

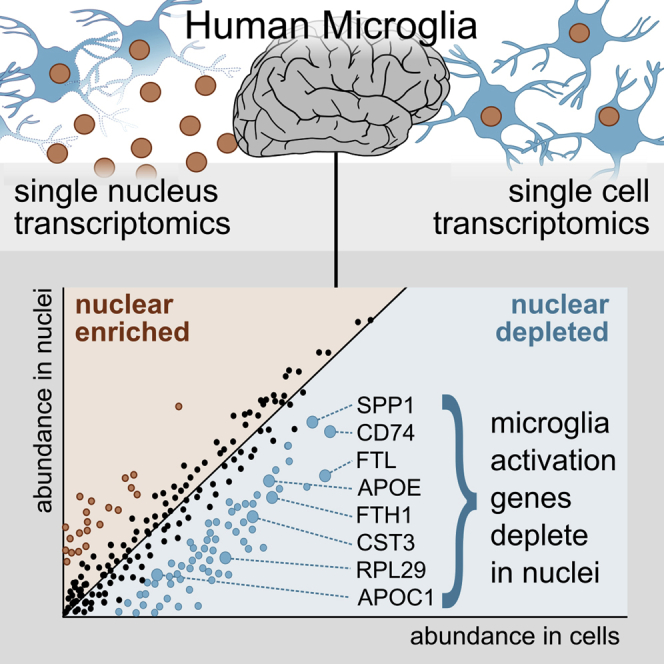
Summary
Single-nucleus RNA sequencing (snRNA-seq) is used as an alternative to single-cell RNA-seq, as it allows transcriptomic profiling of frozen tissue. However, it is unclear whether snRNA-seq is able to detect cellular state in human tissue. Indeed, snRNA-seq analyses of human brain samples have failed to detect a consistent microglial activation signature in Alzheimer’s disease. Our comparison of microglia from single cells and single nuclei of four human subjects reveals that, although most genes show similar relative abundances in cells and nuclei, a small population of genes (∼1%) is depleted in nuclei compared to whole cells. This population is enriched for genes previously implicated in microglial activation, including APOE, CST3, SPP1, and CD74, comprising 18% of previously identified microglial-disease-associated genes. Given the low sensitivity of snRNA-seq to detect many activation genes, we conclude that snRNA-seq is not suited for detecting cellular activation in microglia in human disease.
Keywords: microglia, activation, Alzheimer’s disease, single-nucleus RNA-seq, single-cell RNA-seq, microglial activation, ARM
Graphical Abstract
Highlights
-
•
A small set of genes is depleted in microglial nuclei relative to single cells
-
•
This set is enriched for microglial activation genes, including APOE and SPP1
-
•
This depletion is confirmed in publicly available datasets
-
•
Single-nucleus sequencing is not suited for the detection of human microglial activation
Thrupp et al. demonstrate the depletion of a small population of genes in nuclei relative to cells in human microglia by using single-nucleus and single-cell sequencing. This population is enriched for microglial activation genes, suggesting that single-nucleus sequencing is not suited for the detection of microglial activation in humans.
Introduction
Single-cell approaches allow us to study cell-to-cell heterogeneity (Habib et al., 2017). In brain material, however, it is difficult to dissociate individual cells (Habib et al., 2017; Lake et al., 2016). This difficulty is further complicated if one is interested in studying the human brain, for which often only frozen material is available. One alternative to study cellular transcriptional heterogeneity in brain tissue is single-nucleus transcriptomics. Single-nucleus RNA sequencing (snRNA-seq) studies have shown concordance between single-cell and single-nucleus transcriptome profiles in mice (Bakken et al., 2018; Habib et al., 2017; Lake et al., 2017; Zhou et al., 2020). It is unclear whether a snRNA-seq approach is equally effective for identifying dynamic cellular substates, such as microglial activation in human tissue.
