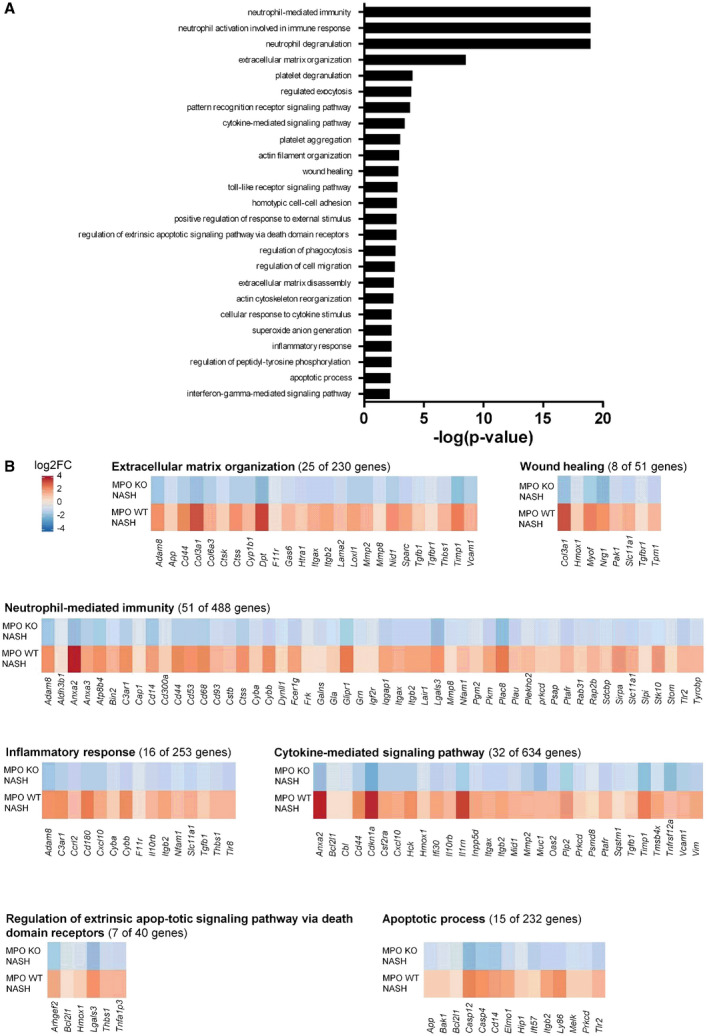
Transcriptome analysis of MPO‐dependent pathways in NASH. Microarray analysis from whole‐liver tissue of hepatic genes differentially (P < 0.05) expressed in WT mice compared with MPO KO animals following 24 weeks of HFCholC feeding. (A) Displayed are the top 25 MPO‐regulated pathways in NASH. (B) Gene‐expression heat maps for selected pathways. The heat maps display genes that were significantly (adjusted P value < 0.05) up‐regulated in the livers of WT mice with HFCholC‐induced NASH compared with chow‐fed WT mice (bottom panel) and down‐regulated in livers of MPO KO mice with HFCholC‐induced NASH compared with HFCholC‐fed WT mice (upper panel). Abbreviations: Adam 8; a disintegrin and metalloproteinase domain‐containing protein 8; Aldh3b1, aldehyde dehydrogenase 3 family, member B1; Anxa, annexin; App, amyloid precursor protein; Atp8b4, ATPase phospholipid transporting 8B4; Bcl2l1, Bcl‐2‐like protein 1; C3ar1, complement component 3a receptor; Cap1, cyclase associated actin cytoskeleton regulatory protein 1; Cbl, Cbl proto‐oncogene; Ccrl2, C‐C chemokine receptor‐like 2; Cd, cluster of differentiation; Cdkn1a, cyclin‐dependent kinase inhibitor 1; Col3a1, α1 type III collagen; Col6a1, α1 type VI collagen, Csf2ra, granulocyte‐macrophage colony‐stimulating factor receptor; Cstb, cystatin b; Ctsk, cathepsin K; Ctss, cathepsin S; Cxcl10, C‐X‐C motif chemokine 10; Cyba; cytochrome b‐245 α chain; Cybb, cytochrome b‐245 β chain; Cyp1b1, cytochrome P450 family 1 subfamily B member 1; Dpt, dermatopontin; Dynll1, dynein light chain LC8‐type 1; F11r, junctional adhesion molecule A; Fcer1g, Fc fragment of IgE, high affinity I, receptor for, gamma polypeptide; Frk, fyn‐related kinase; Galns, N‐acetylgalactosamine‐6‐sulfatase; Gas6, growth arrest specific 6; Gla, galactosidase α; Glipr1, glioma pathogenesis‐related protein 1; Grn, granulin; Hck, tyrosine‐protein kinase HCK; Hmox1, heme oxygenase 1; Htra1, HtrA serine peptidase 1; Ifi30, IFI30 lysosomal thiol reductase; Igf2r, insulin‐like growth factor 2 receptor; Il10rb, interleukin 10 receptor β subunit; Il1rn, interleukin 1 receptor antagonist; Inpp5d, inositol polyphosphate‐5‐phosphatase D; Iqgap1, IQ motif containing GTPase activating protein 1; Itgax, integrin subunit αX; Itgb1, integrin subunit β2; Lair1, leukocyte‐associated immunoglobulin‐like receptor 1; Lama2, laminin subunit α2; Lgals3, galectin 3; Loxl1, lysyl oxidase‐like 1; Mid1, midline 1; Mmp, matrix metalloproteinase; Muc1, mucin 1, cell surface associated; Myof, myoferlin; Nfam1, NFAT activating protein with ITAM motif 1; Nid1, nidogen 1; Nrg1, neuregulin 1; Oas2, 2'‐5'‐oligoadenylate synthetase 2; Pak1, p21‐activated kinase 1; Pgm2, phosphoglucomutase 2; Pkm, pyruvate kinase M1/2; Plac8, placenta‐associated 8; Plau, Urokinase; Plekho2, pleckstrin homology domain containing O2; Plp2, proteolipid protein 2; Prkcd, protein kinase C δ; Psap, prosaposin; Psmd8, proteasome 26S subunit, non‐ATPase 8; Ptafr, platelet‐activating factor receptor; Rab31, Ras‐related protein Rab 31; Rap2b, Ras‐related protein Rap 2b; Sdcbp, syndecan binding protein; Slc11a1, solute carrier family 11 member 1; Slpi, secretory leukocyte peptidase inhibitor; Sparc, osteonectin; Sqstm1, sequestosome 1; Stk10, serine/threonine‐protein kinase 10; Stom, stomatin; Tgfbr, transforming growth factor β receptor; Thbs1, thrombospondin 1; Tlr, toll‐like receptor; Tmsb4x, thymosin β4; Tnfrsf12a, tumor necrosis factor receptor superfamily member 12A; Tpm1, Tropomyosin α1 chain; Tyrobp, transmembrane immune signaling adaptor TYROBP; Vcam1, vascular cell adhesion molecule 1; Vim, vimentin.
