

During infection and inflammation, the innate immune system uses antimicrobial compounds to control bacterial populations. These include toxic metals, like copper, and reactive oxidants, including hypochlorous acid (HOCl). We have now found that RclA, a copper(II) reductase strongly induced by HOCl in proinflammatory Escherichia coli and found in many bacteria inhabiting epithelial surfaces, is required for bacteria to resist killing by the combination of intracellular copper and HOCl and plays an important role in colonization of an animal host. This finding indicates that copper redox chemistry plays a critical and previously underappreciated role in bacterial interactions with the innate immune system.
KEYWORDS: copper, hypochlorous acid, oxidative stress, reactive chlorine
ABSTRACT
Enterobacteria, including Escherichia coli, bloom to high levels in the gut during inflammation and strongly contribute to the pathology of inflammatory bowel diseases. To survive in the inflamed gut, E. coli must tolerate high levels of antimicrobial compounds produced by the immune system, including toxic metals like copper and reactive chlorine oxidants such as hypochlorous acid (HOCl). Here, we show that extracellular copper is a potent detoxifier of HOCl and that the widely conserved bacterial HOCl resistance enzyme RclA, which catalyzes the reduction of copper(II) to copper(I), specifically protects E. coli against damage caused by the combination of HOCl and intracellular copper. E. coli lacking RclA was highly sensitive to HOCl when grown in the presence of copper and was defective in colonizing an animal host. Our results indicate that there is unexpected complexity in the interactions between antimicrobial toxins produced by innate immune cells and that bacterial copper status is a key determinant of HOCl resistance and suggest an important and previously unsuspected role for copper redox reactions during inflammation.
INTRODUCTION
Inflammatory bowel diseases (IBDs), such as Crohn’s disease and ulcerative colitis, are a growing health problem (1) and are associated with dramatic changes in the composition of the gut microbiome (2–6). Patients with IBDs have increased proportions of proteobacteria, especially Escherichia coli and other Enterobacteriaceae, in their gut microbiomes, which is thought to contribute to the progression of disease (7–10). The bloom of enterobacteria in the inflamed gut is driven by increased availability of respiratory terminal electron acceptors (e.g., oxygen, nitrate, and trimethylamine N-oxide) and carbon sources (e.g., ethanolamine and mucin), which E. coli and other facultative anaerobes can use to outcompete the obligate anaerobes (Bacteroides and Clostridia) that dominate a healthy gut microbiome (2, 3). In addition to these nutritional changes in the gut environment, inflammation also leads to infiltration of innate immune cells (e.g., neutrophils) into the lumen of the gut (11) and an associated increased production of antimicrobial compounds by the innate immune system (9, 10, 12), which also impact the bacterial community in the gut. These include antimicrobial peptides, toxic metals (e.g., copper), reactive oxygen species (ROS), reactive nitrogen species, and reactive chlorine species (RCS) (3, 13, 14). Since bacteria living in an inflamed gut are likely to be exposed to substantially increased levels of these toxins, the differential survival of proteobacteria during long-term inflammation suggests that E. coli may have evolved better mechanisms to resist these stresses than other types of commensal bacteria.
Macrophages use copper as an antimicrobial agent and copper levels rise in inflamed tissues, although the exact mechanism(s) by which copper kills bacteria are not yet fully understood (15–17). RCS are highly reactive oxidants produced by neutrophils and are potent antibacterial compounds (18, 19) that have been reported to play a role in controlling intestinal bacterial populations (20–22). E. coli does not efficiently survive being phagocytosed by neutrophils (23, 24), but the release of RCS-generating myeloperoxidase from neutrophils in the inflamed gut (11) means that E. coli is exposed to increased levels of RCS in that environment. The RCS response of E. coli is complex and incompletely understood (19, 25–28) but characteristically involves repair of damaged cellular components, often proteins (19). Protein-stabilizing chaperones are upregulated during RCS stress, including Hsp33 (29, 30) and inorganic polyphosphate (28, 31, 32), and enzymes are expressed that repair oxidized proteins, including periplasmic methionine sulfoxide reductase (MsrPQ) (33) and the chaperedoxin CnoX (34). E. coli has multiple HOCl-sensing regulators, including YedVW (which regulates MsrPQ) (33), NemR (a regulator of NemA and GloA, which detoxify reactive aldehydes) (25), HypT (which regulates cysteine, methionine, and iron metabolism) (27), and RclR, the RCS-specific activator of the rclABC operon (26).
The rclA gene of E. coli encodes a predicted cytoplasmic flavin-dependent oxidoreductase, is upregulated >100-fold in the presence of RCS (26, 35), and protects against killing by HOCl via a previously unknown mechanism (26). RclA is the most phylogenetically conserved protein of the Rcl system (Fig. 1) and is found almost exclusively in bacteria known to colonize epithelial surfaces (see Data Set S1 in the supplemental material), suggesting that it may play an important role in host-microbe interactions in many species. Bacteria encoding RclA homologs include Gram-negative species (e.g., Salmonella enterica), Gram-positive species (e.g., Streptococcus sanguinis), obligate anaerobes (e.g., Clostridium perfringens), facultative anaerobes (e.g., Staphylococcus aureus), pathogens (e.g., Enterococcus faecalis), commensals (e.g., Bacteroides thetaiotaomicron), and probiotics (e.g., Lactobacillus reuteri) (36), suggesting that RclA’s function may be broadly conserved and not specific to a single niche or type of host-microbe interaction. RclR, RclB, and RclC are much less widely conserved and are found only in certain species of proteobacteria, primarily members of the Enterobacteriaceae (Fig. 1; see Data Set S1 in the supplemental material). RclB and RclC both contribute to HOCl resistance, but their mechanistic roles have yet to be determined (26). RclB is predicted to be located in the periplasm, and RclC is predicted to be an integral inner membrane protein.
FIG 1.
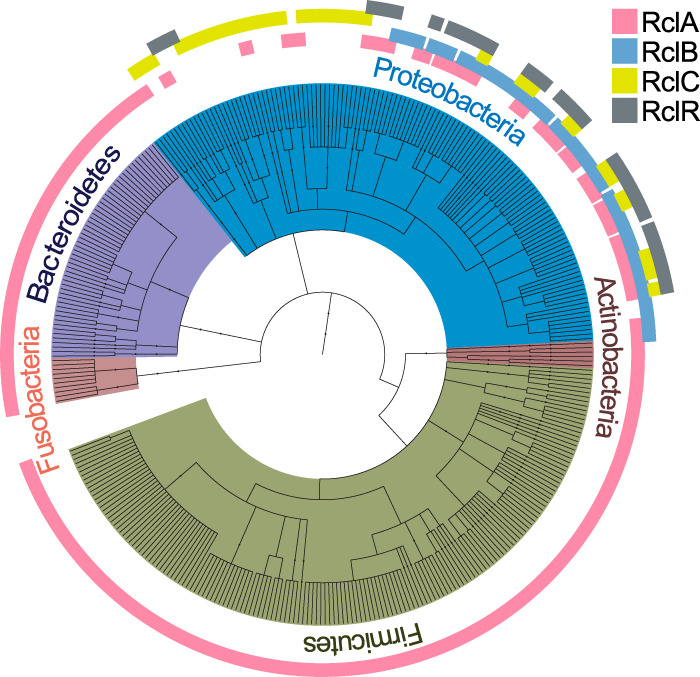
RclA is widely conserved among bacteria that colonize epithelial surfaces. A phylogenetic tree constructed from amino acid sequence alignments of RclA (pink, 284 species), RclB (blue, 61 species), RclC, (yellow, 49 species), and RclR (gray, 43 species), relative to the respective proteins in E. coli MG1655. Phyla are indicated by color: Actinobacteria (brown), Bacteroidetes (purple), Firmicutes (green), Fusobacteria (mauve), and Proteobacteria (blue). See Data Set S1 in the supplemental material for lists of each hit used in the phylogenetic tree. A tree graphic was made using the interactive tree of life (108).
Bacterial species containing homologs of the Rcl proteins of E. coli. Homologs of the RclA, RclB, RclC, and RclR proteins of E. coli included in Fig. 1, as identified by BLAST: RclA (BLAST E-value < 1 × 10−90 in 284 species), RclB (BLAST E-value < 1 × 10−1 in 61 species), RclC, (BLAST E-value < 1 × 10−80 in 49 species), and RclR (BLAST E-value < 1 × 10−40 in 43 species). Download Data Set S1, XLSX file, 0.03 MB (27.5KB, xlsx) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Here, we have now determined that RclA is a thermostable, HOCl-resistant copper(II) reductase that is required for efficient colonization of an animal host and protects E. coli specifically against the combination of HOCl and intracellular copper, possibly by preventing the formation of highly reactive Cu(III). We also found that, surprisingly, extracellular copper effectively protects bacteria against killing by HOCl both in cell culture and in an animal colonization model. These findings reveal a previously unappreciated interaction between two key inflammatory antimicrobial compounds and a novel way in which a commensal bacterium responds to and resists the combinatorial stress caused by copper and HOCl.
(This article was submitted to an online preprint archive [37].)
RESULTS
RclA contributes to HOCl resistance and host colonization.
An rclA mutant of E. coli is more susceptible to HOCl-mediated killing than is the wild type (26). To expand on these results, we utilized here a growth curve-based method to measure sensitivity to sublethal HOCl stress by quantifying changes in the lag-phase extension (LPE) of cultures grown in the presence of HOCl. Using this method, which we have found to be considerably more reproducible than other techniques for assessing bacterial HOCl sensitivity, we observed significant increases in LPE for a ΔrclA mutant strain compared to the wild type grown in the presence of various concentrations of HOCl (see Fig. S1A, B, and C in the supplemental material). We observed similar trends when the assay was performed with stationary- and log-phase cells (see Fig. S1D, E, and G), so stationary-phase cultures were used for subsequent assays. Furthermore, we determined that there was no decrease in CFU after treatment with HOCl at these concentrations, indicating that LPE measures the recovery of cultures from nonlethal stress (see Fig. S1F). These results confirm previous results and further illustrate the importance of RclA in resisting HOCl-mediated oxidative stress in E. coli.
Growth curve method to determine lag-phase extension in the presence of HOCl. Overnight cultures of (A) MG1655 wild-type and (B) MJG0046 (ΔrclA) were normalized to an A600 of 0.8 in MOPS minimal media and diluted to an A600 of 0.08 in MOPS ± HOCl at the indicated concentrations. Cultures were incubated with shaking at 37°C in a Tecan Infinite M1000 plate reader with the A600 being measured every hour for 24 h (n = 3 to 4, ± the SD). Sensitivity was assessed by comparing lag phase extensions (LPE; difference in hours to reach A600 ≥ 0.15 from no HOCl control; vertical dotted lines) for each HOCl concentration. An example LPE calculation for wild-type grown in 148 μM HOCl shown in panel A. (C) Average LPE values for growth curves shown in panels A and B and statistics comparing wild-type and ΔrclA strains at all conditions. Differences in average LPE values between the strains were analyzed using two-way ANOVA with Tukey’s multiple-comparison test. HOCl growth curves were performed as in panels A and B but on log-phase cultures of each strain. To prepare the log-phase cultures, saturated overnight cultures were subcultured in MOPS glucose and grown up at 37°C with shaking at 200 rpm until A600 = 0.5. E. coli lacking rclA (D) took longer to reach an A600 of 0.15 relative to the wild type (E) at all the tested HOCl concentrations. Time for each sample to reach an average A600 of 0.15 is indicated by vertical dashed lines. The observed growth delays of the strains in the presence of HOCl did not correspond with killing, as determined by viability spot plates (C, pictures representative of three biological replicates). Viability spot plates were prepared by collecting 5-μl aliquots from each well used for growth curves 10 min after adding MOPS ± HOCl to the OD-normalized cells. Serial dilutions in PBS of the aliquots were performed, and 5 μl of the dilutions was spotted on LB plates and grown overnight at 37°C. (G) Average LPE values for growth curves shown in D and E and statistics comparing wild-type and ΔrclA strains at all conditions. Differences in average LPE values between the strains were analyzed using two-way ANOVA with Tukey’s multiple-comparison test. Download FIG S1, TIF file, 2.2 MB (2.2MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To directly test the role of RclA in interactions with an animal host, we examined the ability of E. coli to colonize the intestine of Drosophila melanogaster, where the presence of enterobacteria is known to stimulate antimicrobial HOCl production by the dual oxidase Duox (20, 22). Since an E. coli K-12 strain did not efficiently colonize D. melanogaster (see Fig. S2A), we used the colonization-proficient E. coli strain Nissle 1917 (EcN) (38, 39) in these experiments. The genome of EcN encodes homologs of all the known HOCl resistance genes found in E. coli K-12 (40, 41). Unlike K-12, EcN forms robust biofilms (42–45), preventing the accurate measurement of growth curves and LPE (Fig. S2B), but EcN was slightly more sensitive to killing by lethal doses of HOCl than was MG1655 (Fig. S2C and D). EcN ΔrclA mutants had a significant defect in their ability to colonize NP1-GAL4 D. melanogaster flies compared to wild-type EcN at 3 and 8 h postinfection (hpi) (Fig. 2, circles). This shows that rclA is important for EcN tolerance of host responses during early colonization.
FIG 2.
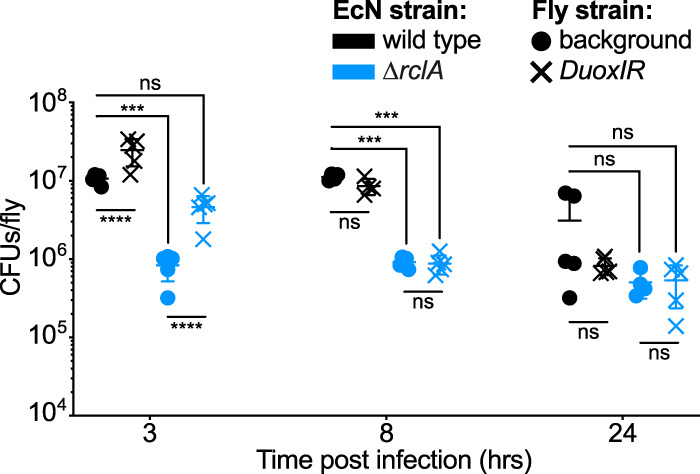
EcN lacking rclA colonizes D. melanogaster less effectively, and early colonization with ΔrclA EcN is improved in the absence of Duox-mediated oxidation. Flies were fed either wild-type or ΔrclA EcN (1 × 1011 CFU/ml), and bacterial loads were measured at the indicated times postinfection (n = 4 to 5, ± the SD). RCS-deficient Duox-RNAi flies were obtained from crosses of UAS-dDuox-RNAi with NP1-GAL4 (enterocyte-specific driver). Statistical analysis was performed using a two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test (****, P < 0.0001; ***, P < 0.001; ns, not significant).