A recent breakthrough in the field of Alzheimer’s disease (AD) using single-cell RNA-seq (scRNA-seq) demonstrated clearly that microglia become activated in response to amyloid plaques in mouse models (Keren-Shaul et al., 2017). This response comprises a transcriptional switch to a state called activation response microglia (ARM) (Sala Frigerio et al., 2019) or disease-associated microglia (DAM, MGnD) (Keren-Shaul et al., 2017; Krasemann et al., 2017). Evidence suggests that this microglial response is also relevant in human AD: microglia are believed to play a role in amyloid clearance (Efthymiou and Goate, 2017) and complement-mediated synapse loss (Fonseca et al., 2017) and histological studies have demonstrated considerable microgliosis around plaques in humans (McGeer et al., 1987). In addition, there is significant overlap between those genes involved in the mouse microglial response and AD risk genes identified in genome-wide association studies (Efthymiou and Goate, 2017; Gosselin et al., 2017; Jansen et al., 2019; Kunkle et al., 2019; Lambert et al., 2013; Marioni et al., 2018), for example, APOE, TREM2, APOC1, and CD33 (Sala Frigerio et al., 2019). Most recently, the engrafting of human microglia into an AD mouse model, followed by scRNA-seq, identified 66 DAM genes relevant to human activation (Hasselmann et al., 2019). Although it is not yet clear how a mouse brain environment may influence the development of human microglia, this study does offer additional evidence for microglial activation in human.
In contrast, a number of high-profile snRNA-seq studies of microglia in human AD (Del-Aguila et al., 2019; Grubman et al., 2019; Mathys et al., 2019) have been unable to recover an equivalent activation signature or, possibly more revealing, a consistent activation signature. A recent cluster analysis by Mathys et al. (2019) of 48 AD patients and controls reported only 28 of 257 orthologous activation genes in common with the DAM signature. Differential expression analysis between AD and control patients revealed 22 genes upregulated in AD patients (5 overlapping with the DAM signature). Of these AD genes, only 8 were also upregulated in another snRNA-seq study of human AD (Grubman et al., 2019), and only 4 were upregulated in a snRNA-seq study of AD TREM2 variants (Zhou et al., 2020). The AD TREM2 variant study also only identified 11 DAM genes enriched in AD patients compared with controls. Del-Aguila et al. (2019) analyzed single-nucleus transcriptomics from 3 AD patients and were unable to recapitulate an activation signature (Del-Aguila et al., 2019). This inconsistency within human studies and across species has made it difficult to assess the true nature of human microglial activation.
Here, we compare the performance of snRNA-seq to scRNA-seq for the analysis of microglia from human cortical biopsies and demonstrate that technical limitations inherent to snRNA-seq provide a likely explanation for this lack of consistency in snRNA-seq studies of AD. We confirm our results using publicly available data.
Results
snRNA-Seq Recovers Major Cell Types from Human Tissue but Not Microglial State
scRNA-seq of FACS-sorted microglia was performed on temporal cortices of four human subjects who had undergone neocortical resection (Mancuso et al., 2019). See Table S1 for subject data. Mean Unique Molecular Identifier (UMI) count per cell was 4,527, and mean number of genes per cell was 2,242.
We generated snRNA-seq libraries from these same subjects. Single-cell and single-nucleus libraries were prepared using the 10X Genomics single-cell gene expression v2 kit. Following quality filtering, principal-component analysis (PCA) analysis, and clustering of 37,060 nuclei, we identified 7 major cell types (Figures S1A and S1B): oligodendrocytes (ODCs; 34.0%), excitatory neurons (27.0%), interneurons (11.2%), ODC precursors (OPCs; 9.4%), microglia (11.3%), astrocytes (6.0%), and endothelial cells (1.1%).
We extracted microglia from the main snRNA-seq dataset (Figures S1A–S1C). After pre-processing, 3,927 microglial nuclei remained, with a mean UMI count of 1,328 and a mean gene count of 879 per nucleus. We first checked whether clustering analysis of single nuclei could recover subpopulations of microglia comparable to the single-cell approach. A comparison of single-nucleus and single-cell clustering suggested that cytokine clusters and macrophage clusters recovered well by using single-nucleus methods; however, differences between other microglial subpopulations were not convincingly recovered (see STAR Methods and Figures S1D–S1F).