In summary, the microarray gene expression analysis indicates that MPO predominantly mediates neutrophil‐mediated inflammation and fibrogenesis in the liver during NASH progression.
MPO Is Not Essential in Nonsteatotic Experimental Liver Fibrosis
For evaluation of MPO as a universal mediator of liver fibrosis driven by injury and inflammation, we analyzed the effect of MPO deficiency in two different nonsteatotic mouse models of liver fibrosis: MDR2 KO mice that spontaneously develop cholestatic liver injury and fibrosis,( 20 ) and liver fibrosis induced by CCl4 treatment, respectively. MDR2‐MPO‐DKO mice and their MDR2‐KO–MPO‐WT littermates developed biliary liver injury and fibrosis after 12 weeks with no significant difference between the genotypes, as shown by similar hepatic expression of fibrosis‐related genes (Fig. 5A) and sirius red staining of hepatic collagen (Fig. 5B). Also, there was no difference in serum alanine aminotransferase (ALT) levels between MDR2‐MPO‐DKO mice and MDR2 KO–MPO WT mice (Fig. 5C), suggesting that MPO was promoting neither liver fibrosis nor liver injury in this model. Similarly, compared with WT littermates, MPO KO mice were not protected from liver fibrosis or injury induced by CCl4 treatment (Fig. 5D‐F). Thus, our data suggest that the role of MPO in hepatic fibrogenesis is specific for steatohepatitis as the underlying disease mechanism.
Fig. 5.
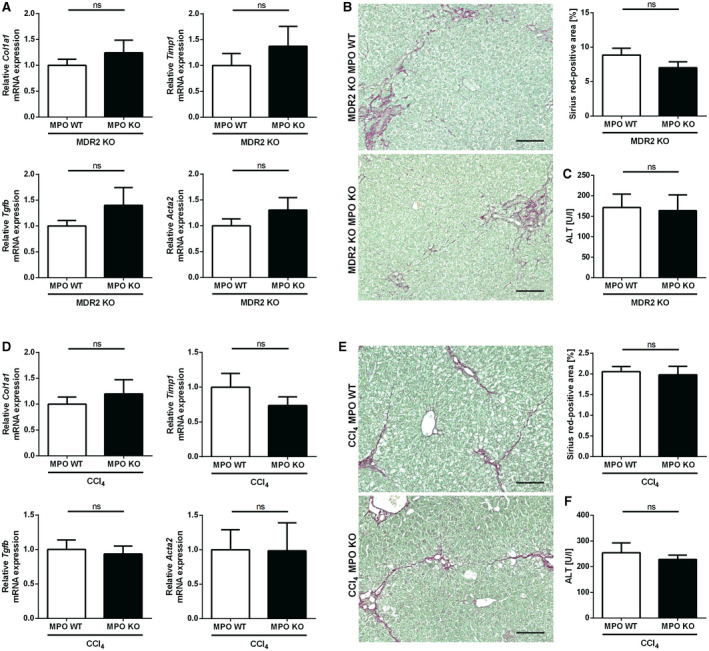
MPO is not relevant in nonsteatotic liver fibrosis models. (A‐C) Spontaneous liver fibrosis was evaluated in 62‐week‐old female MDR2‐MPO‐DKO mice and MDR2 KO–MPO WT littermates (n = 11 each). (A) qPCR of fibrosis‐related genes. (B) Representative hepatic sirius red staining (scale bar: 100 µm) and quantification of stained area. (C) Serum ALT. (D‐F) Toxin‐induced liver fibrosis was analyzed in MPO KO mice (n = 8) and WT littermates (n = 9) after 10 intraperitoneal injections of CCl4 over 5 weeks. (D) qPCR of fibrosis‐related genes. (E) Representative hepatic sirius red staining (scale bar: 100 µm) and quantification of stained area. (F) Serum ALT. Data are presented as mean ± SEM. *P ≤ 0.05. Abbreviation: Tgfb, transforming growth factor β.
MPO Deficiency Has No Effect on Hepatic Carcinogenesis in Mice
Interference with hepatic wound healing and fibrogenesis in NASH could affect hepatic carcinogenesis, and MPO has been associated with HCC.( 12 , 13 ) Therefore, we analyzed the effect of MPO deficiency on hepatic carcinogenesis in mice in two different models. The combination of HFHC feeding and diethylnitrosamine treatment led to the development of liver tumors in almost all HFHC‐fed mice. The tumor load was similar in WT and MPO KO mice (Supporting Fig. S5A).
Hepatic carcinogenesis in a nonsteatotic mouse model was evaluated in female MDR2‐deficient mice that not only spontaneously develop liver injury and fibrosis, but also HCC. However, concomitant MPO deficiency had no effect on the HCC development (Supporting Fig. S5B,C).