Differences in fly colonization and HOCl sensitivity between E. coli K-12 and EcN. (A) EcN colonizes Canton-S D. melanogaster to a greater extent than MG1655. Flies were fed either E. coli K-12 MG1655 (K-12) or Nissle 1917 (EcN) and bacterial loads were measured (n = 5, ± the SD). Statistical analysis was performed using two-way ANOVA with Tukey’s multiple-comparison test (****, P < 0.0001; ns, not significant). (B) EcN growth curves with HOCl. EcN forms adherent growth in 96-well plates when grown with HOCl, which obstructs accurate measurement of absorbance over time. Growth curves performed as in Fig. S1A (n = 3 to 4, ± the SD). (C and D) EcN is more sensitive to HOCl-mediated killing than K-12. Wild-type E. coli MG1655 (K-12) (C) and Nissle 1917 (EcN) (D) were grown overnight in MOPS minimal media. A portion (500 μl) of the overnight culture was pelleted, resuspended in copper-free MOPS, and subcultured into 9.5 ml of copper-free MOPS. The cultures were then grown with shaking at 37°C to mid-log phase (A600 = 0.3 to 0.4). Once mid-log phase was reached, 1 ml of cells was aliquoted into microcentrifuge tubes for each treatment. Cells were treated by adding the indicated amounts HOCl and then incubated on a 37°C heat block for 10 min. After incubation, treatments were serially diluted in PBS, and 5-μl aliquots were spotted on LB agar plates. Plates were dried and incubated at room temperature for 2 days before imaging. Download FIG S2, TIF file, 1.3 MB (1.3MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To investigate the role of host-produced RCS in the colonization defect of the rclA EcN mutant, we reduced the gut-specific expression of Duox in the flies using Duox-RNAi and repeated the colonization experiments. Both strains colonized significantly better at 3 hpi when Duox was knocked down in the flies (Fig. 2, “×” symbols), which was expected because rclA is not the only gene that contributes to HOCl resistance in E. coli (19). Importantly, the colonization defect of ΔrclA EcN at 3 hpi was abrogated in the DuoxIR flies, with CFU/fly not being significantly different from wild-type EcN colonizing flies that are able to express Duox in the gut (Fig. 2). We confirmed that HOCl production was reduced in the DuoxIR flies using the HOCl-sensing fluorescent probe R19-S (see Fig. S3A to C) (46). Taken together, these results show that rclA facilitates early colonization of an animal host and indicate that rclA relieves stress caused by host-produced oxidation in early stages of colonization. Duox activation and HOCl production are rapid host immune responses that occur at early stages (minutes to first few hours) of bacterial colonization of the gut (20). However, ROS and RCS production are not the only antimicrobial responses in Drosophila, which may explain why rclA is only required in early colonization. As the course of infection progresses, additional antimicrobial effectors, such as antimicrobial peptides regulated by NF-κB signaling, become more abundant (27). EcN infection induced robust production of antimicrobial peptide production in our fly model (Fig. S3D, E, and F).
Fly response to EcN colonization. (A) Host-produced HOCl production is silenced by Duox RNAi. Prior to treatment, flies were starved for 2 h at 29°C. (A) As an uninfected control, NP1-GAL4 female flies fed on a mixture of LB, and 100 μM R19-S was prepared in 5% sucrose. RCS production in response to EcN infection was determined by feeding a mixture of EcN (5 × 107 cells) and 100 μM R19-S to NP1-GAL4 female flies (B) and NP1-GAL4-UAS-DuoxIR female flies (C). After 30 min of feeding on the designated mixture, all treatments were switched to a sucrose/R19-S mixture. At 90 min after the initial infection, guts were dissected in PBS and fixed in 4% formaldehyde for 50 min. Guts were mounted in Vectashield/DAPI and visualized on a Leica SP8 confocal microscope with a red emission range of 530 to 603 nm. Images presented are representative of a total of three gut tissues imaged per treatment. (D to F) The antimicrobial peptide Diptericin (Dpt) is expressed in response to oral infection with EcN in DD;NP1 flies. Adult female flies were fed EcN (E) or a sucrose (D) control for 6 h, and gut tissues were dissected and stained with X-Gal to reveal regions of Dpt expression, method described previously (C. Neyen, A. J. Bretscher, O. Binggeli, and B. Lemaitre, Methods 68:116–128, 2014, https://doi.org/10.1016/j.ymeth.2014.02.023). (F) β-Galactosidase activity of adult female gut tissues at various time points after oral infection with EcN. The relative β-galactosidase activity is expressed as a percentage of baseline activity observed in flies fed a sterile sucrose solution. Download FIG S3, TIF file, 2.7 MB (2.7MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RclA is homologous to mercuric reductase and copper response genes are upregulated after HOCl stress in EcN.
Although the fact that rclA protects E. coli from RCS was previously known (26), the mechanism by which it does so was not. Based on its homology to other flavin-dependent disulfide oxidoreductases (47), we hypothesized that RclA catalyzed the reduction of an unknown cellular component oxidized by RCS. RclA is homologous to mercuric reductase (MerA), an enzyme that reduces Hg(II) to Hg(0) (48). These sequences are particularly well conserved at the known active site of MerA, a CXXXXC motif (Fig. 3A). However, MerA has an extra N-terminal domain and two additional conserved cysteine pairs used in metal binding (48), while RclA only has one conserved cysteine pair (see Fig. S4). This indicates that if RclA does interact with metal(s), the interactions must be mediated through mechanisms different from those of MerA. While the present manuscript was in revision, Baek et al. published a crystal structure of RclA (49), and alignment of this structure with that of MerA (50) clearly illustrates the homology between these two proteins as well as the location of the MerA-specific domains (Fig. 3B).
FIG 3.

RclA and mercuric reductase (MerA) share a conserved active site and are structurally homologous. (A) Active site alignment of E. coli RclA and MerA amino acid sequences from seven bacterial species (Escherichia coli, Staphylococcus aureus, Salmonella enterica, Listeria monocytogenes, Klebsiella pneumoniae, and Serratia marcescens). Alignment was made using CLUSTAL O (1.2.4), and a graphic was made using WEBLOGO. (B) Structural alignment of MerA (orange) and RclA (cyan) with active sites in purple and nonconserved regions in gray. Polymers represented as ribbons, and ligands (FAD, SO4, glycerol, and Cl–) are represented in ball-and-stick form. Comparison and graphics made using the “sequence and structure alignment” tool (jFATCAT_rigid algorithm with input sequences 1ZK7.A and 6KGY. (B) Provided by the RCSB (109).
Alignment of full-length RclA with MerA sequences from representative bacterial species. Alignment of E. coli RclA (ADC80840.1) and MerA amino acid sequences from seven species (Escherichia coli, ADC80840.1; Staphylococcus aureus, AKA87329.1; Salmonella enterica, ABQ57371.1; Listeria monocytogenes, PDA94520.1; Klebsiella pneumoniae, ABY75610.1; Serratia marcescens, ADM52740.1). Alignment, conservation scoring, and graphic were made using PRALINE. Download FIG S4, TIF file, 2.6 MB (2.7MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
HOCl-stressed E. coli K-12 downregulates genes encoding iron import systems (e.g., fepABCD and fhuACDF) and upregulates genes for zinc and copper resistance (e.g., copA, cueO, cusC, zntA, and zupT) (25), also suggesting metals may play some role in RCS resistance. The genome of EcN encodes the same complement of known copper resistance genes as is found in E. coli K-12 (40, 41). Transcriptomic profiling of EcN after treatment with a sublethal dose of HOCl confirmed the regulation of metal stress response genes by HOCl, including the upregulation of several genes encoding proteins involved in response to copper toxicity (Fig. 4; see also Data Set S2 in the supplemental material), despite the very small amounts of copper present in the media used in that experiment (9 nM) (51). These included members of the Cus and Cue export systems, which are factors appreciated for their role in preventing copper toxicity and importance for E. coli colonization within mammalian hosts (52–55). The expression of the rcl operon is not regulated by any of the known Cu-sensing transcription factors of E. coli (56) and is not affected by changes in media copper concentrations (57, 58). Our results suggested that copper might play an important role during HOCl stress for EcN. The homology between RclA and MerA and the indication that copper and HOCl responses may be connected in E. coli led us to investigate the role of rclA in resisting HOCl stress under growth conditions containing different amounts of copper.
FIG 4.
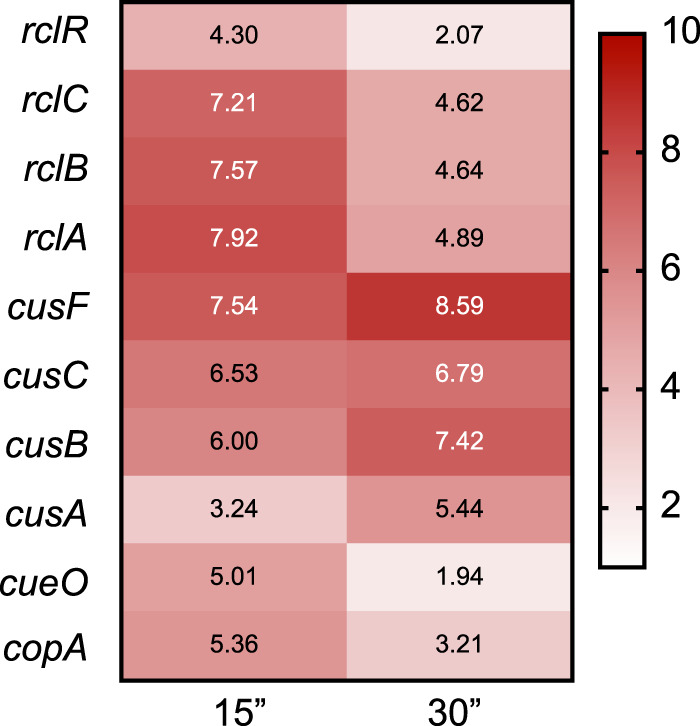
EcN upregulates copper export proteins during HOCl stress. Log2-fold change in EcN gene expression at 15 and 30 min after nonlethal HOCl treatment (0.4 mM), relative to a control sample taken directly before treatment. mRNA counts were measured using RNA sequencing and analyzed with Bowtie2.
Results of RNA sequencing of HOCl-treated EcN. RPKM and log2-fold change values for all genes in E. coli Nissle before and 15 and 30 min after treatment with 0.4 mM HOCl. Raw data are deposited under GEO series accession number GSE144068. Download Data Set S2, XLSX file, 1.4 MB (1.4MB, xlsx) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Extracellular CuCl2 protects both wild-type and ΔrclA E. coli strains against HOCl.
How the presence of copper influences bacterial sensitivity to RCS has not been investigated before this study. However, it is important to note that Cu chemically catalyzes the decomposition of HOCl to nontoxic O2 and Cl– (59–62). We first used growth curves in the presence of copper and HOCl to identify how combinations of HOCl and extracellular copper influenced the sensitivity of wild-type and ΔrclA mutant E. coli (Fig. 5A and B). As noted above, the ΔrclA mutant was more sensitive to HOCl in MOPS, but the sensitivity to HOCl of both strains was greatly increased when Cu was removed from the media, indicating that the low concentration of Cu present in morpholinepropanesulfonic acid (MOPS) medium (9 nM) (51) was enough to react with the added HOCl and change the sensitivity of our strains. Consistent with this, addition of 10 μM CuCl2 to HOCl-containing media greatly decreased sensitivity of both the wild type and the rclA-null mutant to sublethal HOCl stress (Fig. 5A and B; see also Fig. S5A, B, and C in the supplemental material). That copper is uniformly protective for both strains makes it likely that extracellular copper had reacted with and detoxified the HOCl before cells were inoculated into the media. To confirm this result, we tested whether the addition of exogenous copper (0.5 mM CuCl2) could protect E. coli against killing by a very high concentration of HOCl (1 mM). We found that treatment with 1 mM HOCl resulted in complete killing of both strains (Fig. 5C). Both strains survived several orders of magnitude better when 0.5 mM CuCl2 was added to the media immediately before HOCl stress (Fig. 5C). In addition, colonization of flies by EcN was enhanced when their diet was supplemented with copper (see Fig. S5D), showing that copper can influence the microbiome in vivo, and consistent with the model that copper detoxifies HOCl in the gut. Taken together, these results illustrate that the presence of exogenous copper strongly protects E. coli against HOCl.
FIG 5.
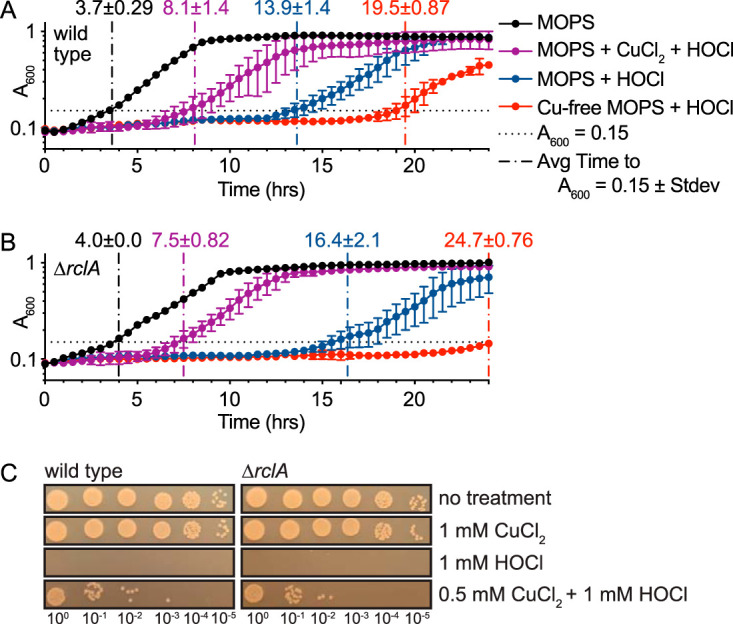
Extracellular CuCl2 protects both wild-type and ΔrclA E. coli strains against HOCl. Growth curves of E. coli K-12 wild-type (A) and ΔrclA (B) strains in MOPS with the addition of various combinations of HOCl (132 μM) and CuCl2 (10 μM) (n = 3, ± the SD for each treatment). Time (means ± the SD) for each sample to reach an A600 of 0.15 is indicated by vertical dashed lines. Only one of several HOCl concentrations tested is shown for simplicity (see Fig. S5A to C in the supplemental material for lag-phase extension calculations, a summary of all the conditions tested using the growth curve method, and statistics comparing LPE values shown in this figure). (C) Exogenous copper protects both wild-type and ΔrclA E. coli strains from lethal HOCl stress (10 min at 1 mM). Cells were treated as described in Fig. S2C and D.