Gene Expression Profiling of Human Nuclei and Cells
To compare gene abundance in single microglial cells (14,823 cells) and nuclei (3,940 nuclei), we performed a differential abundance analysis between cells and nuclei from the 4 subjects (Figure 1A). As demonstrated in previous studies (Bakken et al., 2018; Gerrits et al., 2019; Habib et al., 2017; Lake et al., 2017), most genes showed similar normalized abundance levels in cells and nuclei, with 98.6% of genes falling along the diagonal in Figure 1A (Pearson’s correlation coefficient = 0.92, p < 2.2e-16). However, we identified a group of 246 genes (1.1% of all genes detected) that was less abundant in nuclei (fold change < −2, adjusted p value (padj) < 0.05; blue in Figure 1A). A second population of 67 genes (0.3%) was found to be more abundant in nuclei (fold change > 2, padj < 0.05; red in Figure 1A). Additionally, 3,248 genes were only detected in cells, and 5,068 genes were exclusively detected in nuclei.
Figure 1.

Gene Abundance in Single Microglial Cells versus Single Microglial Nuclei of Human Cortical Tissue
(A) Mean normalized gene abundance in cells (x axis) and nuclei (y axis). A total of 3,721 nuclei and 14,435 cells were extracted from the cortical tissue of 4 human patients. Red, genes with significantly higher abundance in nuclei (padj < 0.05, fold change > 2); blue, genes that are significantly less abundance in nuclei (padj < 0.05, fold change < −2). Genes were normalized to read depth (per cell), scaled by 10,000, and log-transformed using the natural log. MALAT1 (which had normalized abundance levels of 6.0 and 6.9, respectively, in cells and nuclei) has been removed for visualization purposes. The black dashed line represents no fold change; the gray dotted lines represent 2- and 4-fold differences between cells and nuclei. FC, fold change; R2, correlation coefficient. Full results are available in Table S1.
(B) Scatterplot as in (A), per patient (with the same genes highlighted).
(C) Each bar represents a comparison between two datasets (X versus Y), with the bootstrapped Z scores representing the extent to which cell-enriched genes (top panel) and nuclear-enriched genes (bottom panel) have lower specificity for microglia in dataset Y relative to that in dataset X. Larger Z scores indicate greater depletion of genes, and red bars indicate a statistically significant depletion (padj < 0.05, by bootstrapping). KI, Karolinska Institutet; AIBS, Allen Institute for Brain Science. See also Figure S1 and S2A and Table S1.
The observed differences in abundance between cells and nuclei were consistent across all four subjects (Figure 1B; Figure S2A). Downsampling cellular reads indicated that differences in abundance were not the result of different sequencing depths (Figure S2B), and downsampling the number of cells indicated that differences in abundance were not a result of different numbers of cells/nuclei (Figure S2C). The full differential abundance results can be found in Table S1.
To assess the robustness of this differential abundance, we used our nuclei-abundant genes and cell-abundant genes to compare enrichment across all pairs of 8 publicly available single-cell or single-nucleus datasets (Table S1; Figure 1C). We consistently found our nuclei-underrepresented (cell-abundant) genes to be depleted in other single-nucleus microglia compared to single-cell microglia (mean microglial Z score of cell-abundant genes was 7.95 when comparing cells to nuclei, whereas cell-to-cell comparisons yielded a mean of 0.01, and nuclei-to-nuclei comparisons yielded a mean of 0.81, for Z scores with padj < 0.05). We also found our nuclei-abundant genes to be consistently enriched in other microglial nuclei compared with microglial cells (mean microglial Z score of −2.99 compared to −2.33 in nuclei against nuclei, no significant enrichment was found in cell-to-cell comparisons with padj < 0.05).