Pharmacological MPO Inhibition Attenuates NASH Progression and Liver Fibrosis in Mice
Having demonstrated that MPO deficiency attenuates NASH‐induced liver fibrosis, we wanted to explore the therapeutic potential of MPO inhibition in already established NASH. For this purpose, male WT mice were fed the HFHC diet rich in fructose and medium‐chain trans fats( 21 ) to induce NASH, along with obesity and insulin resistance (Supporting Fig. S6), typical comorbidities of human NASH. The strategy for MPO inhibition by AZM198 (ad libitum in diet) with early and late intervention is presented in the feeding scheme (Fig. 6A). AZM198 serum levels of inhibitor‐treated mice were approximately 1 µmol/L (Fig. 6B), which corresponds to more than 95% inhibition in in vitro cell‐free assays. Interestingly, AZM198 treatment significantly reduced serum MPO levels (Fig. 6B).
Fig. 6.
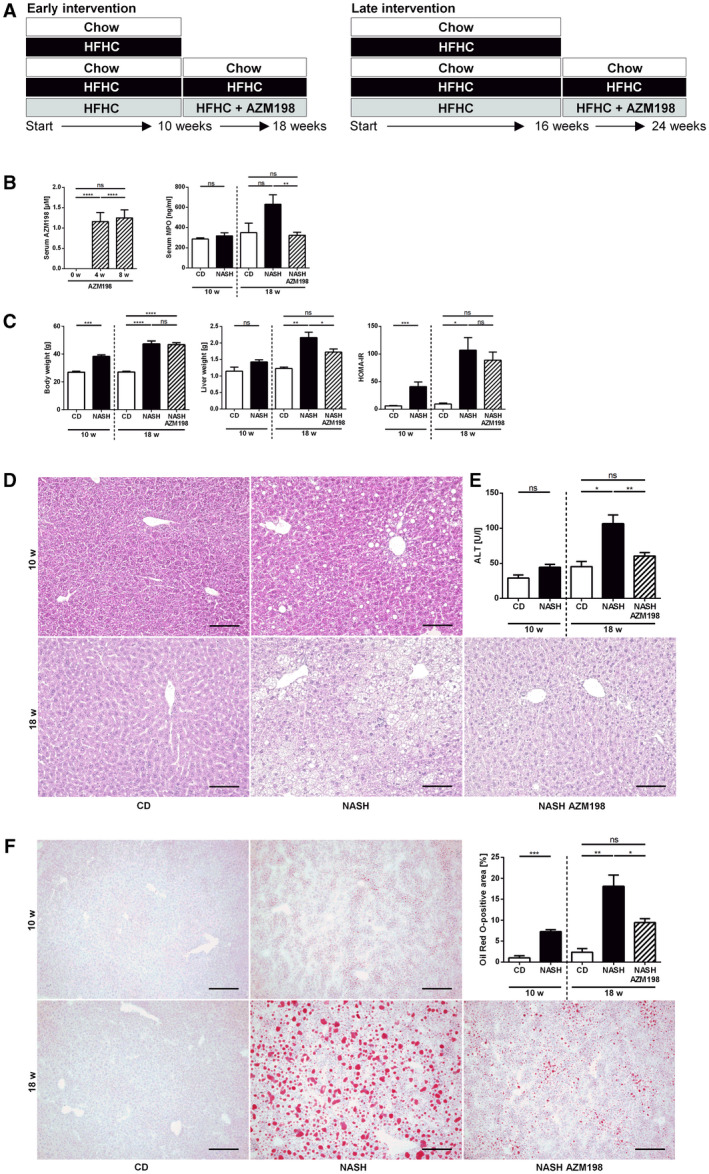
Pharmacological MPO inhibition attenuates NASH progression in mice. Liver injury, steatosis, and metabolic parameters were evaluated in C57BL/6J mice fed with chow diet (“CD”) or HFHC diet with (“NASH AZM198”) or without (“NASH”) the MPO inhibitor, AZM198 (nCD 10w = 4; nNASH 10w = 13; nCD 18w = 4; nNASH 18w = 13‐14; nNASH AZM198 = 12‐14). (A) Experimental design. (B) AZM198 serum concentration after 0, 4, and 8 weeks of HFHC + AZM198 feeding and MPO serum concentration. (C) Body weight, liver weight, and HOMA‐IR. (D) Representative hepatic hematoxylin and eosin stainings (scale bar: 100 µm). (E) Serum ALT. (F) Hepatic steatosis was evaluated by Oil Red O staining (scale bar: 200 µm). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Mice fed with a HFHC diet for 10 weeks displayed significant weight gain and increased HOMA‐IR, indicating insulin resistance (Fig. 6C), and liver histology showed hepatic steatosis (Fig. 6D). To investigate the role of MPO in early disease progression, these mice were treated with the MPO inhibitor AZM198 for another 8 weeks, while being continued on the HFHC diet. Untreated mice were continued on the HFHC diet without AZM198 or on chow diet (Fig. 6A). HFHC feeding resulted in significantly increased liver weight after 18 weeks (Fig. 6C), matching the diet‐induced hepatocellular steatosis (Fig. 6D). AZM198 treatment of HFHC‐fed mice significantly reduced liver weight (Fig. 6C) and hepatocellular lipid accumulation, as demonstrated by Oil Red O staining (Fig. 6F). There was no effect on body weight or HOMA‐IR scores, however (Fig. 6C). AZM198 significantly reduced NASH‐induced liver injury, as demonstrated by decreased ALT levels (Fig. 6E) and reduction of hepatic 3‐chlorotyrosine, a marker of MPO‐mediated oxidative protein damage (Fig. 7A). Quantification of hepatic HNE did not show a difference between control mice and MPO inhibitor‐treated mice, however (Supporting Fig. S7A). Importantly, MPO inhibitor treatment significantly blunted NASH‐induced hepatic increase of fibrosis‐related genes such as Col1a1 and Timp1 (Fig. 7B). Moreover, hepatic α‐smooth muscle actin (α‐SMA) protein staining as a marker of hepatic stellate cell activation and liver fibrosis was significantly reduced by AZM198 (Supporting Fig. S7B). As liver fibrosis was very mild after 18 weeks HFHC feeding, we did not detect quantifiable sirius red staining (Supporting Fig. S7C). For this reason, we repeated this experiment with extended HFHC feeding, initiating the MPO inhibitor treatment after 16 weeks of HFHC diet. Indeed, this created a more robust NASH fibrosis phenotype with significant collagen deposition, as shown by sirius red staining (Fig. 7C). Histological evaluation of sirius red and α‐SMA staining confirmed attenuation of liver fibrosis by MPO inhibitor treatment at this later time point during NASH progression (Fig. 7C,D). Consistent with the previous experiment, MPO inhibition decreased diet‐induced hepatic steatosis (Supporting Fig. S8A) and 3‐chlorotyrosine staining (Supporting Fig. S8B). Moreover, HNE measurement demonstrated significantly decreased hepatic lipid peroxidation in AZM198‐treated mice (Supporting Fig. S8C). MPO inhibitor treatment did not reduce the amount of F4/80‐positive macrophages in NASH livers (Supporting Fig. S9A) or corresponding hepatic Adgre1 messenger RNA (mRNA) expression (Supporting Fig. S9B). Moreover, MPO inhibition did not decrease hepatic recruitment of PMNs in NASH (Supporting Fig. S9C).