Extracellular CuCl2 reduces LPE for both wild-type and ΔrclA E. coli grown in the presence of HOCl. (A) Average LPE values for growth curves shown in Fig. 5 and statistics comparing wild-type and ΔrclA at all conditions. Differences in average LPE values between the strains were analyzed using two-way ANOVA with Tukey’s multiple-comparison test. LPE determination for wild-type (B) and ΔrclA (C) strains grown in the presence of the indicated HOCl and extracellular CuCl2 conditions. Growth curves were performed as described in Fig. S1 but with supplementing the MOPS glucose with the indicated CuCl2 concentrations. HOCl and CuCl2 were mixed in MOPS minimal media prior to diluting cells A600-normalized to 0.08 into each media type. LPEs were calculated by determining the difference in time (h) to reach A600 > 0.15 for each condition relative to no stress controls. Sensitivity was assessed within strains by comparing LPEs for each HOCl/CuCl2 concentration (n = 4, ± the SD) at each HOCl concentration (two-way ANOVA for each strain with Dunnett’s multiple comparison using the no CuCl2 control for each HOCl concentration, respectively (****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, not significant). (D) EcN colonization is improved when flies are fed copper. D. melanogaster Canton-S females fed for 24 h on a mixture of sucrose and 1 mM CuCl2 or sterile water (as a control), which was applied to a filter disk placed on the fly food surface. Flies were then transferred to new food tubes containing sucrose and either EcN wild-type (WT) or EcN ΔrclA. Individuals were collected, surface sterilized, homogenized, and plated on LB agar at various time points to determine bacterial load (CFUs/fly). Statistical analysis was performed with GraphPad Prism using a multiple t test with Sidak-Bonferroni correction for multiple comparisons (n = 60). Download FIG S5, TIF file, 1.3 MB (1.3MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RclA protects E. coli against the combination of HOCl and intracellular copper.
Next, we sought to investigate how intracellular copper affects the HOCl resistance of E. coli. To address this, we grew wild-type and ΔrclA mutant E. coli strains overnight in minimal media with or without copper before inoculating the strains into copper-free media to perform HOCl-stress growth curves. Growing overnight cultures in media lacking copper was expected to starve the cells for this metal, thereby reducing the concentration of intracellular copper in those cultures. A broad range of HOCl concentrations were assayed to account for quenching of the oxidant by media components.
Consistent with the results shown in Fig. 5, E. coli was more sensitive to inhibition by HOCl in media without copper. The ΔrclA mutant was more sensitive to HOCl than the wild type when grown overnight in copper-containing media, but this phenotype was lost when cells were starved for copper before stress (Fig. 6). The wild-type strain was also slightly more sensitive to some concentrations of HOCl in the presence of intracellular copper (see Fig. S6B and C), but this difference was much more subtle than in the ΔrclA mutant. These results suggest that the physiological role of RclA is to resist the stress resulting from the combination of HOCl and copper in the cytoplasm. They are not consistent, however, with a model where RclA uses Cu to detoxify RCS or other oxidants in the cytoplasm (49), since in that case we would expect the sensitivity of the wild type to decrease to match that of the ΔrclA mutant in the absence of Cu, the opposite of what we actually observed (Fig. 6B).
FIG 6.
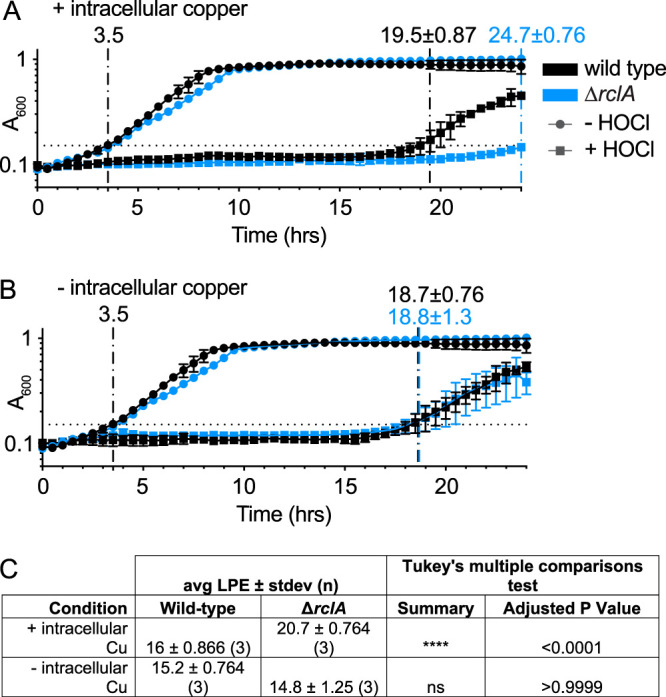
rclA is required to resist the combination of HOCl and intracellular Cu. HOCl (132 μM) growth curves (n = 3, ± the SD) in copper-free MOPS of wild-type and ΔrclA E. coli after being grown overnight in MOPS with (A) or without (B) copper. The time for each sample to reach an average A600 of 0.15 is indicated by vertical dashed lines. Copper-free MOPS was prepared by treating the media with Chelex-100 chelating resin (Bio-Rad 1421253) and adding back all the metals (metal stock solutions prepared in metal-free water, Optima LC/MS Grade, Fisher Chemical W6-1), except for copper, to the published concentrations (51). In cells containing intracellular copper (A), the ΔrclA mutant has delayed growth relative to the wild type; there is no difference between the strains when the cells were starved for intracellular copper (B). One HOCl concentration is shown for simplicity (see Fig. S6 in the supplemental material for growth curves showing more HOCl concentrations and the statistical analysis comparing LPE values between the strains at each condition). (C) Average LPE values for growth curves shown in panels A and B and statistics comparing wild-type and ΔrclA strains under all conditions. Differences in average LPE values between the strains were analyzed using two-way ANOVA with Tukey’s multiple-comparison test.
Comparing LPEs of strains stressed with or without intracellular Cu. (A) Average LPE values for growth curves shown in Fig. 6 and in panels B to E. Differences in average LPE values between the strains were analyzed using a two-way ANOVA with Tukey’s multiple-comparison test. Wild type (B and C) and ΔrclA (C and D) E. coli were grown up overnight in MOPS glucose with (D and E) or without copper (A and C). The time for each sample to reach an average A600 of 0.15 is indicated by vertical dashed lines. HOCl growth curves were performed as described in Fig. 6. Download FIG S6, TIF file, 1.6 MB (1.6MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RclA reduces copper(II) to copper(I).
Based on the effect of copper starvation on the HOCl sensitivity of the ΔrclA mutant, the sequence homology between RclA and MerA, and the predicted oxidoreductase activity of RclA (47), we hypothesized that the substrate of RclA might be copper. The reaction between copper and HOCl is known to generate strong oxidizing intermediates, most likely highly reactive Cu(III) (59–63). HOCl is also capable of oxidizing other transition metals, including iron (64, 65) and manganese (66, 67). We therefore measured the specific activity (SA) of purified RclA in the presence of a panel of biologically relevant metals. We also included mercury in the panel of metals because of RclA’s homology to MerA, although it is unlikely to be physiologically relevant since we do not expect E. coli to encounter this metal in its environment under normal conditions. We note that the oxidized forms of many transition metals are insoluble in aqueous solution, which limited the set of substrates we could test with this experiment.
In the absence of any metal, RclA slowly oxidized NADH (0.0303 μmole NAD+ min−1 mg−1 RclA), consistent with the background activity of other flavin-dependent oxidoreductases in the absence of their specific substrates (68, 69). Three of the metals we tested significantly affected RclA SA, as measured by NADH oxidation. Copper and mercury both significantly increased the SA of RclA, whereas zinc caused a decrease in SA (Fig. 7A). As mentioned above, while the present manuscript was in revision, Baek et al. (49) reported crystal structures of E. coli RclA in the presence or absence of bound copper, along with testing a similar panel of metals as the substrates. The results from that study are consistent with ours, supporting our identification of RclA as Cu(II) reductase. Copper is a potent inhibitor of MerA activity (70), further emphasizing the distinct nature of these two enzymes. RclA oxidized NADPH at similar rates to NADH in the absence of metals, but there was no significant increase to SA when copper was added to the reactions. The addition of exogenous thiols, commonly added as β-mercaptoethanol (BME), is required for MerA activity (71). To determine whether exogenous thiols increase the reaction rate of RclA, we added 1 mM BME to the RclA reactions. BME rapidly reduced Cu(II) to Cu(I) in the absence of RclA but had no effect on the SA of NADH reduction by RclA with or without the addition of copper (see Fig. S7A and B).
FIG 7.
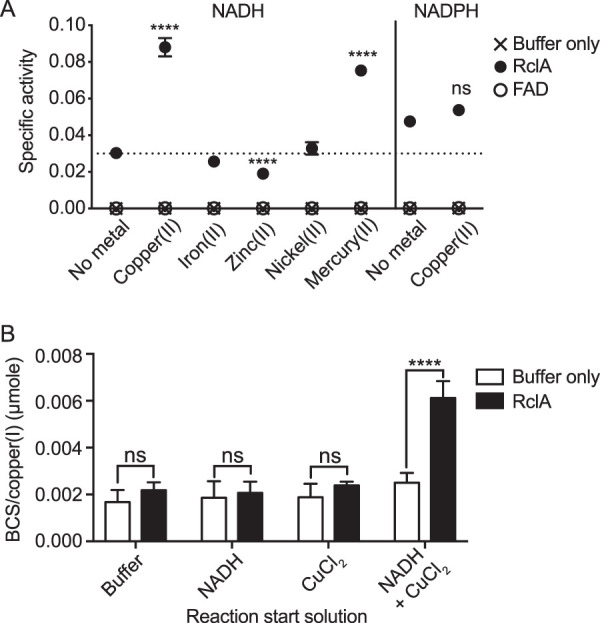
RclA reduces Cu(II) to Cu(I). (A) RclA specific activity (SA) increases in the presence of Cu(II) and Hg(II). SA [μmol of NAD(P)+ min−1 mg−1 RclA] of RclA was assayed by measuring NAD(P)H oxidation over time spectrophotometrically (n = 6 for each reaction type, ± the SD). Reactions were started by adding 100 μl of NAD(P)H or NADH and each indicated metal (both to 200 μM final) to 100 μl of RclA (3 μM final) at 37°C using the injector system of a Tecan Infinite M1000 plate reader. All reactions were carried out in 20 mM HEPES–100 mM NaCl (pH 7). NADH absorbance at 340 nm was measured each minute for 5 min. “Buffer only” denotes NAD(P)H oxidation in the presence of the indicated metals, and the “FAD” reactions were performed with 3 μM FAD to control for any possible free cofactor that may contribute to metal reduction in the RclA positive reactions. Differences in SA in the presence of each metal were analyzed using a two-way ANOVA with Dunnett’s multiple-comparison test using the no metal reaction as the control (****, P < 0.0001; **, P < 0.01). (B) Cu(I) accumulates after the RclA and NADH/copper (II) reaction, as measured by BCS/Cu(I)-complex absorption. Each reaction described in panel A was then stopped at 5 min with 10 μl of a BCS (400 μM final) and EDTA (1 mM final) solution using the injector system of the plate reader. The stopped reaction mixtures were incubated at 37°C for 5 min, with the absorbance of BCS/Cu(I) complex being measured at 483 nm each minute to ensure saturation of BCS. Differences in the amount of BCS/Cu(I) complex between the buffer-only and RclA reactions were analyzed using a two-way ANOVA with Dunnett’s multiple-comparison test using the buffer-only sample as the control for each reaction start solution (****, P < 0.0001; ns, not significant).