To assess functional enrichment among genes found to be more abundant in cells or nuclei, we ranked all genes according to log fold change (genes with a low abundance in nuclei had a negative log fold change) and performed a gene set enrichment analysis (GSEA; Subramanian et al., 2005) against gene markers from previous studies (Figure 2A). For these analyses, a positive normalized enrichment score (NES) represented nuclear enrichment, and a negative NES represented nuclear depletion. As expected, cytoplasmic RNA (defined by Bahar Halpern et al., 2015) was clearly enriched among genes found to be more abundant in cells (NES = −2.03, padj = 3.6e-05), as was mitochondrial mRNA (NES = −1.69, padj = 2.7e-04, gene set extracted from Ensembl’s BioMart; Zerbino et al., 2018). mRNA found to be more abundant in the nucleus by Bahar Halpern et al. (2015) tended toward enrichment in nuclei but was not significant (NES = 0.86, padj = 8.7e-01), which is to be expected as scRNA-seq captures both nuclear and cytoplasmic RNA. RNA of genes coding for ribosomal proteins was also depleted in nuclei (NES = −2.37, padj = 3.6e-05), as previously described (Habib et al., 2017). Genes with shorter coding sequences (CDSs) were depleted in nuclei (NES = −1.88, padj = 3.6e-05), whereas longer CDSs were enriched (NES = 1.91, padj = 3.6e-05), as already observed in earlier snRNA-seq studies (Bakken et al., 2018). GC content was not enriched in either direction—low GC content NES was −0.99 (padj = 5.0e-01) and high GC content was −1.16 (padj = 1.7e-01) —suggesting no role for the differences in abundance between cells and nuclei. Finally, the genes defined by Gerrits et al. (2019) as cellular enriched in a differential analysis of microglial cells versus (fresh) nuclei in humans were also enriched in cells in our data, showing a NES score of −1.92 (padj = 3.6e-05).
Figure 2.

Functional Analysis of Genes That Are Enriched or Depleted in Nuclei
(A) Gene set enrichment analysis (GSEA) of gene sets related to cellular location and gene coding sequence (CDS) length. Background genes were ranked according to log fold change of nuclei (3,721 nuclei) versus cells (14,435 cells). Red, higher normalized enrichment score (NES), i.e., more genes associated with nuclear enrichment; blue, negative NES scores (depletion in nuclei). ∗∗∗ represents significance (padj < 0.0005). GC, GC content.
(B) GSEA of super-Gene Ontology gene sets against ranked nucleus-cell log fold changes. Only top and bottom categories (according to NES) are shown. Colors as in (A). MHCI, major histocompatibility complex class I.
(C) GSEA of selected gene sets from previous studies of microglial activation, against log fold change as in (A). ∗∗∗ represents significance (padj < 0.0005). Mic0, markers of microglial cluster 0 in human brain tissue; Mic1, markers of microglial cluster 1 (activation response to plaques) defined by Mathys et al., 2019 in human brain tissue. ARM, activation response microglia (Sala Frigerio et al., 2019); DAM, disease-associated microglia (Keren-Shaul et al., 2017); LPS, lipopolysaccharide (Gerrits et al., 2019).
(D) Scatterplot as in Figure 1A, highlighting in green the DAM genes. A regression line for the highlighted genes is shown in green (slope = 0.60).
(E) Scatterplot as in (D), highlighting in green the ARM genes. A regression line for the highlighted genes is shown in green (slope = 0.64).
(F) Scatterplot as in (D), highlighting the DAM genes recovered in the study of human activation in AD (Mathys et al., 2019). Purple, DAM genes not recovered in their study; orange, DAM genes recovered in their study.
(G) Scatterplot as in (D); green, human activation marker genes defined by Mathys et al. (2019). Gene sets, results of GO clustering, and results of GSEA analysis are available in Table S1. See also Figures S2B–S2G.