Fig. 7.
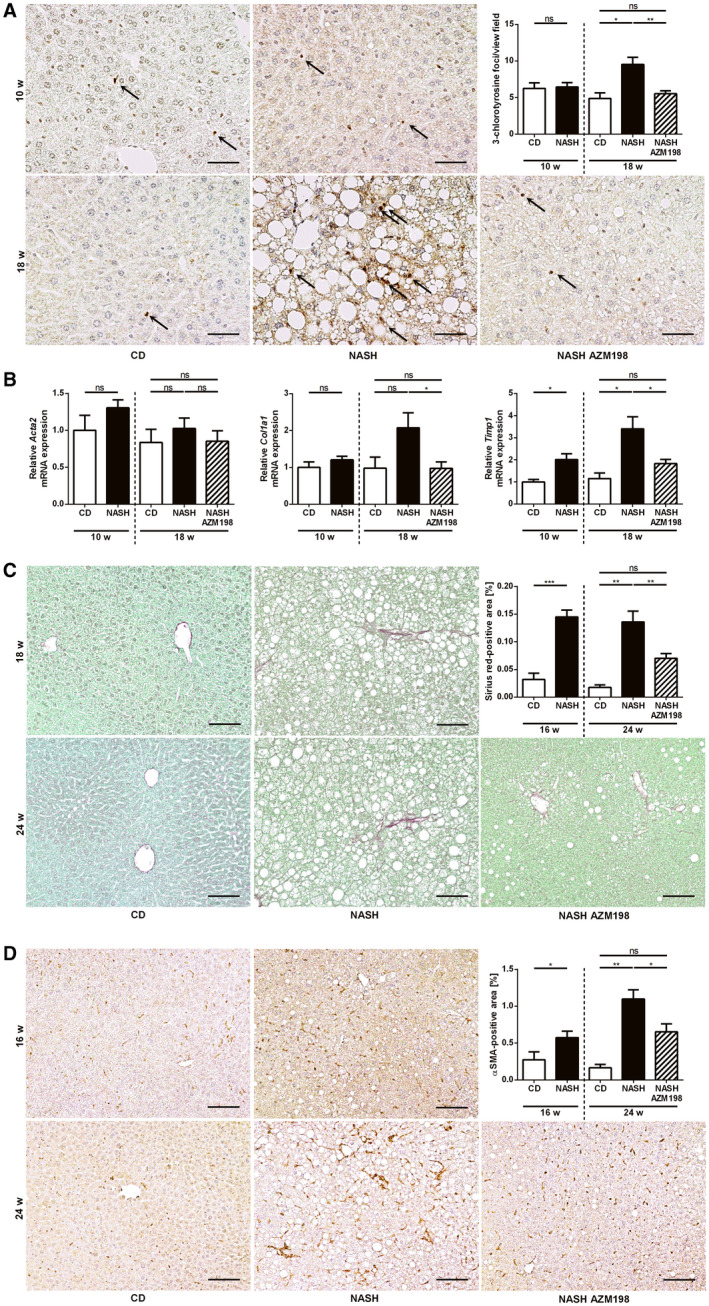
Pharmacological MPO inhibition attenuates NASH‐induced liver fibrosis in mice. MPO‐mediated tissue damage and NASH‐induced liver fibrosis were assessed in C57BL/6J mice, fed with chow diet (“CD”) or HFHC diet with (“NASH AZM198”) or without (“NASH”) the MPO inhibitor, AZM198, at different time points: (A,B) Early intervention (nCD 10w = 3‐4; nNASH 10w = 10‐12; nCD 18w = 4; nNASH 18w = 13‐14; nNASH AZM198 = 12‐14). (C,D) Late intervention (nCD 16w = 5; nNASH 16w = 15; nCD 24w = 4; nNASH 24w = 15‐16; nNASH AZM198 = 16). (A) Representative 3‐chlorotyrosine immunostainings of liver sections (scale bar: 50 µm), quantification of stained foci. (B) qPCR of fibrosis‐related genes. Liver fibrosis was quantified histologically by sirius red staining (C) (scale bar: 100 µm) and α‐SMA immunohistochemistry (D) (scale bar: 100 µm). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
In summary, we could demonstrate that an orally ingested MPO inhibitor reduces hepatic steatosis, oxidative liver injury, and fibrogenesis induced by a HFHC diet.