Control experiments for RclA activity. (A) Adding BME to the start solutions of RclA reactions does not increase SA and causes Cu(I) accumulation. (A) RclA reactions performed as in Fig. 7 but with the addition of 1 mM BME for indicated samples (n = 6, ± the SD). SA (A) and BCS/Cu(I) (B) concentration determined as in Fig. 7. Differences in SA in the presence of each metal were analyzed using a two-way ANOVA with Dunnett’s multiple-comparison test using the buffer-only sample as the control for each start solution (****, P < 0.0001). (C and D) Changes in NADH and copper (I) concentrations over time during the RclA reaction. NADH oxidation (C) and BCS/Cu(I) complex formation (D) were measured over time spectrophotometrically with and without RclA. Reactions were started by adding 100 μl of NADH (200 μM final) or NADH and CuCl2 (both 200 μM final) to 100 μl of RclA (3 μM final) at 37°C using the injector system of a Tecan Infinite M1000 plate reader. All reactions were carried out in 20 mM HEPES–100 mM NaCl (pH 7). NADH absorbance at 340 nm was measured each minute for the indicated reaction times. Each reaction was then stopped at the indicated times with 10 μl of a BCS (400 μM final) and EDTA (1 mM final) solution using the injector system of the plate reader (n = 6 for each reaction type, ± the SD). The stopped reaction mixtures were incubated at 37°C for 5 min, with the absorbance of BCS/Cu(I) complex being measured at 483 nm each minute to ensure saturation of BCS. Reported BCS/Cu(I) values were calculated using the average of the total absorbance measurements over the 5-min incubation because the values were stable over that time. Differences in the amount of NADH or BCS/Cu(I) complex between the buffer-only and RclA reactions were analyzed using two-way ANOVA with Sidak’s multiple-comparison test (****, P < 0.0001; ***, P < 0.001; *, P < 0.05; ns, not significant). (E) The copper reductase activity of RclA is maintained under anaerobic conditions. BCS/Cu(I) complex concentrations were determined as in Fig. 7, but all reactions were performed in an anaerobic chamber (n = 6, ± the SD). Differences in the amount of BCS/Cu(I) complex between the buffer-only and RclA (3 μM) reactions were analyzed using a two-way ANOVA with Tukey’s multiple-comparison test (****, P < 0.0001; ns, not significant). (F) The melting temperature (Tm) of RclA is ∼10°C higher than the average Tm of the E. coli proteome. The CD spectra of RclA were measured at the indicated temperatures; 222 nm is indicated by a vertical dashed line. Download FIG S7, TIF file, 1.6 MB (1.6MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Since RclA is an NADH oxidase, the results shown in Fig. 7A strongly suggested that this enzyme was concurrently reducing copper. Copper exists in four possible oxidation states, Cu(I), Cu(II), and the less common and highly reactive Cu(III) and Cu(IV) states (72). The copper salt used in our RclA SA determinations was CuCl2, suggesting that RclA was reducing this Cu(II) species to Cu(I). This was initially surprising to us, since Cu(I) is often thought of as a toxic species that causes oxidative stress (54, 73). We therefore first sought to validate that RclA was in fact reducing Cu(II) to Cu(I) while oxidizing NADH to NAD+. We measured Cu(I) accumulation in RclA reactions directly using the Cu(I)-specific chelator bathocuproinedisulfonic acid (BCS) (74). NADH spontaneously reduces Cu(II) (75) at rates too slow to impact the measurements made here, but BCS increases the rate of this nonenzymatic copper reduction by shifting the equilibrium of the reaction toward Cu(I) (76). Stopping RclA reactions with a mixture of BCS and EDTA, to chelate any remaining Cu(II), allowed us to observe RclA-dependent Cu(I) accumulation (see Fig. S7C and D). We observed a significant increase in BCS/Cu(I) complex formation only in reaction mixtures containing RclA, NADH, and Cu(II) and not in reaction mixtures lacking any single component (Fig. 7B; see also Fig. S7C and D in the supplemental material). Furthermore, we validated that the copper reductase activity of RclA was maintained when the reactions were performed in an anaerobic chamber (see Fig. S7E) and that FAD alone did not catalyze Cu(II) reduction (see Fig. S7A and B). Taken together, our results show that RclA has Cu(II) reductase activity and directly demonstrate that RclA generates Cu(I) as a product. This is consistent with the findings of Baek et al. (49), who also used site-directed mutagenesis to clarify the roles of the active site cysteines of RclA in Cu(II) reductase activity and binding sensitivity.
RclA is thermostable and resistant to denaturation by HOCl and urea.
We hypothesized that the copper reductase activity of RclA was likely to be relatively stable under denaturing conditions because it must remain active during exposure to HOCl stress, which is known to cause extensive protein misfolding and aggregation in vivo (19, 27, 28, 32–34). To test this hypothesis, we first measured RclA activity after treatment with protein denaturing agents (HOCl and urea) in vitro. HOCl treatment (with 0-, 5-, 10-, and 20-fold molar ratios of HOCl to RclA) was performed on ice for 30 min, and urea treatment (0, 2, 4, and 6 M) was carried out at room temperature for 24 h. RclA retained full copper reductase activity at all HOCl levels tested, indicating that it is highly resistant to treatment with HOCl (Fig. 8A). By comparison, the NADH oxidase activity of lactate dehydrogenase was significantly decreased after treatment with a 5-fold excess of HOCl (Fig. 8B). RclA also retained a remarkable 35.8% of full activity after being equilibrated in 6 M urea (Fig. 8C). Finally, we used circular dichroism (CD) spectroscopy to measure the melting temperature (Tm) of RclA, which was 65°C (Fig. 8D; see also Fig. S7F), indicating that RclA is thermostable relative to the rest of the E. coli proteome, which has an average Tm of 55°C (standard deviation [SD] = 5.4°C) (60, 77).
FIG 8.
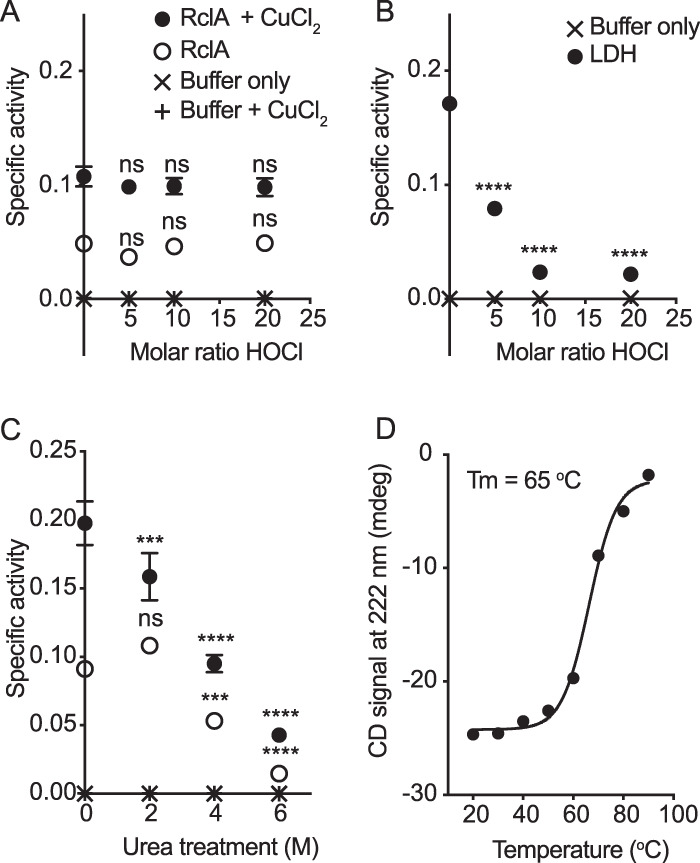
RclA is resistant to denaturation. (A) SA of RclA (μmol NAD+ min−1 mg RclA−1), with or without CuCl2, after being treated with the indicated molar ratios of HOCl to RclA. (B) SA of lactate dehydrogenase (LDH) (μmol of NAD+ min−1 U LDH−1) used as control reactions for HOCl degradation of enzymatic activity. (C) RclA with or without CuCl2 (200 μM final) after being treated with the indicated concentrations of urea. (D) CD signals at 222 nm (mdeg) (raw data are shown in Fig. S7F in the supplemental material) at each temperature used to determine the Tm of RclA (65°C). Differences in SA (n = 6, ± the SD) after treatment were analyzed using two-way ANOVA with Sidak’s multiple-comparison test for HOCl treatment (A and B) and Dunnett’s test using the buffer-only reaction as the control for the urea-treated samples (C) (****, P < 0.0001; *, P < 0.05; ns, not significant).
RclA influences copper homeostasis, but HOCl stress does not lead to copper export in wild-type E. coli.
Since the copper exporters of E. coli (copA and cusCFBA) are upregulated by HOCl treatment (Fig. 4; see also Data Set S2 in the supplemental material) (25) and only transport Cu(I) (54, 55, 78, 79), one possible model for how RclA protects against HOCl is that RclA might facilitate the rapid export of cytoplasmic copper, allowing it to react with and eliminate HOCl outside the cell. To test whether the Cu(II) reductase activity of RclA is important for exporting copper during HOCl stress, we measured intracellular copper concentrations in E. coli MG1655 before and after HOCl stress with ICP mass spectrometry (see Fig. S8A). The copper content of the wild type did not change upon HOCl stress, indicating that copper export is not dramatically upregulated under these conditions. We did find that the ΔrclA mutant contained, on average, more intracellular copper before HOCl stress than did the wild type but that both strains contained similar amounts of copper after HOCl stress. This suggested that RclA has a role in copper homeostasis under nonstress conditions but that any copper export stimulated by HOCl was RclA independent and, in fact, only occurred in the absence of RclA. To attempt to further probe the effect of intracellular copper on HOCl survival, we constructed mutants lacking copA, which is reported to result in increased intracellular copper (55), but unexpectedly found that the copA rclA double mutant had a substantial growth defect in copper-free media, even in the absence of HOCl (see Fig. S8B and C). We do not yet know the explanation for this intriguing result, since there are no known essential copper-containing proteins in E. coli (80), although perhaps the most likely candidate under our growth conditions is the Cu-containing cytochrome bo3 ubiquinol oxidase CyoB, which is involved in aerobic respiration at high O2 concentrations (81, 82). Our results suggest that there is considerable complexity in the interactions between Cu homeostasis and RclA under different growth conditions, and future work in our laboratory is focused on exploring these interactions in more detail. However, our current results clearly indicate that Cu is important to understanding bacterial HOCl sensitivity and that the Cu(II) reductase RclA is involved in modulating that process.
Measuring intracellular copper concentrations during HOCl stress and growth defect of a rclA copA double mutant. (A) Intracellular copper concentrations do not change after HOCl stress in wild-type E. coli. E. coli MG1655 wild-type (wt) and ΔrclA were grown overnight in MOPS. The next day, both strains were subcultured into MOPS, grown at 37°C with shaking to A600 = 0.6, and stressed with 400 μM HOCl for 30 min at 37°C with shaking. Intracellular copper before and after HOCl stress of the strains was determined using ICP-MS. Differences between intracellular copper analyzed using two-way ANOVA with Tukey’s multiple-comparison test (*, P < 0.05; ns, not significant). (B) A double mutant of rclA and copA has a growth defect when grown with (B) or without copper (C). HOCl growth curves on cells with or without intracellular copper were performed as described in Fig. 6 with copper-free MOPS glucose (n = 3). Download FIG S8, TIF file, 0.7 MB (777.3KB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
The antimicrobial function of copper in host-microbe interactions is well established (15, 17, 52, 83), although the exact mechanism(s) by which copper kills bacteria remain incompletely known (84, 85). In the present study, we identified a new way in which copper toxicity contributes to host-bacterium interactions via its reactions with RCS. We identified RclA as a highly stable Cu(II) reductase (Fig. 7). This is consistent with the simultaneous report by Baek et al. (49), who also found that both Cu(II) and Hg(II) increase the rate of NADH oxidation by RclA. Importantly, we have now shown that RclA is required in vivo for resisting killing by the combination of HOCl and intracellular copper in E. coli (Fig. 6). In the absence of rclA, E. coli had a significant defect in initial colonization that was partially eliminated when production of HOCl by the host was reduced (Fig. 2). The amount of copper in bacterial cells is low (15, 16), but how much is unbound by protein and its redox state under different conditions are unknown (15). Given the broad conservation of RclA among host-associated microbes, we propose that there is likely to be a common and previously unsuspected role for copper redox reactions in interactions between bacteria and the innate immune system.
Copper accumulates in host tissues during inflammation (86, 87), as do RCS (88, 89). Our discovery that even very low concentrations of extracellular copper can protect bacteria against RCS both in vitro and in vivo adds a new and important facet to understanding copper’s role in innate immunity. Since a large proportion of host tissue damage during inflammation is due to HOCl (90, 91), the presence of copper in inflamed tissues may play an important role not only in killing bacteria but potentially also in protecting host cells, although this hypothesis will require further testing. Our results also show that media copper concentrations are a key variable in experiments testing the sensitivity of cells to HOCl and that care must be taken to account for media copper content and use metal-free culture vessels in such experiments.
Both HOCl and copper can cause oxidative stress in bacteria and Cu(I) is generally considered more toxic than Cu(II) (15, 19, 52, 83, 84, 92), so we were initially surprised that a Cu(II) reductase protected E. coli against HOCl. Copper reacts with the ROS hydrogen peroxide (H2O2) to form highly reactive hydroxyl radicals in vitro (17, 53, 73, 85), but there is also strong evidence that oxidation is not the major cause of Cu toxicity in E. coli (85, 93). CusRS and CueR are exceptionally sensitive to changes in Cu concentrations in the periplasm and cytoplasm, respectively (94, 95). CueR, for example, has zeptomolar Cu binding affinity (95). The upregulation of the CusRS and CueR regulons under HOCl stress (Fig. 4) indicates that free Cu is increasing in both the cytoplasm and the periplasm, which could plausibly result from the oxidation of cysteine and histidine residues in Cu-binding proteins by HOCl (96, 97). Redox proteomics of RCS-stressed E. coli (98, 99) have not identified oxidized Cu-binding proteins, but the methods used in those studies to date are limited to detection of the most common proteins in the cell. Detection of oxidation in less abundant proteins will require more specialized methods (30).
How the presence of copper influences bacterial sensitivity to RCS has not been investigated before this study, but the chemistry of reactions between HOCl and copper is complicated and different from that of reactions between ROS and Cu. HOCl can oxidize Cu(II) to highly reactive Cu(III) (59–63), and both Cu(I) and Cu(II) are known to catalyze the breakdown of HOCl (59–62). At nearly neutral pH, similar to that in the large intestine or bacterial cytoplasm, Cu(I) accelerates the rate of decomposition of HOCl to O2 and chloride ions by as much as 108-fold (60). One possibility to explain the protective effect of RclA is that it might facilitate an HOCl-degrading Cu(I)/Cu(II) redox cycle in the cytoplasm. Alternatively, Baek et al. (49) note the increased O2 consumption by RclA in the presence of Cu(II), and propose that RclA protects against oxidative stress by lowering O2 levels. If either of these were the case, however, RclA would require copper to drive HOCl resistance and the ΔrclA mutant would become more sensitive to HOCl in the absence of copper, the opposite of what we actually observed (Fig. 6). We therefore propose that RclA-catalyzed reduction of Cu(II) to Cu(I) may act to limit the production of Cu(III) in the cytoplasm (Fig. 9). Uncontrolled production of Cu(III) could greatly potentiate the ability of HOCl to kill bacterial cells Alternatively, RclA may also ensure that intracellular copper remains in the Cu(I) state, where it can be bound by Cu chaperones like CopA(Z) (100). All of the known proteins involved in Cu homeostasis and export in the E. coli cytoplasm are specific to Cu(I) (52, 54, 55). Although our data (see Fig. S8A in the supplemental material) indicate that Cu export is not detectably upregulated in HOCl-stressed wild-type cells, we cannot rule out a role for RclA in maintaining Cu homeostasis under nonstress conditions, especially given the unexpected phenotype of a copA rclA double mutant (see Fig. S8B and C). This is an active area of research in our lab, and we are currently exploring how RclA and the various Cu homeostasis mechanisms of E. coli interact both under nonstress conditions and in the presence of HOCl.