To further characterize genes with higher or lower abundance in nuclei compared with cells, we performed GSEA, using Gene Ontology (GO) terms extracted from the Molecular Signatures Database (MSigDb, Liberzon et al., 2011) against the ranked log fold change. We selected the 100 terms with the highest NES and the 100 terms with the lowest NES (padj < 0.05). Given the high overlap in terms, we clustered ontology terms based on the number of shared genes to define super-GO clusters. We repeated the GSEA analysis by using these super-GO clusters (Figure 2B; Table S1) and observed an enrichment of neuronal and synaptic terms in nuclei-abundant genes (also shown in the red population in Figure 1A). We suspect a synaptosome contamination during centrifugation. This is supported by the enrichment of synaptosome genes (NES = 1.82, padj = 3.6e-05, Figure 2A; Hafner et al., 2019) and ambient RNA—mRNA originating not from intact cells/nuclei but from free-floating transcripts (Macosko et al., 2015)—(NES = 1.71, padj = 9.2e-05; Figure 2A; Figure S2D) within the nucleus-abundant genes. The two gene sets share a strong overlap (Table S1). These genes, although enriched in nuclei compared with cellular levels, still show low abundance (most of these genes show a normalized abundance of no more than 2; Figure 1A). Removal of this ambient mRNA from our dataset did not affect normalized gene abundance comparisons between cells and nuclei abundance; we detected 238 genes that were significantly less abundant in cells (fold change < −2, padj < 0.05; blue in Figure S2E), in comparison to the original 246 genes. No changes were seen in the number of genes that were underrepresented in nuclei (67).
Activation Genes Identified in Mouse Models of AD Are Depleted in Human Nuclei
More interesting was the depletion of immune-related genes in nuclei (Figure 2B). We therefore tested whether microglial activation genes were also depleted in nuclei (Figure 2C; Table S1). Remarkably, we found a strong depletion of genes associated with mouse microglial activation: 45 of 257 orthologous DAM genes (Keren-Shaul et al., 2017; NES = −2.16, padj = 3.6e-05; Figures 2C and 2D) and 28 of 200 orthologous ARM genes (Sala Frigerio et al., 2019; NES = −2.01, padj = 3.6e-05; Figures 2C and 2E), confirming that mouse microglial activation genes were less abundant in nuclei. Genes upregulated by lipopolysaccharide (LPS) stimulation in mice (Gerrits et al., 2019) also showed depletion in nuclei (NES = −1.86, padj = 3.6e-05; Figure 2C; Figure S2F).
As we observed an enrichment for shorter genes in the nuclei-depleted population, we wanted to test whether activation genes were also shorter. We performed a differential expression analysis of ARM genes against homeostatic genes (Sala Frigerio et al., 2019) and ranked genes according to this log fold change. We then performed a GSEA, taking the 200 genes with the shortest and the longest CDS and the 200 genes with the highest and lowest GC content. Figure S2G shows that these activation genes are not enriched for GC content, but they do tend to be shorter than the general gene population (NES = 1.85, padj = 9.1e-05).
Activation Genes Identified in Human Studies of AD Are Depleted in Human Nuclei
We next examined genes that were identified as markers of the human microglial response to AD in the recent snRNA-seq study by Mathys et al. (2019) (Figures 2C, 2F, and 2G). Markers of this response (referred to by Mathys et al., 2019 as “Mic1”) had a NES score of −2.14 (padj = 3.6e-05), indicating that they were depleted in nuclei (Figure 2C). The study identified 28 DAM genes as marker genes of the Mic1 response cluster (shown in orange in Figure 2F); however, most DAM genes were not identified as activation genes using their snRNA-seq protocol (purple in Figure 2F). Figure 2G shows in green all the markers of the human activation cluster Mic1. Clearly, DAM genes and other Mic1 markers showed a higher abundance in cells relative to nuclei (confirming the NES score observed in Figure 2C). Furthermore, it seems likely that the recovered DAM genes (orange in Figure 2F) and Mic1 markers in general (green in Figure 2G) were detected in the original snRNA-seq experiment owing to their higher nuclear abundance than the nuclear abundance of other genes, including those DAM genes that were not recovered (purple in Figure 2F).
Comparisons with Previous Studies
A recent comparison of snRNA-seq and scRNA-seq in human microglia (Gerrits et al., 2019) found little difference in abundance between nuclei and cells, in contrast to our results. In Gerrits et al. (2019), they FACS-sorted microglia from fresh postmortem tissue and performed scRNA-seq and snRNA-seq on fresh nuclei. In addition, they extracted and froze adjacent tissue, and then they isolated nuclei from it. We downloaded their raw data and examined the differences in abundance between cells and fresh nuclei, as well as between cells and frozen nuclei. Frozen nuclei contained multiple cell types. After clustering of the frozen nuclei (Figure S3A), we classified clusters into cell types based on the expression of know markers (Figure S3B) and isolated 2,659 microglial nuclei.