AZM198 Prevents MPO‐Induced Hepatocyte Death In Vitro
To confirm our hypothesis that MPO mediates hepatocyte death as a driving force behind hepatic fibrogenesis, we incubated the human hepatoma cell line Hep‐G2 with fatty acids with or without recombinant human MPO. As expected, MPO induced cell death in Hep‐G2 cells, as demonstrated by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay. Concomitant treatment with the MPO inhibitor AZM198 completely prevented MPO‐induced hepatocyte death (Fig. 8), corroborating our in vivo findings.
Fig. 8.
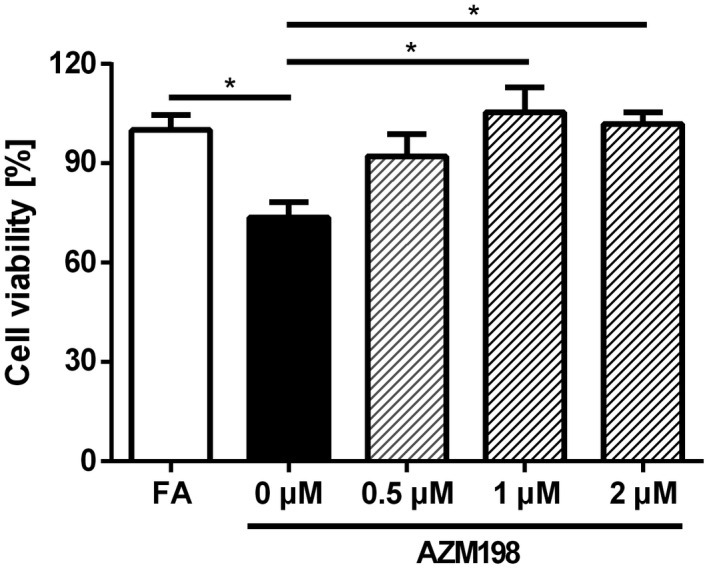
AZM198 prevents MPO‐induced cell death in vitro. To analyze the influence of MPO and AZM198 on hepatocytes, Hep‐G2 cells were incubated for 24 hours with 1‐mM fatty acids. Then, they were left untreated or incubated with 1 U/L MPO, 1 mU/L glucose oxidase, and either DMSO (0 µM AZM198) or AZM198 at a concentration of 0.5 µM, 1 µM, or 2 µM for 24 hours. Cell viability was measured by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay (n = 8). Data are representative of three experiments and are presented as mean ± SEM. *P ≤ 0.05. Abbreviation: FA, fatty acid.
Pharmacological MPO Inhibition but Not MPO Deficiency Significantly Changes the Gut Microbiome During NASH Progression
As the intestinal microbiome may modulate liver inflammation and injury in NAFLD, we analyzed whether MPO inhibition during NASH progression affected the gut microbiome. Indeed, the hepatoprotective effects of MPO inhibition were accompanied by significant changes in gut microbiota at a phylum level (Fig. 9A and Supporting Fig. S10A). Of note, AZM198 treatment induced a significant suppression of the Firmicutes/Bacteroides ratio (Fig. 9B and Supporting Fig. S10B) in two independent experiments. On a species level, MPO inhibitor treatment induced a strong and significant expansion of Akkermansia muciniphila (Fig. 9C and Supporting Fig. S10C). Contrasting these results, MPO‐deficient mice on a NASH diet displayed a similar gut microbiome compared with their WT littermates (Fig. 9D‐F).
Fig. 9.
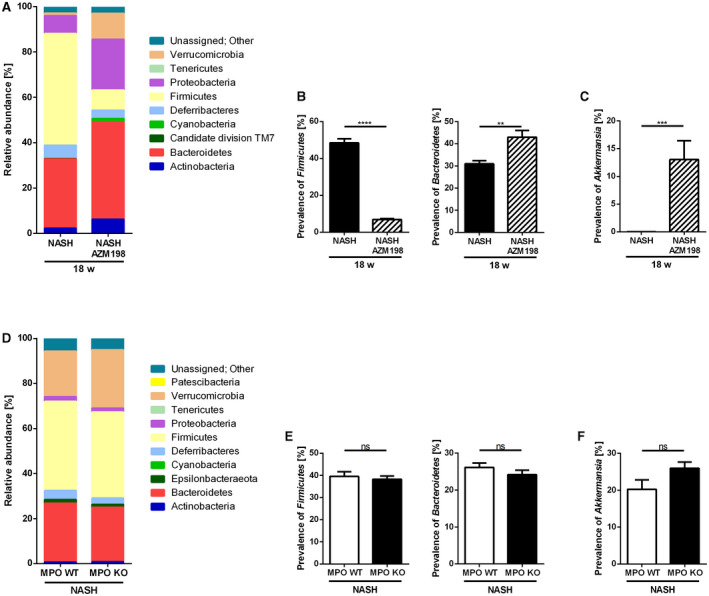
Pharmacological MPO inhibition in NASH progression affects microbiome in mice. Analysis of the gut microbiome by 16S ribosomal RNA sequencing in caecum samples of C57BL/6 mice fed with HFHC diet with (“NASH AZM198”) or without (“NASH”) MPO inhibitor, AZM198 (nNASH = 14; nNASH AZM198 = 14) (A‐C), and in samples of MPO KO and WT littermates fed with a HFCholC diet (“NASH”) for 24 weeks (nMPO WT NASH = 12; nMPO KO NASH = 16) (D‐F). (A,D) Composition of the gut microbiome represented by relative abundance of phylae. (B,E) Ratio of the phylae Firmicutes and Bacteroidetes. (C,F) Relative abundance of Verrucomicrobia species Akkermansia muciniphila. Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Discussion
NAFLD represents an increasing therapeutic challenge, given its high prevalence and the absence of approved pharmacological treatment options. Lifestyle intervention can effectively reduce histopathological features of NASH, but only if significant weight reduction is achieved, which, even in the setting of a clinical trial, succeeds only in the minority of patients.( 22 )
MPO activity has been associated with the progression of several liver diseases, including NASH.( 10 , 12 , 13 , 14 ) In our present study, we demonstrate evidence that MPO is not only functionally relevant for NASH progression, but also represents a potential therapeutic target for the pharmacological treatment of NASH.