FIG 9.
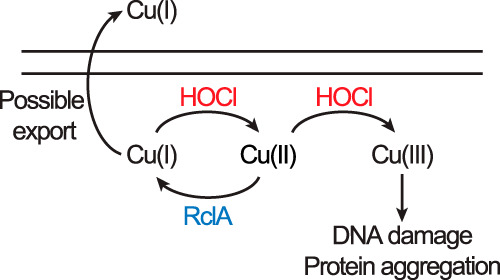
Proposed model for RclA activity in reducing the toxicity of HOCl. Oxidation of Cu by HOCl can result in the production of the highly-reactive and unstable Cu(III) (72, 110). Limiting the amount of cytoplasmic Cu(II) available would prevent the accumulation of Cu(III) during HOCl stress, thereby reducing the toxicity. Converting intracellular Cu(II) to Cu(I) may also facilitate Cu export via CopA(Z) mediated export systems (55).
The rate at which RclA oxidized NADH in the presence of copper in vitro was slow (approximately 4.4 min−1) (Fig. 7) (49), suggesting that we have not yet identified optimal reaction conditions for this enzyme. However, expression of rclA is rapidly induced >100-fold after sublethal doses of HOCl in E. coli (Fig. 4; see Data Set S2 in the supplemental material) (26), which could compensate in vivo for the low rate of NADH turnover we observed in vitro.
While RclA itself is widely conserved, the rclABCR locus as a whole is restricted to certain enteric proteobacteria, including E. coli, Salmonella, Citrobacter, Raoultella, Serratia, and Shigella. These genera are notable for their close association with gut inflammation and the ability of pathogenic strains to bloom to very high levels in the gut in disease states (2, 3, 7–10, 101, 102). We hypothesize that the ability to survive increased levels of antimicrobial compounds (including RCS) in the inflamed gut is important for the ability of enterobacteria to exploit this niche, and our in vivo results with the ΔrclA mutant generally support this idea (Fig. 2). Many noninflammatory commensal bacteria do encode rclA homologs (Fig. 1; see Data Set S1 in the supplemental material), including members of the Bacteroidetes, Clostridiaceae, and Lactobacillaceae, where their physiological roles are unknown. Expression of the rclA homolog of the probiotic Lactobacillus reuteri is induced modestly by HOCl, but an L. reuteri rclA mutant is not sensitive to HOCl stress (36). It is unclear, however, whether this is because RclA has a different physiological function in L. reuteri (possibly related to Cu homeostasis) or because RclA requires either strong induction or the presence of RclB and RclC to protect against HOCl under laboratory growth conditions. We do not currently know the physiological roles of RclB, which is a small predicted periplasmic protein, or RclC, which is a predicted inner membrane protein, although deletion of either of these genes results in increased HOCl sensitivity in E. coli (26). We hypothesize that they may form a complex with RclA in vivo and enhance its copper-dependent protective activity and are currently pursuing experiments to test this idea.
MATERIALS AND METHODS
Strain and plasmid construction.
E. coli strain MJG0586 [F– λ– rph-1 ΔilvG rfb-50 ΔrclA λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5)] was generated from MJG0046 (F– λ– rph-1 ΔilvG rfb-50 ΔrclA) (26) using the Novagen DE3 lysogenization kit, according to the manufacturer’s instructions. The ΔcopA767::kan+ allele from the Keio collection (103) was transduced into E. coli strains MG1655 and MJG0046 by P1 transduction (104), yielding strains MJG1759 (F– λ– rph-1 ΔilvG rfb-50 ΔcopA767::kan+) and MJG1760 (F– λ– rph-1 ΔilvG rfb-50 ΔrclA ΔcopA767::kan+) The RclA coding sequence (1374 bp) was amplified from E. coli MG1655 genomic DNA with the primers 5′ CTC GGT CTC CAA TGA ATA AAT ATC AGG CAG TGA 3′ and 5′ CTC GGT CTC AGC GCT TTA TTT GAC TAA TGA AAA TAG ATC A 3′ and cloned into the Eco31I (BsaI) sites of plasmid pPR-IBA101 (IBA Life Sciences) to yield plasmid pRCLA10. pRCLA11 was generated by using QuikChange site directed mutagenesis (Agilent), modified to use a single mutagenic primer (5′ CTA TTT TCA TTA GTC AAA AGC GCT TGG AGC CAC CC 3′), to remove the stop codon between the RclA coding sequence and the twin-strep tag sequence in pRCLA10. E. coli Nissle 1917 ΔrclA::cat+ strain MJG0846 was made by recombineering (105) using primers 5′ CGT CTA TAG TCA TGA TGT CAA ATG AAC GCG TTT CGA CAG GAA ATC ATC ATG GTG TAG GCT GGA GCT GCT TC 3′ and 5′ CTT TTC TCT GAG ACG CCA GAA TAT TTG TTC TGG CGT CTG ATT TTG AGT TTA CAT ATG AAT ATC CTC CTT AG 3′ to amplify the chloramphenicol resistance cassette from pKD3. The cat+ insertion was resolved using pCP20 resulting in the E. coli Nissle 1917 ΔrclA strain MJG0860.
Protein expression and purification.
Expression of twin-strep-tagged RclA was done in MJG0586 containing pRCLA11 (MJG1338) in M9 minimal media (106) containing 2 g liter−1 glucose and 100 μg ml−1 ampicillin. Overnight cultures of MJG1338 were diluted 1:100 and grown to an A600 of 0.4 at 37°C with shaking. When the A600 reached 0.4, expression was induced with IPTG (isopropyl-β-d-thiogalactopyranoside; 1 mM final concentration) and allowed to continue for 12 to 18 h at 20°C. Purification of recombinant RclA was achieved to high purity using a 1-ml StrepTrap HP column (GE, 28-9136-30 AC) according to the manufacturer’s instructions. Purified protein was subsequently saturated with FAD cofactor by incubating the protein preparation with a 10-fold molar excess of FAD at room temperature for 45 min. Excess FAD and elution buffer were dialyzed away with three exchanges of 1 liter of RclA storage buffer (50 mM Tris-HCl [pH 7.5], 0.5 M NaCl, 2 mM dithiothreitol, 10% glycerol) at 4°C overnight.
Colonization of D. melanogaster with E. coli Nissle 1917.
(i) D. melanogaster stocks and husbandry. Canton-S flies were used as a wild-type line. Duox-RNAi flies were obtained from crosses of UAS-dDuox-RNAi with NP1-GAL4 (gut-specific driver). Unless otherwise noted, the flies were reared on cornmeal medium (per liter of medium: 50 g yeast, 70 g cornmeal, 6 g agar, 40 g sucrose, 1.25 ml methyl-Paraben, and 5 ml 95% ethanol).
(ii) Oral infections. Adult female flies were starved for 2 h at 29°C prior to being fed a 1:1 suspension of bacteria (optical density at 600 nm [OD600] = 200) and 2.5% sucrose applied to a filter paper disk on the surface of fly food. Suspensions of bacteria at an OD600 of 200 were consistently found to contain 1 × 1011 CFU/ml. A negative control was prepared by substituting Luria-Bertani (LB) medium for the bacterial suspension. Infections were maintained at 29°C.
(iii) CFU determination. Flies were surface sterilized in 70% ethanol and rinsed in sterile phosphate-buffered saline (PBS). Individual flies were homogenized in screw-top bead tubes containing 600 μl of PBS. Dilution series of homogenates were prepared with a range of (100 to 10−5). Spots (3 μl) of each dilution were applied to LB plates and grown overnight at 30°C. E. coli colonies were identified by morphology and counted to determine the CFU/fly.
RNA sequencing.
E. coli Nissle 1917 was grown anaerobically (90% N2, 5% CO2, and 5% H2, maintained with a Coy Laboratory Products anaerobic chamber) at 37°C in MOPS minimal media (Teknova) in sealed Hungate tubes to an A600 of 0.25, then treated anaerobically with 0.4 mM HOCl. Samples (7 ml) were harvested into 7 ml of ice-cold isopropanol immediately before and 15 or 30 min after HOCl addition, harvested by centrifugation, and stored at –80°C until use. RNA was purified using a RiboPure-Bacteria kit (Ambion), according to the manufacturer’s instructions. mRNA sequencing was performed at the UAB Heflin Center for Genomic Sciences using an Illumina NextSeq 500 as described by the manufacturer (Illumina, Inc.). Use of an Agilent SureSelect strand-specific mRNA library kit (Agilent) and ribosome reduction with the RiboMinus protocol for Gram-negative and Gram-positive bacteria (Life Technologies) were performed as described by the manufacturers. The resulting mRNA was randomly fragmented with cations and heat, followed by first-strand synthesis using random primers. Second-strand cDNA production was done with standard techniques, and cDNA libraries were quantitated using qPCR in a Roche LightCycler 480 with a Kapa Biosystems kit for Illumina library quantitation prior to cluster generation, which was performed according to the manufacturers’ recommendations for onboard clustering (Illumina). We generated approximately 8 million double-stranded 50-bp reads per sample. Data were analyzed using Bowtie2 and DEseq2. RNA sequencing data have been deposited in NCBI’s Gene Expression Omnibus (107) and are accessible through GEO series accession number GSE144068.
Growth curves for measuring sensitivity to HOCl.
The molar HOCl concentration from concentrated sodium hypochlorite (Sigma-Aldrich, catalog no. 425044) was quantified by measuring the A292 of the stock solution diluted in 10 mM NaOH. Copper-free MOPS was prepared by removing total metals from MOPS minimal media (Teknova) containing 2 g liter−1 glucose and 1.32 mM K2HPO4 by treating prepared media with a universal chelator (Chelex 100 chelating resin; Bio-Rad catalog no. 1421253), filtering sterilizing away the Chelex resin, and adding back the metals normally present in MOPS suspended in metal-free water, except for copper. Overnight cultures of MG1655 and MJG0046 were grown up overnight in MOPS with or without copper. The overnight cultures were normalized to an A600 of 0.8 in and diluted to an A600 of 0.08 in MOPS minimal medium without copper using the indicated combinations and/or concentrations of HOCl and CuCl2. Cultures were incubated with shaking at 37°C in a Tecan Infinite M1000 plate reader with the A600 being measured every 30 min for 27 h. Sensitivity was subsequently determined by comparing lag-phase extensions (the difference in hours to reach A600 ≥ 0.15 from the no HOCl treatment control under the same CuCl2 condition) for each stress condition.
Determining the effect of copper on lethal HOCl stress.
Wild-type and ΔrclA E. coli MG1655 strains were grown overnight in MOPS minimal media. Next, 500 μl of the overnight culture was pelleted, resuspended in copper-free MOPS, and subcultured into 9.5 ml of copper-free MOPS, followed by growth with shaking at 37°C to mid-log phase (A600 = 0.3 to 0.4). Once mid-log phase was reached, 1 ml of cells was aliquoted into microcentrifuge tubes for each treatment. The cells were treated by adding the indicated amounts of copper (II) chloride, followed by 1 mM HOCl, and then incubated on a 37°C heat block for 10 min. After incubation, treatments were serially diluted in PBS, and 5-μl aliquots were spotted onto LB agar plates. Plates were dried and incubated at room temperature for 2 days.
Measuring intracellular copper after HOCl stress.
Wild-type and ΔrclA E. coli strains were grown to an A600 of 0.6 in MOPS medium and stressed with 400 μM HOCl for 30 min at 37°C with shaking. After 30 min, the cultures were diluted 2-fold with MOPS medium to quench HOCl and then pelleted. The mass of pellets was determined after three rinses with PBS. The amount of copper was measured via inductively coupled plasma mass spectrometry (7700× ICP-MS; Agilent Technologies, Santa Clara, CA) after suspension of the collected pellets in concentrated nitric acid and dilution of the suspensions to a 2% nitric acid matrix. Samples were filtered through 0.22-μm polytetrafluoroethylene filters to remove any particulates before running metal determinations. Copper concentrations were determined by comparison to a standard curve (Agilent, catalog no. 5188-6525) as calculated by Agilent software (ICP-MS MassHunter v4.3). Values determined by ICP-MS were normalized to pellet mass and dilution factor.
NADH oxidase activity.
Activity of purified RclA was assayed in 20 mM HEPES–100 mM NaCl (pH 7) by measuring NADH oxidation over time spectrophotometrically. Purified recombinant RclA was preloaded into wells of a 96-well plate (100 μl of 6 μM RclA). Reactions were started by adding 100 μl of NADH (200 μM final) with the indicated metal salts (200 μM final) to RclA (3 μM final) at 37°C. The absorbance of NADH (A340) was measured kinetically each minute for 5 min in a Tecan Infinite M1000 plate reader. A340 values were then used to calculate the μmol of NADH (ε340 = 6,300 M−1 cm−1) and NADPH (ε340 = 6,220 M−1 cm−1) at each time point. The absolute values of slopes of NAD(P)H (μmol) over time (min) were divided by the amount (mg) of RclA used to determine the reported specific activities [SA; μmol NAD(P)+ min−1 mg−1 RclA] for each condition.
Copper(I) quantification.
Cu(I) accumulation after the course of the RclA reaction was measured by using bathocuproinedisulfonic acid disodium salt (BCS; Sigma-Aldrich, B1125). NADH oxidation reactions were carried out as in the previous section but were started with either NADH (200 μM final), CuCl2 (200 μM final), NADH and CuCl2 (both at 200 μM final), or reaction buffer as a negative control to observe levels of background copper in the reagents used. Each reaction was stopped at 5 min with a BCS (400 μM final) and EDTA (1 mM final) solution using the injector system of a Tecan Infinite M1000 plate reader. The stopped reaction mixtures were incubated at 37°C, with the absorbance of BCS/Cu(I) complex being measured at 483 nm every minute for 5 min to ensure complete saturation of the BCS. The amount of the BCS/Cu(I) complex after 5 min was determined using the molar extinction coefficient 13,000 M−1 cm−1 (74).