In agreement with the results of Gerrits et al. (2019), when we plotted the normalized abundance of fresh nuclei versus cells, we saw very little difference between the two populations (Figure S3C). However, when we compared frozen nuclei to fresh cells, we again observed the same set of nuclei-depleted genes showing a lower relative abundance in frozen nuclei of Gerrits et al. (2019) (Figure S3D).
Discussion
In our comparison of nuclear (snRNA-seq) and total cellular transcriptomes (scRNA-seq) of human microglia, we have identified a set of genes (1.1% of the gene population) with at least 2-fold lower abundance in nuclei compared to their cellular levels (Figures 1A and 1B). This small set is strongly enriched for genes previously associated with microglial activation in mouse models of AD, for example, APOE, CST3, FTL, SPP1, B2M, PLD3, and CD74 (Figures 2B–2E). Many these genes have been implicated in AD, for example, through genetic risk studies (Efthymiou and Goate, 2017), but have not been consistently detected in snRNA-seq studies of AD (Del-Aguila et al., 2019; Grubman et al., 2019; Mathys et al., 2019). Thus, although our work agrees with previous experiments demonstrating that snRNA-seq can determine cell type (Figures S1A and S1B), we argue that a methodology that is unable to assess the dysregulation of these genes in humans is not suitable to answer questions relating to cellular state in human microglia. These limitations are likely responsible for the difficulty in identifying a consistent activation signature in the human brain in snRNA-seq-based studies. We identified similar patterns of depletion in publicly available single-nucleus microglial datasets (Figure 1C).
Re-examination of data from the Mathys et al. (2019) study of human nuclei in AD shows that only genes with higher nuclear abundance levels were detected (Figures 2C, 2F, and 2G). This further suggests that the discordance between human and mouse microglial activation is at least in part a consequence of limitations in the technology, rather than biological differences between the species as current snRNA-seq suggest. Deeper sequencing (or increased sample size) could possibly compensate for this lack of sensitivity. Both our study and the Mathys et al. (2019) study relied on the 10X Genomics single-cell expression v2 kit. Further improvements in snRNA-seq library preparation will possibly result in better sensitivity and better resolution of the nuclear transcriptome. The improved v3 chemistry yields a higher number of reads and genes per cell, as shown in a comparison using peripheral blood mononuclear cells (PBMCs) (Ding et al., 2020). The study, however, did not assess the ability of this deeper sequencing to detect additional cell populations or states. The sparse nature of snRNA-seq and the high level of heterogeneity in human samples, combined with the fact that many relevant genes have a more than 2-fold lower abundance in nuclei (e.g., APOE fold change = 2.57, CST3 fold change = 3.44, FTL fold change = 6.53), suggest that this lack of sensitivity may remain a problem.
It appears that CDS may play a role in the ability of snRNA-seq to capture genes (genes underrepresented in nuclei tended to be shorter; Figure 2A). However, we are unable to attribute the differences in cellular and nuclear abundance to a biological mechanism (for instance, mRNA trafficking) or a technical artifact of the methodology. Droplet-based methods do not appear to suffer from length bias (Phipson et al., 2017); thus, we cannot not attribute differences in capture efficiency to a preference for capturing longer genes. For our single-cell protocol, we performed dissociation at 4°C to mitigate potential activation (Mancuso et al., 2019), so it is unlikely the dissociation procedures account for the differences. Our lab has previously used the same protocol to study microglial activation in mice, and no activation due to technical artifacts was seen (Sala Frigerio et al., 2019).
One caveat of the present study is that we have used non-diseased tissue, and we cannot dismiss the possibility that mRNA localization distributions change in more activated populations of microglia.