First, we could show that MPO is increased in the blood of patients with NASH compared with healthy control subjects and patients with NAFL, as well as in mice that were fed a NASH‐inducing HFHC diet. Thus, we could establish that systemic abundance of MPO is associated with the presence of NASH. This is in line with studies showing elevated MPO plasma levels in severely obese patients with NASH compared to subjects with simple steatosis( 10 ) and in severely obese patients compared to healthy controls.( 16 ) Elevated plasma MPO levels have also been found to be independently associated with arterial hypertension, especially in the presence of hyperglycemia.( 15 ) Obesity, arterial hypertension, and hyperglycemia are not only features of the metabolic syndrome but have also been shown to be associated with NASH progression.( 1 , 23 , 24 ) Thus, increased systemic MPO levels are associated with risk factors of NASH progression and with the presence of NASH itself. The relevance of MPO in NASH progression is further supported by our finding that, in patients, hepatic expression of MPO significantly correlated with BMI and HbA1c as risk factors for NAFLD progression. Moreover, we could show that MPO‐positive cells are recruited to the liver in several mouse models of liver fibrosis, including diet‐induced NASH. This parallels a study showing that in patients, hepatic MPO activity is correlated with NASH severity.( 10 ) In summary, our data and published evidence demonstrate that NASH progression takes place in the presence of increased systemic and hepatic MPO activity.
Importantly, we could establish that MPO activity is not only associated with NAFLD/NASH, but also functionally drives disease progression, as liver injury, inflammation, and hepatic fibrogenesis induced by HFCholC diet were significantly attenuated in MPO‐deficient mice. Similar findings have been described by two other studies,( 25 , 26 ) both supporting a role for MPO in NASH‐induced liver fibrosis. One group used low‐density lipoprotein receptor–deficient mice to enhance the NASH phenotype in MPO‐deficient and MPO‐positive bone marrow chimera that were fed a high‐fat diet for 8 weeks.( 26 ) The other group used a methionine and choline–deficient diet for 4 weeks for rapid NASH induction in MPO KO mice and C57 WT control animals.( 25 ) Although this nutrient deprivation model efficiently produces a NASH phenotype, the rapid disease progression and concomitant massive weight loss are somewhat unphysiological. For our study, we deliberately avoided a genetic disease model in favor of an overnutrition model of slow disease progression and used littermate controls to reduce confounding influences and to mimic the situation in patients more closely.
Having demonstrated that MPO is required for NASH‐induced hepatic fibrogenesis, we explored the therapeutic potential of MPO inhibition in NASH, by treating mice with pre‐existing NASH with an orally administered MPO inhibitor. Consistent with our results from MPO KO mice, we could show that pharmacological MPO inhibition significantly attenuated NASH‐induced hepatic fibrogenesis. This therapeutic effect is particularly important, as liver fibrosis in NASH is an independent predictor of mortality( 5 , 6 ) and fibrosis reduction as a surrogate marker for prognosis improvement is an accepted endpoint in clinical NASH trials.( 27 )
The mechanism by which MPO promotes hepatic fibrogenesis has yet to be defined. In fact, previous studies using toxic or cholestatic fibrosis models have stated that PMNs, the main MPO‐expressing cell type, are recruited to the injured liver but are not functionally relevant for hepatic fibrogenesis.( 28 , 29 ) This is confirmed by our findings showing that MPO deficiency had no effect on steatosis‐independent models of liver fibrosis, but exclusively attenuated NASH progression. Thus, it is unlikely that MPO is a stereotypical mediator of liver fibrogenesis, regardless of the underlying disease, even if in vitro data suggest that MPO can directly activate hepatic stellate cells,( 25 ) the main profibrogenic cell type in the liver.( 30 ) Our results suggest that the profibrogenic effect of MPO is a specific consequence of NAFLD‐driven liver injury. According to our microarray analysis, MPO activates pathways relevant for both neutrophil‐mediated inflammation and liver fibrosis (extracellular matrix reorganization and wound healing) during NASH progression. Indeed, we observed that MPO deficiency and MPO inhibition attenuated NASH‐induced liver injury. Consistent with this, Pulli et al. had observed that MPO directly induced hepatocyte death in vitro and in mice exposed to a methionine and choline–deficient diet.( 25 ) Moreover, treatment with the MPO inhibitor AZM198 decreased hepatic 3‐chlorotyrosine, a specific marker of MPO‐catalyzed oxidative protein damage that was up‐regulated during NASH progression. Also, hepatic lipid peroxidation in murine steatohepatitis was significantly decreased in the absence of MPO. Thus, we conclude that MPO directly induces oxidative damage of steatotic liver tissue in NASH. In support of this concept, it has been previously shown that hepatocytes isolated from mice with NASH were more sensitive against MPO‐induced oxidative cell death in vitro, possibly due to reduced antioxidative capacity.( 25 , 31 ) This was further confirmed in our finding that steatotic human hepatoma cells underwent cell death in response to MPO treatment, which, in agreement with our in vivo data, could be completely prevented by AZM198. Taken together, our findings suggest that specifically in the steatotic liver, MPO induces oxidative hepatocellular injury and subsequent injury‐driven hepatic fibrogenesis.