NADH oxidase activity after treatment with urea and HOCl.
The SA of RclA after being treated with denaturing agents was measured by determining the SA as described above after incubation of RclA with either urea or HOCl. Urea treatment was done on 3 μM RclA for 24 h at room temperature with increasing concentrations of urea (0, 2, 4, and 6 M) in reaction buffer (20 mM HEPES, 100 mM NaCl [pH 7]). HOCl treatment was done by mixing increasing molar ratios of HOCl (0, 5, 10, and 20×) with concentrated RclA or l-lactate dehydrogenase (LDH; Sigma, LLDH-RO 10127230001; 35 μM) in oxidation buffer (50 mM sodium phosphate [pH 6.8], 150 mM NaCl) and incubating the mixtures on ice for 30 min. HOCl treatment was quenched after 30 min by diluting the RclA solutions to 6 μM and the LDH solutions to 1 μM in reaction buffer (20 mM HEPES, 100 mM NaCl [pH 7]). NADH oxidation reactions with LDH were performed under the same conditions as the RclA reactions but were started with 1.2 mM NADH and 1.2 mM pyruvate.
Melting temperature determination.
CD spectra were obtained on a Jasco J815 circular dichroism spectrometer. CD spectra were collected on purified recombinant RclA exchanged into 20 mM HEPES and 100 mM NaCl (pH 7.5). Room temperature CD spectra in the range of 260 to 190 nm were obtained in 0.1-mm demountable quartz cells. Thermal CD data between 30 and 90°C were obtained in standard 1.0-mm quartz cells. All data were collected with a 1.0-nm step size, an 8-s averaging time per point, and a 2-nm bandwidth. Data were baseline corrected against the appropriate buffer solution and smoothed with Jasco software.
Data analysis and bioinformatics.
(i) Statistics. All statistical analyses were performed using GraphPad Prism (v7.0a).
(ii) Phylogenetic tree. An RclA conservation tree was made from amino acid sequence alignments of RclA (BLAST E-value < 1 × 10−90 in 284 species), RclB (BLAST E-value < 1 × 10−1 in 61 species), RclC, (BLAST E-value < 1 × 10−80 in 49 species), and RclR (BLAST E-value < 1 × 10−40 in 43 species). BLAST searches were done by comparing to each respective protein in E. coli MG1655. A tree graphic was made using the Interactive Tree of Life (108) (https://itol.embl.de/).
(iii) Amino acid sequence alignments. Active site alignment of E. coli RclA (ADC80840.1) and MerA amino acid sequences was done for seven bacterial species (Escherichia coli, ADC80840.1; Staphylococcus aureus, AKA87329.1; Salmonella enterica, ABQ57371.1; Listeria monocytogenes, PDA94520.1; Klebsiella pneumoniae, ABY75610.1; and Serratia marcescens, ADM52740.1). Alignment was done using CLUSTAL O (1.2.4; www.ebi.ac.uk/Tools/msa/clustalo/), and a graphic was prepared using WEBLOGO (weblogo.berkeley.edu/logo.cgi). A full-length alignment of E. coli RclA (ADC80840.1) and MerA (ADC80840.1) was also prepared. Alignment, conservation scoring, and graphic were done using PRALINE (www.ibi.vu.nl/programs/pralinewww/).
Data accessibility.
All strains generated in the course of this study are available from the authors upon request.
ACKNOWLEDGMENTS
We thank Peter Prevelige (UAB Department of Microbiology) for helpful discussions, Emily Schwessinger (University of Michigan) for preliminary characterization of RclA, Michael Jablonsky (UAB Chemistry Department) for assistance with CD spectrum acquisition, Won-Jae Lee (Seoul National University) for providing the D. melanogaster Duox RNAi line, and William Van Der Pol and Ranjit Kumar at the UAB Center for Clinical and Translational Science for their help with the RNA sequencing analysis.
This project was supported by University of Alabama at Birmingham Department of Microbiology startup funds, NIH grant R35GM124590 (to M.J.G.), NIH grant R35GM128871 (to N.A.B.), and NIH training grant T90DE022736 (to R.M.D.).
We have no conflicts of interest to declare.
R.M.D., A.J.B., C.E.B., and I.F.C. performed research, and R.M.D., A.J.B., S.E.L., N.A.B., and M.J.G. contributed to designing research, analyzing data, and writing the paper.
Footnotes
Citation Derke RM, Barron AJ, Billiot CE, Chaple IF, Lapi SE, Broderick NA, Gray MJ. 2020. The Cu(II) reductase RclA protects Escherichia coli against the combination of hypochlorous acid and intracellular copper. mBio 11:e01905-20. https://doi.org/10.1128/mBio.01905-20.
REFERENCES
- 1.Kaplan GG. 2015. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol 12:720–727. doi: 10.1038/nrgastro.2015.150. [DOI] [PubMed] [Google Scholar]
- 2.Litvak Y, Byndloss MX, Tsolis RM, Baumler AJ. 2017. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol 39:1–6. doi: 10.1016/j.mib.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Stecher B. 2015. The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol Spectr 3. doi: 10.1128/microbiolspec.MBP-0008-2014. [DOI] [PubMed] [Google Scholar]
- 4.Manichanh C, Borruel N, Casellas F, Guarner F. 2012. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 9:599–608. doi: 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 5.Casen C, et al. . 2015. Deviations in human gut microbiota: a novel diagnostic test for determining dysbiosis in patients with IBS or IBD. Aliment Pharmacol Ther 42:71–83. doi: 10.1111/apt.13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickson I. 2017. Gut microbiota: diagnosing IBD with the gut microbiome. Nat Rev Gastroenterol Hepatol 14:195. doi: 10.1038/nrgastro.2017.25. [DOI] [PubMed] [Google Scholar]
- 7.Litvak Y, Mon KKZ, Nguyen H, Chanthavixay G, Liou M, Velazquez EM, Kutter L, Alcantara MA, Byndloss MX, Tiffany CR, Walker GT, Faber F, Zhu Y, Bronner DN, Byndloss AJ, Tsolis RM, Zhou H, Bäumler AJ. 2019. Commensal Enterobacteriaceae protect against Salmonella colonization through oxygen competition. Cell Host Microbe 25:128–139 e125. doi: 10.1016/j.chom.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Zhu W, Winter MG, Byndloss MX, Spiga L, Duerkop BA, Hughes ER, Büttner L, de Lima Romão E, Behrendt CL, Lopez CA, Sifuentes-Dominguez L, Huff-Hardy K, Wilson RP, Gillis CC, Tükel Ç, Koh AY, Burstein E, Hooper LV, Bäumler AJ, Winter SE. 2018. Precision editing of the gut microbiota ameliorates colitis. Nature 553:208–211. doi: 10.1038/nature25172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. 2007. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2:119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Winter SE, Lopez CA, Baumler AJ. 2013. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep 14:319–327. doi: 10.1038/embor.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wera O, Lancellotti P, Oury C. 2016. The dual role of neutrophils in inflammatory bowel diseases. J Clin Med 5:118. doi: 10.3390/jcm5120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JH, Peyrin-Biroulet L, Eisenhut M, Shin JI. 2017. IBD immunopathogenesis: a comprehensive review of inflammatory molecules. Autoimmun Rev 16:416–426. doi: 10.1016/j.autrev.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Chami B, Martin NJJ, Dennis JM, Witting PK. 2018. Myeloperoxidase in the inflamed colon: a novel target for treating inflammatory bowel disease. Arch Biochem Biophys 645:61–71. doi: 10.1016/j.abb.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Pavlick KP, Laroux FS, Fuseler J, Wolf RE, Gray L, Hoffman J, Grisham MB. 2002. Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease. Free Radic Biol Med 33:311–322. doi: 10.1016/S0891-5849(02)00853-5. [DOI] [PubMed] [Google Scholar]
- 15.Braymer JJ, Giedroc DP. 2014. Recent developments in copper and zinc homeostasis in bacterial pathogens. Curr Opin Chem Biol 19:59–66. doi: 10.1016/j.cbpa.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheldo JR, Skaar EP. 2019. Metals as phagocyte antimicrobial effectors. Curr Opin Immunol 60:1–9. doi: 10.1016/j.coi.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Besold AN, Culbertson EM, Culotta VC. 2016. The yin and yang of copper during infection. J Biol Inorg Chem 21:137–144. doi: 10.1007/s00775-016-1335-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winterbourn CC, Kettle AJ. 2013. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal 18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 19.Gray MJ, Wholey WY, Jakob U. 2013. Bacterial responses to reactive chlorine species. Annu Rev Microbiol 67:141–160. doi: 10.1146/annurev-micro-102912-142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim SH, Lee WJ. 2014. Role of DUOX in gut inflammation: lessons from Drosophila model of gut-microbiota interactions. Front Cell Infect Microbiol 3:116. doi: 10.3389/fcimb.2013.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ha EM, Oh CT, Bae YS, Lee WJ. 2005. A direct role for dual oxidase in Drosophila gut immunity. Science 310:847–850. doi: 10.1126/science.1117311. [DOI] [PubMed] [Google Scholar]
- 22.Lee K-A, Kim S-H, Kim E-K, Ha E-M, You H, Kim B, Kim M-J, Kwon Y, Ryu J-H, Lee W-J. 2013. Bacterial-derived uracil as a modulator of mucosal immunity and gut-microbe homeostasis in Drosophila. Cell 153:797–811. doi: 10.1016/j.cell.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Staudinger BJ, Oberdoerster MA, Lewis PJ, Rosen H. 2002. mRNA expression profiles for Escherichia coli ingested by normal and phagocyte oxidase-deficient human neutrophils. J Clin Invest 110:1151–1163. doi: 10.1172/JCI0215268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofman P, Piche M, Far DF, Le Negrate G, Selva E, Landraud L, Alliana-Schmid A, Boquet P, Rossi B. 2000. Increased Escherichia coli phagocytosis in neutrophils that have transmigrated across a cultured intestinal epithelium. Infect Immun 68:449–455. doi: 10.1128/iai.68.2.449-455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray MJ, Wholey WY, Parker BW, Kim M, Jakob U. 2013. NemR is a bleach-sensing transcription factor. J Biol Chem 288:13789–13798. doi: 10.1074/jbc.M113.454421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker BW, Schwessinger EA, Jakob U, Gray MJ. 2013. The RclR protein is a reactive chlorine-specific transcription factor in Escherichia coli. J Biol Chem 288:32574–32584. doi: 10.1074/jbc.M113.503516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drazic A, Miura H, Peschek J, Le Y, Bach NC, Kriehuber T, Winter J. 2013. Methionine oxidation activates a transcription factor in response to oxidative stress. Proc Natl Acad Sci U S A 110:9493–9498. doi: 10.1073/pnas.1300578110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray MJ, Wholey W-Y, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JCA, Jakob U. 2014. Polyphosphate is a primordial chaperone. Mol Cell 53:689–699. doi: 10.1016/j.molcel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahl JU, Gray MJ, Jakob U. 2015. Protein quality control under oxidative stress conditions. J Mol Biol 427:1549–1563. doi: 10.1016/j.jmb.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winter J, Ilbert M, Graf PC, Ozcelik D, Jakob U. 2008. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell 135:691–701. doi: 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoo NG, Dogra S, Meinen BA, Tse E, Haefliger J, Southworth DR, Gray MJ, Dahl J-U, Jakob U. 2018. Polyphosphate stabilizes protein unfolding intermediates as soluble amyloid-like oligomers. J Mol Biol 430:4195–4208. doi: 10.1016/j.jmb.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gray MJ, Jakob U. 2015. Oxidative stress protection by polyphosphate: new roles for an old player. Curr Opin Microbiol 24:1–6. doi: 10.1016/j.mib.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gennaris A, Ezraty B, Henry C, Agrebi R, Vergnes A, Oheix E, Bos J, Leverrier P, Espinosa L, Szewczyk J, Vertommen D, Iranzo O, Collet J-F, Barras F. 2015. Repairing oxidized proteins in the bacterial envelope using respiratory chain electrons. Nature 528:409–412. doi: 10.1038/nature15764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goemans CV, Vertommen D, Agrebi R, Collet JF. 2018. CnoX is a chaperedoxin: a holdase that protects its substrates from irreversible oxidation. Mol Cell 70:614–627. doi: 10.1016/j.molcel.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Deng K, Zaremba S, Deng X, Lin C, Wang Q, Tortorello ML, Zhang W. 2009. Transcriptomic response of Escherichia coli O157:H7 to oxidative stress. AEM 75:6110–6123. doi: 10.1128/AEM.00914-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basu Thakur P, Long AR, Nelson BJ, Kumar R, Rosenberg AF, Gray MJ. 2019. Complex responses to hydrogen peroxide and hypochlorous acid by the probiotic bacterium Lactobacillus reuteri. mSystems 4 doi: 10.1128/mSystems.00453-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derke RM, Barron AJ, Billiot CE, Chaple IF, Lapi SE, Broderick NA, Gray MJ. 2020. The Cu(II) reductase RclA protects Escherichia coli against the combination of hypochlorous acid and intracellular copper. bioRxiv 690669. doi: 10.1101/690669v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grozdanov L, Raasch C, Schulze J, Sonnenborn U, Gottschalk G, Hacker J, Dobrindt U. 2004. Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle 1917. J Bacteriol 186:5432–5441. doi: 10.1128/JB.186.16.5432-5441.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wassenaar TM. 2016. Insights from 100 years of research with probiotic Escherichia coli. Eur J Microbiol Immunol 6:147–161. doi: 10.1556/1886.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun J, Gunzer F, Westendorf AM, Buer J, Scharfe M, Jarek M, Gössling F, Blöcker H, Zeng A-P. 2005. Genomic peculiarity of coding sequences and metabolic potential of probiotic Escherichia coli strain Nissle 1917 inferred from raw genome data. J Biotechnol 117:147–161. doi: 10.1016/j.jbiotec.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 41.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD. 2014. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang Y, Kong Q, Roland KL, Wolf A, Curtiss R III.. 2014. Multiple effects of Escherichia coli Nissle 1917 on growth, biofilm formation, and inflammation cytokines profile of Clostridium perfringens type A strain CP4. Pathog Dis 70:390–400. doi: 10.1111/2049-632X.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hancock V, Vejborg RM, Klemm P. 2010. Functional genomics of probiotic Escherichia coli Nissle 1917 and 83972, and UPEC strain CFT073: comparison of transcriptomes, growth and biofilm formation. Mol Genet Genomics 284:437–454. doi: 10.1007/s00438-010-0578-8. [DOI] [PubMed] [Google Scholar]