Our plasma and serum data in patients and mice clearly indicate that increased MPO in NAFLD is a systemic phenomenon and not limited to the liver. In our study, pharmacological MPO inhibition reduced MPO serum levels, consistent with the concept that MPO facilitates PMN recruitment and migration.( 32 , 33 ) In view of this, MPO inhibition or deficiency could reduce the extrahepatic risk factors of NASH progression, obesity and insulin resistance.( 1 , 24 , 34 ) However, neither absence nor inhibition of MPO affected body weight or HOMA scores in our mice on a NASH diet. Thus, it is more likely that MPO promotes NASH progression by a liver‐specific mechanism.
We found that MPO deficiency blunted HFCholC‐diet‐induced up‐regulation of the hepatic expression of Tnfα and Ccl2, which had previously been implicated with inflammation‐driven hepatic fibrogenesis and NASH progression, respectively.( 35 , 36 ) However, MPO inhibition or MPO deficiency had no effect on hepatic recruitment of macrophages or PMNs in murine NASH. It is unlikely, therefore, that hepatoprotection by MPO inhibition is mediated by blocking the chemotactic effects of MPO in the liver, which rather emphasizes MPO‐induced tissue injury as a relevant driver of liver fibrosis in NASH. Interestingly, NASH‐induced hepatic expression of proapoptotic genes was significantly decreased in MPO KO mice. However, despite concordant results regarding liver injury and fibrosis, we could not reproduce these gene expression patterns in AZM198‐treated mice. A possible explanation may be the long duration of our dietary NASH models, as it is unclear exactly when during NASH progression transcriptional changes occur that precede the biological effects observed.
Interestingly, pharmacological MPO inhibition reduced not only injury and fibrosis but also hepatic steatosis in murine NASH, while this effect was not observed in MPO‐deficient mice. This may be explained by the AZM198‐induced alteration of the intestinal microbiota, which included a significant expansion of Akkermansia muciniphila and a reduction of the Firmicutes/Bacteroidetes ratio, a compositional change of intestinal microbiota that has been associated with weight reduction in obesity( 37 ) and absence of experimental NAFLD.( 38 ) MPO deficiency had no effect on gut microbiota, possibly because MPO KO mice were co‐housed with WT littermates. Thus, it is possible that AZM198‐specific microbial effects mediate additional hepatoprotection. This is supported by studies demonstrating that Akkermansia muciniphila reduces high‐fat diet–induced metabolic dysregulation,( 39 , 40 ) and hepatic lipid content and inflammation in murine alcoholic steatohepatitis.( 41 ) The additional antisteatogenic effect of MPO inhibition may also be related to potential off‐target effects of AZM198. AZM198 has been profiled in selectivity screens, and the concentration observed in the current study corresponds to approximately 95% inhibition of MPO, 6% inhibition of thyroid peroxidase, and negligible effects on other molecular targets assessed in vitro.( 42 )
Several studies indicate that MPO and PMNs may play a role in hepatic carcinogenesis.( 12 , 13 , 43 , 44 ) However, in our study, neither a mouse model of NASH‐related HCC nor the NASH‐independent MDR2 model showed any effects of MPO on HCC development, indicating that MPO is not the essential mediator of PMN‐driven carcinogenesis. On the other hand, MPO is not required for hepatic tumor suppression, so therapeutic MPO inhibition is unlikely to increase the risk of HCC promotion.
In summary, we establish that MPO is functionally relevant in NASH progression, and we provide evidence that AZM198, an oral MPO inhibitor, attenuates hepatic injury and fibrogenesis in a mouse model of NASH. MPO may be a particularly attractive therapeutic target, as several lines of evidence connect MPO activity to cardiovascular disease,( 45 , 46 , 47 ) a common comorbidity of NASH of high prognostic relevance( 6 ) that may also therapeutically benefit from MPO inhibition.( 42 ) Our present work warrants future studies to evaluate whether beneficial effects of MPO inhibition in murine NASH can be translated into humans.
Supporting information
Supplementary Material
Acknowledgments
We gratefully acknowledge Alexandra Peric of AstraZeneca for the quantification of AZM198, Nicole Fischer and Jiabin Huang for the microbiome analyses, and Martina Fahl for the support in cell culture.
Potential conflict of interest: Dr. Michaelsson owns stock in, is employed by, and owns intellectual property rights in AstraZeneca.
Supported by the Deutsche Forschungsgemeinschaft (Grant/Award No. SFB841).
References
Author names in bold designate shared co‐first authorship.