- 44.Hancock V, Dahl M, Klemm P. 2010. Probiotic Escherichia coli strain Nissle 1917 outcompetes intestinal pathogens during biofilm formation. J Med Microbiol 59:392–399. doi: 10.1099/jmm.0.008672-0. [DOI] [PubMed] [Google Scholar]
- 45.Luterbach CL, Forsyth VS, Engstrom MD, Mobley HLT. 2018. TosR-mediated regulation of adhesins and biofilm formation in uropathogenic Escherichia coli. mSphere 3:e00222-18. doi: 10.1128/mSphere.00222-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Lee K-A, Ren X, Ryu J-C, Kim G, Ryu J-H, Lee W-J, Yoon J. 2016. Synthesis of a highly HOCl-selective fluorescent probe and its use for imaging HOCl in cells and organisms. Nat Protoc 11:1219–1228. doi: 10.1038/nprot.2016.062. [DOI] [PubMed] [Google Scholar]
- 47.Macheroux P, Kappes B, Ealick SE. 2011. Flavogenomics: a genomic and structural view of flavin-dependent proteins. FEBS J 278:2625–2634. doi: 10.1111/j.1742-4658.2011.08202.x. [DOI] [PubMed] [Google Scholar]
- 48.Lian P, Guo H-B, Riccardi D, Dong A, Parks JM, Xu Q, Pai EF, Miller SM, Wei D-Q, Smith JC, Guo H. 2014. X-ray structure of a HgII+ complex of mercuric reductase (MerA) and quantum mechanical/molecular mechanical study of HgII+ transfer between the C-terminal and buried catalytic site cysteine pairs. Biochemistry 53:7211–7222. doi: 10.1021/bi500608u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baek Y, Kim J, Ahn J, Jo I, Hong S, Ryu S, Ha N-C. 2020. Structure and function of the hypochlorous acid-induced flavoprotein RclA from Escherichia coli. J Biol Chem 295:3202–3212. doi: 10.1074/jbc.RA119.011530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ledwidge R, Patel B, Dong A, Fiedler D, Falkowski M, Zelikova J, Summers AO, Pai EF, Miller SM. 2005. NmerA, the metal binding domain of mercuric ion reductase, removes HgII+ from proteins, delivers it to the catalytic core, and protects cells under glutathione-depleted conditions. Biochemistry 44:11402–11416. doi: 10.1021/bi050519d. [DOI] [PubMed] [Google Scholar]
- 51.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. doi: 10.1128/JB.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hodgkinson V, Petris MJ. 2012. Copper homeostasis at the host-pathogen interface. J Biol Chem 287:13549–13555. doi: 10.1074/jbc.R111.316406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ladomersky E, Petris MJ. 2015. Copper tolerance and virulence in bacteria. Metallomics 7:957–964. doi: 10.1039/c4mt00327f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rensing C, Grass G. 2003. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol Rev 27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 55.Petersen C, Moller LB. 2000. Control of copper homeostasis in Escherichia coli by a P-type ATPase, CopA, and a MerR-like transcriptional activator, CopR. Gene 261:289–298. doi: 10.1016/S0378-1119(00)00509-6. [DOI] [PubMed] [Google Scholar]
- 56.Santos-Zavaleta A, Salgado H, Gama-Castro S, Sánchez-Pérez M, Gómez-Romero L, Ledezma-Tejeida D, García-Sotelo JS, Alquicira-Hernández K, Muñiz-Rascado LJ, Peña-Loredo P, Ishida-Gutiérrez C, Velázquez-Ramírez DA, Del Moral-Chávez V, Bonavides-Martínez C, Méndez-Cruz C-F, Galagan J, Collado-Vides J. 2019. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in Escherichia coli K-12. Nucleic Acids Res 47:D212–D220. doi: 10.1093/nar/gky1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamamoto K, Ishihama A. 2005. Transcriptional response of Escherichia coli to external copper. Mol Microbiol 56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- 58.Kershaw CJ, Brown NL, Constantinidou C, Patel MD, Hobman JL. 2005. The expression profile of Escherichia coli K-12 in response to minimal, optimal and excess copper concentrations. Microbiology (Reading) 151:1187–1198. doi: 10.1099/mic.0.27650-0. [DOI] [PubMed] [Google Scholar]
- 59.Edward RWT, Gray T, Margerum DW. 1977. Kinetics and mechanisms of the copper-catalyzed decomposition of hypochlorite and hypobromite: properties of a dimeric copper(III) hydroxide intermediate. Inorg Chem 16:3047–3055. doi: 10.1021/ic50178a014. [DOI] [Google Scholar]
- 60.Liu C, von Gunten U, Croue JP. 2012. Enhanced bromate formation during chlorination of bromide-containing waters in the presence of CuO: catalytic disproportionation of hypobromous acid. Environ Sci Technol 46:11054–11061. doi: 10.1021/es3021793. [DOI] [PubMed] [Google Scholar]
- 61.Church JA. 1994. Kinetics of the uncatalyzed and Cu(II)-catalyzed decomposition of sodium hypochlorite. Ind Eng Chem Res 33:239–245. doi: 10.1021/ie00026a010. [DOI] [Google Scholar]
- 62.Lister MW. 1956. Decomposition of sodium hypochlorite: the uncatalyzed reaction. Can J Chem 34:465–478. doi: 10.1139/v56-068. [DOI] [Google Scholar]
- 63.Pham AN, Xing G, Miller CJ, Waite TD. 2013. Fenton-like copper redox chemistry revisited: hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J Catalysis 301:54–64. doi: 10.1016/j.jcat.2013.01.025. [DOI] [Google Scholar]
- 64.Folkes LK, Candeias LP, Wardman P. 1995. Kinetics and mechanisms of hypochlorous acid reactions. Arch Biochem Biophys 323:120–126. doi: 10.1006/abbi.1995.0017. [DOI] [PubMed] [Google Scholar]
- 65.Conocchioli TJ, Hamilton EJ Jr, Sutin N. 1965. The formation of iron(IV) in the oxidation of iron(II). J Am Chem Soc 87 87:926–927. doi: 10.1021/ja01082a050. [DOI] [Google Scholar]
- 66.Hao OJ, Davis AP, Chang PH. 1991. Kinetics of manganese(II) oxidation with chlorine. J Environ Eng 117:359–374. doi: 10.1061/(ASCE)0733-9372(1991)117:3(359). [DOI] [Google Scholar]
- 67.Allard S, Fouche L, Dick J, Heitz A, von Gunten U. 2013. Oxidation of manganese(II) during chlorination: role of bromide. Environ Sci Technol 47:8716–8723. doi: 10.1021/es401304r. [DOI] [PubMed] [Google Scholar]
- 68.Holmgren A. 1976. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc Natl Acad Sci U S A 73:2275–2279. doi: 10.1073/pnas.73.7.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang X, Ma K. 2010. Characterization of a thioredoxin-thioredoxin reductase system from the hyperthermophilic bacterium Thermotoga maritima. J Bacteriol 192:1370–1376. doi: 10.1128/JB.01035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rinderle SJ, Booth JE, Williams JW. 1983. Mercuric reductase from R-plasmid NR1: characterization and mechanistic study. Biochemistry 22:869–876. doi: 10.1021/bi00273a025. [DOI] [PubMed] [Google Scholar]
- 71.Miller SM, Ballou DP, Massey V, Williams CH Jr, Walsh CT. 1986. Two-electron reduced mercuric reductase binds Hg(II) to the active site dithiol but does not catalyze Hg(II) reduction. J Biol Chem 261:8081–8084. [PubMed] [Google Scholar]
- 72.Sutton HC, Winterbourn CC. 1989. On the participation of higher oxidation states of iron and copper in Fenton reactions. Free Radic Biol Med 6:53–60. doi: 10.1016/0891-5849(89)90160-3. [DOI] [PubMed] [Google Scholar]
- 73.Gunther MR, Hanna PM, Mason RP, Cohen MS. 1995. Hydroxyl radical formation from cuprous ion and hydrogen peroxide: a spin-trapping study. Arch Biochem Biophys 316:515–522. doi: 10.1006/abbi.1995.1068. [DOI] [PubMed] [Google Scholar]
- 74.Johnson DK, Stevenson MJ, Almadidy ZA, Jenkins SE, Wilcox DE, Grossoehme NE. 2015. Stabilization of Cu(I) for binding and calorimetric measurements in aqueous solution. Dalton Trans 44:16494–16505. doi: 10.1039/c5dt02689j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morgenstern S, Flor R, Kessler, Klein GB. 1966. The automated determination of NAD-coupled enzymes. II. Serum lactate dehydrogenase. Clin Chem 12:274–281. doi: 10.1093/clinchem/12.5.274. [DOI] [PubMed] [Google Scholar]
- 76.Oikawa S, Kawanishi S. 1996. Site-specific DNA damage induced by NADH in the presence of copper(II): role of active oxygen species. Biochemistry 35:4584–4590. doi: 10.1021/bi9527000. [DOI] [PubMed] [Google Scholar]
- 77.Mateus A, Bobonis J, Kurzawa N, Stein F, Helm D, Hevler J, Typas A, Savitski MM. 2018. Thermal proteome profiling in bacteria: probing protein state in vivo. Mol Syst Biol 14:e8242. doi: 10.15252/msb.20188242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mealman TD, Blackburn NJ, McEvoy MM. 2012. Metal export by CusCFBA, the periplasmic Cu(I)/Ag(I) transport system of Escherichia coli. Curr Top Membr 69:163–196. doi: 10.1016/B978-0-12-394390-3.00007-0. [DOI] [PubMed] [Google Scholar]
- 79.Su CC, Long F, Yu EW. 2011. The Cus efflux system removes toxic ions via a methionine shuttle. Protein Sci 20:6–18. doi: 10.1002/pro.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Juhas M, Reuss DR, Zhu B, Commichau FM. 2014. Bacillus subtilis and Escherichia coli essential genes and minimal cell factories after one decade of genome engineering. Microbiology 160:2341–2351. doi: 10.1099/mic.0.079376-0. [DOI] [PubMed] [Google Scholar]
- 81.Au DC, Lorence RM, Gennis RB. 1985. Isolation and characterization of an Escherichia coli mutant lacking the cytochrome o terminal oxidase. J Bacteriol 161:123–127. doi: 10.1128/JB.161.1.123-127.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jones SA, Chowdhury FZ, Fabich AJ, Anderson A, Schreiner DM, House AL, Autieri SM, Leatham MP, Lins JJ, Jorgensen M, Cohen PS, Conway T. 2007. Respiration of Escherichia coli in the mouse intestine. Infect Immun 75:4891–4899. doi: 10.1128/IAI.00484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Samanovic MI, Ding C, Thiele DJ, Darwin KH. 2012. Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe 11:106–115. doi: 10.1016/j.chom.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Letelier ME, Sanchez-Jofre S, Peredo-Silva L, Cortes-Troncoso J, Aracena-Parks P. 2010. Mechanisms underlying iron and copper ions toxicity in biological systems: pro-oxidant activity and protein-binding effects. Chem Biol Interact 188:220–227. doi: 10.1016/j.cbi.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 85.Macomber L, Rensing C, Imlay JA. 2007. Intracellular copper does not catalyze the formation of oxidative DNA damage in Escherichia coli. J Bacteriol 189:1616–1626. doi: 10.1128/JB.01357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beveridge SJ, Garrett IR, Whitehouse MW, Vernon-Roberts B, Brooks PM. 1985. Biodistribution of 64Cu in inflamed rats following administration of two anti-inflammatory copper complexes. Agents Actions 17:104–111. doi: 10.1007/BF01966692. [DOI] [PubMed] [Google Scholar]
- 87.Achard MES, Stafford SL, Bokil NJ, Chartres J, Bernhardt PV, Schembri MA, Sweet MJ, McEwan AG. 2012. Copper redistribution in murine macrophages in response to Salmonella infection. Biochem J 444:51–57. doi: 10.1042/BJ20112180. [DOI] [PubMed] [Google Scholar]
- 88.Casciaro M, Di Salvo E, Pace E, Ventura-Spagnolo E, Navarra M, Gangemi S. 2017. Chlorinative stress in age-related diseases: a literature review. Immun Ageing 14:21. doi: 10.1186/s12979-017-0104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. 2006. Biomarkers of oxidative damage in human disease. Clin Chem 52:601–623. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 90.Klebanoff SJ. 2005. Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 91.Franceschi C, Campisi J. 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 92.Djoko KY, McEwan AG. 2013. Antimicrobial action of copper is amplified via inhibition of heme biosynthesis. ACS Chem Biol 8:2217–2223. doi: 10.1021/cb4002443. [DOI] [PubMed] [Google Scholar]
- 93.Macomber L, Imlay JA. 2009. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci U S A 106:8344–8349. doi: 10.1073/pnas.0812808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Affandi T, McEvoy MM. 2019. Mechanism of metal ion-induced activation of a two-component sensor kinase. Biochem J 476:115–135. doi: 10.1042/BCJ20180577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Changela A, Chen K, Xue Y, Holschen J, Outten CE, O’Halloran TV, Mondragón A. 2003. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science 301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 96.Katz AK. 2003. Copper-binding motifs: structural and theoretical aspects. Helvetica 86:1320–1338. doi: 10.1002/hlca.200390120. [DOI] [Google Scholar]
- 97.Cook NL, Pattison DI, Davies MJ. 2012. Myeloperoxidase-derived oxidants rapidly oxidize and disrupt zinc–cysteine/histidine clusters in proteins. Free Radic Biol Med 53:2072–2080. doi: 10.1016/j.freeradbiomed.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 98.Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, Jakob U. 2008. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci U S A 105:8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arnitz R, Sarg B, Ott HW, Neher A, Lindner H, Nagl M. 2006. Protein sites of attack of N-chlorotaurine in Escherichia coli. Proteomics 6:865–869. doi: 10.1002/pmic.200500054. [DOI] [PubMed] [Google Scholar]
- 100.Meydan S, Klepacki D, Karthikeyan S, Margus T, Thomas P, Jones JE, Khan Y, Briggs J, Dinman JD, Vázquez-Laslop N, Mankin AS. 2017. Programmed ribosomal frameshifting generates a copper transporter and a copper chaperone from the same gene. Mol Cell 65:207–219. doi: 10.1016/j.molcel.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rivera-Chavez F, Baumler AJ. 2015. The pyromaniac inside you: Salmonella metabolism in the host gut. Annu Rev Microbiol 69:31–48. doi: 10.1146/annurev-micro-091014-104108. [DOI] [PubMed] [Google Scholar]