- 1. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11‐20. [DOI] [PubMed] [Google Scholar]
- 3. Lindenmeyer CC, McCullough AJ. The natural history of nonalcoholic fatty liver disease—an evolving view. Clin Liver Dis 2018;22:11‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology 2015;61:1547‐1554. [DOI] [PubMed] [Google Scholar]
- 7. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander‐Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467‐2474. [DOI] [PubMed] [Google Scholar]
- 8. van der Veen BS, de Winther MP, Heeringa P. Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal 2009;11:2899‐2937. [DOI] [PubMed] [Google Scholar]
- 9. Zhang R, Brennan ML, Shen Z, MacPherson JC, Schmitt D, Molenda CE, et al. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem 2002;277:46116‐46122. [DOI] [PubMed] [Google Scholar]
- 10. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol 2009;175:1473‐1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown KE, Brunt EM, Heinecke JW. Immunohistochemical detection of myeloperoxidase and its oxidation products in Kupffer cells of human liver. Am J Pathol 2001;159:2081‐2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nahon P, Sutton A, Rufat P, Charnaux N, Mansouri A, Moreau R, et al. A variant in myeloperoxidase promoter hastens the emergence of hepatocellular carcinoma in patients with HCV‐related cirrhosis. J Hepatol 2012;56:426‐432. [DOI] [PubMed] [Google Scholar]
- 13. Nahon P, Sutton A, Rufat P, Ziol M, Akouche H, Laguillier C, et al. Myeloperoxidase and superoxide dismutase 2 polymorphisms comodulate the risk of hepatocellular carcinoma and death in alcoholic cirrhosis. Hepatology 2009;50:1484‐1493. [DOI] [PubMed] [Google Scholar]
- 14. Osterreicher CH, Datz C, Stickel F, Hellerbrand C, Penz M, Hofer H, et al. Association of myeloperoxidase promotor polymorphism with cirrhosis in patients with hereditary hemochromatosis. J Hepatol 2005;42:914‐919. [DOI] [PubMed] [Google Scholar]
- 15. Van der Zwan LP, Scheffer PG, Dekker JM, Stehouwer CD, Heine RJ, Teerlink T. Hyperglycemia and oxidative stress strengthen the association between myeloperoxidase and blood pressure. Hypertension 2010;55:1366‐1372. [DOI] [PubMed] [Google Scholar]
- 16. Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) 2009;17:2014‐2018. [DOI] [PubMed] [Google Scholar]
- 17. Worthmann A, John C, Ruhlemann MC, Baguhl M, Heinsen FA, Schaltenberg N, et al. Cold‐induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat Med 2017;23:839‐849. [DOI] [PubMed] [Google Scholar]
- 18. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group . Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 2010;160:1577‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet‐induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 2013;305:G483‐G495. [DOI] [PubMed] [Google Scholar]
- 20. Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004;127:261‐274. [DOI] [PubMed] [Google Scholar]
- 21. Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, et al. High‐fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010;52:934‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vilar‐Gomez E, Martinez‐Perez Y, Calzadilla‐Bertot L, Torres‐Gonzalez A, Gra‐Oramas B, Gonzalez‐Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015;149:367‐378.e5; quiz e14‐5. [DOI] [PubMed] [Google Scholar]
- 23. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol 2015;13:643‐654.e9; quiz e39‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patel YA, Gifford EJ, Glass LM, McNeil R, Turner MJ, Han B, et al. Risk factors for biopsy‐proven advanced non‐alcoholic fatty liver disease in the Veterans Health Administration. Aliment Pharmacol Ther 2018;47:268‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase‐hepatocyte‐stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid Redox Signal 2015;23:1255‐1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri‐Sverdlov R, et al. Neutrophil‐derived myeloperoxidase aggravates non‐alcoholic steatohepatitis in low‐density lipoprotein receptor‐deficient mice. PLoS One 2012;7:e52411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Younossi ZM, Loomba R, Rinella ME, Bugianesi E, Marchesini G, Neuschwander‐Tetri BA, et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2018;68:361‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saito JM, Bostick MK, Campe CB, Xu J, Maher JJ. Infiltrating neutrophils in bile duct‐ligated livers do not promote hepatic fibrosis. Hepatol Res 2003;25:180‐191. [DOI] [PubMed] [Google Scholar]
- 29. Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL‐2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol 2014;60:782‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reiniers MJ, van Golen RF, van Gulik TM, Heger M. Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia‐reperfusion injury in the fatty liver. Antioxid Redox Signal 2014;21:1119‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haegens A, Heeringa P, van Suylen RJ, Steele C, Aratani Y, O'Donoghue RJ, et al. Myeloperoxidase deficiency attenuates lipopolysaccharide‐induced acute lung inflammation and subsequent cytokine and chemokine production. J Immunol 2009;182:7990‐7996. [DOI] [PubMed] [Google Scholar]
- 33. Klinke A, Nussbaum C, Kubala L, Friedrichs K, Rudolph TK, Rudolph V, et al. Myeloperoxidase attracts neutrophils by physical forces. Blood 2011;117:1350‐1358. [DOI] [PubMed] [Google Scholar]
- 34. European Association for the Study of the Liver , European Association for the Study of Diabetes , European Association for the Study of Obesity . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol 2016;64:1388‐1402. [DOI] [PubMed] [Google Scholar]
- 35. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013;58:1461‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP‐1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012;61:416‐426. [DOI] [PubMed] [Google Scholar]
- 37. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022‐1023. [DOI] [PubMed] [Google Scholar]
- 38. Gomez‐Zorita S, Aguirre L, Milton‐Laskibar I, Fernandez‐Quintela A, Trepiana J, Kajarabille N, et al. Relationship between changes in microbiota and liver steatosis induced by high‐fat feeding—a review of rodent models. Nutrients 2019;11:2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross‐talk between Akkermansia muciniphila and intestinal epithelium controls diet‐induced obesity. Proc Natl Acad Sci U S A 2013;110:9066‐9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med 2017;23:107‐113. [DOI] [PubMed] [Google Scholar]
- 41. Grander C, Adolph TE, Wieser V, Lowe P, Wrzosek L, Gyongyosi B, et al. Recovery of ethanol‐induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2018;67:891‐901. [DOI] [PubMed] [Google Scholar]
- 42. Rashid I, Maghzal GJ, Chen YC, Cheng D, Talib J, Newington D, et al. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high‐risk atherosclerotic plaque. Eur Heart J 2018;39:3301‐3310. [DOI] [PubMed] [Google Scholar]
- 43. Schults MA, Nagle PW, Rensen SS, Godschalk RW, Munnia A, Peluso M, et al. Decreased nucleotide excision repair in steatotic livers associates with myeloperoxidase‐immunoreactivity. Mutat Res 2012;736:75‐81. [DOI] [PubMed] [Google Scholar]
- 44. Wilson CL, Jurk D, Fullard N, Banks P, Page A, Luli S, et al. NFkappaB1 is a suppressor of neutrophil‐driven hepatocellular carcinoma. Nat Commun 2015;6:6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA 2001;286:2136‐2142. [DOI] [PubMed] [Google Scholar]
- 46. Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, et al. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med 2003;349:1595‐1604. [DOI] [PubMed] [Google Scholar]
- 47. Teng N, Maghzal GJ, Talib J, Rashid I, Lau AK. Stocker Rl. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep 2017;22:51‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material