- 102.Shin NR, Whon TW, Bae JW. 2015. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 33:496–503. doi: 10.1016/j.tibtech.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 103.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Silhavy TJ, Berman ML, Enquist LW. 1985. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 105.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 107.Edgar R, Domrachev M, Lash AE. 2002. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prlić A, Bliven S, Rose PW, Bluhm WF, Bizon C, Godzik A, Bourne PE. 2010. Pre-calculated protein structure alignments at the RCSB PDB website. Bioinformatics 26:2983–2985. doi: 10.1093/bioinformatics/btq572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meyerstein D, Copper T. 1971. A pulse radiolytic study of the formation and decomposition of amino complexes. Inorg Chem 10:2244–2249. doi: 10.1021/ic50104a031. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial species containing homologs of the Rcl proteins of E. coli. Homologs of the RclA, RclB, RclC, and RclR proteins of E. coli included in Fig. 1, as identified by BLAST: RclA (BLAST E-value < 1 × 10−90 in 284 species), RclB (BLAST E-value < 1 × 10−1 in 61 species), RclC, (BLAST E-value < 1 × 10−80 in 49 species), and RclR (BLAST E-value < 1 × 10−40 in 43 species). Download Data Set S1, XLSX file, 0.03 MB (27.5KB, xlsx) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth curve method to determine lag-phase extension in the presence of HOCl. Overnight cultures of (A) MG1655 wild-type and (B) MJG0046 (ΔrclA) were normalized to an A600 of 0.8 in MOPS minimal media and diluted to an A600 of 0.08 in MOPS ± HOCl at the indicated concentrations. Cultures were incubated with shaking at 37°C in a Tecan Infinite M1000 plate reader with the A600 being measured every hour for 24 h (n = 3 to 4, ± the SD). Sensitivity was assessed by comparing lag phase extensions (LPE; difference in hours to reach A600 ≥ 0.15 from no HOCl control; vertical dotted lines) for each HOCl concentration. An example LPE calculation for wild-type grown in 148 μM HOCl shown in panel A. (C) Average LPE values for growth curves shown in panels A and B and statistics comparing wild-type and ΔrclA strains at all conditions. Differences in average LPE values between the strains were analyzed using two-way ANOVA with Tukey’s multiple-comparison test. HOCl growth curves were performed as in panels A and B but on log-phase cultures of each strain. To prepare the log-phase cultures, saturated overnight cultures were subcultured in MOPS glucose and grown up at 37°C with shaking at 200 rpm until A600 = 0.5. E. coli lacking rclA (D) took longer to reach an A600 of 0.15 relative to the wild type (E) at all the tested HOCl concentrations. Time for each sample to reach an average A600 of 0.15 is indicated by vertical dashed lines. The observed growth delays of the strains in the presence of HOCl did not correspond with killing, as determined by viability spot plates (C, pictures representative of three biological replicates). Viability spot plates were prepared by collecting 5-μl aliquots from each well used for growth curves 10 min after adding MOPS ± HOCl to the OD-normalized cells. Serial dilutions in PBS of the aliquots were performed, and 5 μl of the dilutions was spotted on LB plates and grown overnight at 37°C. (G) Average LPE values for growth curves shown in D and E and statistics comparing wild-type and ΔrclA strains at all conditions. Differences in average LPE values between the strains were analyzed using two-way ANOVA with Tukey’s multiple-comparison test. Download FIG S1, TIF file, 2.2 MB (2.2MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differences in fly colonization and HOCl sensitivity between E. coli K-12 and EcN. (A) EcN colonizes Canton-S D. melanogaster to a greater extent than MG1655. Flies were fed either E. coli K-12 MG1655 (K-12) or Nissle 1917 (EcN) and bacterial loads were measured (n = 5, ± the SD). Statistical analysis was performed using two-way ANOVA with Tukey’s multiple-comparison test (****, P < 0.0001; ns, not significant). (B) EcN growth curves with HOCl. EcN forms adherent growth in 96-well plates when grown with HOCl, which obstructs accurate measurement of absorbance over time. Growth curves performed as in Fig. S1A (n = 3 to 4, ± the SD). (C and D) EcN is more sensitive to HOCl-mediated killing than K-12. Wild-type E. coli MG1655 (K-12) (C) and Nissle 1917 (EcN) (D) were grown overnight in MOPS minimal media. A portion (500 μl) of the overnight culture was pelleted, resuspended in copper-free MOPS, and subcultured into 9.5 ml of copper-free MOPS. The cultures were then grown with shaking at 37°C to mid-log phase (A600 = 0.3 to 0.4). Once mid-log phase was reached, 1 ml of cells was aliquoted into microcentrifuge tubes for each treatment. Cells were treated by adding the indicated amounts HOCl and then incubated on a 37°C heat block for 10 min. After incubation, treatments were serially diluted in PBS, and 5-μl aliquots were spotted on LB agar plates. Plates were dried and incubated at room temperature for 2 days before imaging. Download FIG S2, TIF file, 1.3 MB (1.3MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fly response to EcN colonization. (A) Host-produced HOCl production is silenced by Duox RNAi. Prior to treatment, flies were starved for 2 h at 29°C. (A) As an uninfected control, NP1-GAL4 female flies fed on a mixture of LB, and 100 μM R19-S was prepared in 5% sucrose. RCS production in response to EcN infection was determined by feeding a mixture of EcN (5 × 107 cells) and 100 μM R19-S to NP1-GAL4 female flies (B) and NP1-GAL4-UAS-DuoxIR female flies (C). After 30 min of feeding on the designated mixture, all treatments were switched to a sucrose/R19-S mixture. At 90 min after the initial infection, guts were dissected in PBS and fixed in 4% formaldehyde for 50 min. Guts were mounted in Vectashield/DAPI and visualized on a Leica SP8 confocal microscope with a red emission range of 530 to 603 nm. Images presented are representative of a total of three gut tissues imaged per treatment. (D to F) The antimicrobial peptide Diptericin (Dpt) is expressed in response to oral infection with EcN in DD;NP1 flies. Adult female flies were fed EcN (E) or a sucrose (D) control for 6 h, and gut tissues were dissected and stained with X-Gal to reveal regions of Dpt expression, method described previously (C. Neyen, A. J. Bretscher, O. Binggeli, and B. Lemaitre, Methods 68:116–128, 2014, https://doi.org/10.1016/j.ymeth.2014.02.023). (F) β-Galactosidase activity of adult female gut tissues at various time points after oral infection with EcN. The relative β-galactosidase activity is expressed as a percentage of baseline activity observed in flies fed a sterile sucrose solution. Download FIG S3, TIF file, 2.7 MB (2.7MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Alignment of full-length RclA with MerA sequences from representative bacterial species. Alignment of E. coli RclA (ADC80840.1) and MerA amino acid sequences from seven species (Escherichia coli, ADC80840.1; Staphylococcus aureus, AKA87329.1; Salmonella enterica, ABQ57371.1; Listeria monocytogenes, PDA94520.1; Klebsiella pneumoniae, ABY75610.1; Serratia marcescens, ADM52740.1). Alignment, conservation scoring, and graphic were made using PRALINE. Download FIG S4, TIF file, 2.6 MB (2.7MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Results of RNA sequencing of HOCl-treated EcN. RPKM and log2-fold change values for all genes in E. coli Nissle before and 15 and 30 min after treatment with 0.4 mM HOCl. Raw data are deposited under GEO series accession number GSE144068. Download Data Set S2, XLSX file, 1.4 MB (1.4MB, xlsx) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Extracellular CuCl2 reduces LPE for both wild-type and ΔrclA E. coli grown in the presence of HOCl. (A) Average LPE values for growth curves shown in Fig. 5 and statistics comparing wild-type and ΔrclA at all conditions. Differences in average LPE values between the strains were analyzed using two-way ANOVA with Tukey’s multiple-comparison test. LPE determination for wild-type (B) and ΔrclA (C) strains grown in the presence of the indicated HOCl and extracellular CuCl2 conditions. Growth curves were performed as described in Fig. S1 but with supplementing the MOPS glucose with the indicated CuCl2 concentrations. HOCl and CuCl2 were mixed in MOPS minimal media prior to diluting cells A600-normalized to 0.08 into each media type. LPEs were calculated by determining the difference in time (h) to reach A600 > 0.15 for each condition relative to no stress controls. Sensitivity was assessed within strains by comparing LPEs for each HOCl/CuCl2 concentration (n = 4, ± the SD) at each HOCl concentration (two-way ANOVA for each strain with Dunnett’s multiple comparison using the no CuCl2 control for each HOCl concentration, respectively (****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, not significant). (D) EcN colonization is improved when flies are fed copper. D. melanogaster Canton-S females fed for 24 h on a mixture of sucrose and 1 mM CuCl2 or sterile water (as a control), which was applied to a filter disk placed on the fly food surface. Flies were then transferred to new food tubes containing sucrose and either EcN wild-type (WT) or EcN ΔrclA. Individuals were collected, surface sterilized, homogenized, and plated on LB agar at various time points to determine bacterial load (CFUs/fly). Statistical analysis was performed with GraphPad Prism using a multiple t test with Sidak-Bonferroni correction for multiple comparisons (n = 60). Download FIG S5, TIF file, 1.3 MB (1.3MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparing LPEs of strains stressed with or without intracellular Cu. (A) Average LPE values for growth curves shown in Fig. 6 and in panels B to E. Differences in average LPE values between the strains were analyzed using a two-way ANOVA with Tukey’s multiple-comparison test. Wild type (B and C) and ΔrclA (C and D) E. coli were grown up overnight in MOPS glucose with (D and E) or without copper (A and C). The time for each sample to reach an average A600 of 0.15 is indicated by vertical dashed lines. HOCl growth curves were performed as described in Fig. 6. Download FIG S6, TIF file, 1.6 MB (1.6MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Control experiments for RclA activity. (A) Adding BME to the start solutions of RclA reactions does not increase SA and causes Cu(I) accumulation. (A) RclA reactions performed as in Fig. 7 but with the addition of 1 mM BME for indicated samples (n = 6, ± the SD). SA (A) and BCS/Cu(I) (B) concentration determined as in Fig. 7. Differences in SA in the presence of each metal were analyzed using a two-way ANOVA with Dunnett’s multiple-comparison test using the buffer-only sample as the control for each start solution (****, P < 0.0001). (C and D) Changes in NADH and copper (I) concentrations over time during the RclA reaction. NADH oxidation (C) and BCS/Cu(I) complex formation (D) were measured over time spectrophotometrically with and without RclA. Reactions were started by adding 100 μl of NADH (200 μM final) or NADH and CuCl2 (both 200 μM final) to 100 μl of RclA (3 μM final) at 37°C using the injector system of a Tecan Infinite M1000 plate reader. All reactions were carried out in 20 mM HEPES–100 mM NaCl (pH 7). NADH absorbance at 340 nm was measured each minute for the indicated reaction times. Each reaction was then stopped at the indicated times with 10 μl of a BCS (400 μM final) and EDTA (1 mM final) solution using the injector system of the plate reader (n = 6 for each reaction type, ± the SD). The stopped reaction mixtures were incubated at 37°C for 5 min, with the absorbance of BCS/Cu(I) complex being measured at 483 nm each minute to ensure saturation of BCS. Reported BCS/Cu(I) values were calculated using the average of the total absorbance measurements over the 5-min incubation because the values were stable over that time. Differences in the amount of NADH or BCS/Cu(I) complex between the buffer-only and RclA reactions were analyzed using two-way ANOVA with Sidak’s multiple-comparison test (****, P < 0.0001; ***, P < 0.001; *, P < 0.05; ns, not significant). (E) The copper reductase activity of RclA is maintained under anaerobic conditions. BCS/Cu(I) complex concentrations were determined as in Fig. 7, but all reactions were performed in an anaerobic chamber (n = 6, ± the SD). Differences in the amount of BCS/Cu(I) complex between the buffer-only and RclA (3 μM) reactions were analyzed using a two-way ANOVA with Tukey’s multiple-comparison test (****, P < 0.0001; ns, not significant). (F) The melting temperature (Tm) of RclA is ∼10°C higher than the average Tm of the E. coli proteome. The CD spectra of RclA were measured at the indicated temperatures; 222 nm is indicated by a vertical dashed line. Download FIG S7, TIF file, 1.6 MB (1.6MB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Measuring intracellular copper concentrations during HOCl stress and growth defect of a rclA copA double mutant. (A) Intracellular copper concentrations do not change after HOCl stress in wild-type E. coli. E. coli MG1655 wild-type (wt) and ΔrclA were grown overnight in MOPS. The next day, both strains were subcultured into MOPS, grown at 37°C with shaking to A600 = 0.6, and stressed with 400 μM HOCl for 30 min at 37°C with shaking. Intracellular copper before and after HOCl stress of the strains was determined using ICP-MS. Differences between intracellular copper analyzed using two-way ANOVA with Tukey’s multiple-comparison test (*, P < 0.05; ns, not significant). (B) A double mutant of rclA and copA has a growth defect when grown with (B) or without copper (C). HOCl growth curves on cells with or without intracellular copper were performed as described in Fig. 6 with copper-free MOPS glucose (n = 3). Download FIG S8, TIF file, 0.7 MB (777.3KB, tif) .
Copyright © 2020 Derke et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
All strains generated in the course of this study are available from the authors upon request.