In a previous comparison of human microglial cells and nuclei (Gerrits et al., 2019), little difference in abundance between genes was observed comparing fresh cells with fresh nuclei. However, using their data to compare fresh cells to frozen nuclei (Figure S3D), we observed a depletion of nuclear genes, similar to the depletion seen in our data. The reason for this difference between fresh and frozen nuclei is unclear. It may be the freezing/thawing process; however, Gerrits et al. (2019) also employ a different extraction protocol for fresh and frozen nuclei. We elected to compare frozen nuclei because such archival frozen tissue remains the main source of material for transcriptomic studies of AD. For their cluster analysis, Gerrits et al. (2019) scaled cell and nuclei expression to mitochondrial reads. Such methods may inadvertently mask differences between cells and nuclei, thereby making it difficult to assess their claim that single nuclei are a good proxy for single cells based on the ability of (frozen and fresh) nuclei to co-cluster with cells. A more suitable confirmation would be to determine whether, using single nuclei, the same level of clustering resolution can be achieved as when using single cells.
Alternative approaches may be more suitable for generating a brain atlas of human disease such as AD, particularly where we are limited to frozen material. In situ spatial transcriptomics (STs) negates issues related to tissue dissociation and cell or nucleus isolation (Ståhl et al., 2016), while at the same time retaining spatial information. This approach has recently been applied to examine transcriptomic changes and identify genes that are co-expressed across multiple cell types in the amyloid plaque niche of the mouse brain (Chen et al., 2020). In humans, a similar methodology was recently applied to identify pathway dysregulation and regional differences in cellular states of the postmortem spinal tissue of amyotrophic lateral sclerosis (ALS) patients (Maniatis et al., 2019). Its application to AD patients may shed light on transcriptomic changes occurring in microglia that localize near plaques and may also provide insights into the crosstalk occurring between neighboring cells.
In conclusion, although snRNA-seq offers a viable alternative to scRNA-seq for the identification of cell types in tissue for which cell dissociation is problematic, caution should be applied when using snRNA-seq for the assessment of cellular states in disease.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Resected cortical brain tissue | Mancuso et al., 2019 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-(+)-Sucrose, ultrapure DNase RNase free | VWR | 0335; CAS 57-50-1 |
| Calcium chloride | Sigma-Aldrich | 449709; CAS 10043-52-4 |
| Magnesium acetate tetrahydrate | Sigma-Aldrich | M5661; CAS 16674-78-5 |
| Tris (1 M), pH 8.0, RNase-free | Invitrogen | AM9855G |
| Ethylenediaminetetraacetic acid disodium salt solution | Sigma-Aldrich | E7889; CAS 139-33-3 |
| IGEPAL CA-630 | Sigma-Aldrich | I8896; CAS 9002-93-1 |
| Phenylmethylsulfonylfluoride | Thermo Fisher Scientific | #36978; CAS 329-98-6 |
| 2-Mercaptoethanol (50 mM) | Thermo Fisher Scientific | 31350010; CAS 60-24-2 |
| UltraPure DNase/RNase-Free Distilled Water | Invitrogen | 10977035 |
| OptiPrep | Stemcell | #07820 |
| Potassium chloride | Sigma-Aldrich | P4504; CAS 7447-40-7 |
| Magnesium chloride | Sigma-Aldrich | M8266; CAS 7786-30-3 |
| PBS - Phosphate-Buffered Saline (10X) pH 7.4, RNase-free | Invitrogen | AM9624 |
| Bovine serum albumin (BSA) | VWR | 0332; CAS 9048-46-8 |
| RNasin Plus RNase Inhibitor | Promega | N2615 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3′ Library & Gel, Bead Kit v2, 16 rxns | 10x Genomics | 120237 |
| Deposited Data | ||
| Raw count data and fastq files | This paper | GSE153807 |
| Raw count data and fastq files | Mancuso et al., 2019 | GSE137444 |
| Software and Algorithms | ||
| Cellranger v2.1.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| R v3.6.3 | https://cran.r-project.org/ | https://cran.r-project.org/ |
| Seurat v3.0.2 | Butler et al., 2018; Stuart et al., 2019 | https://satijalab.org/seurat/install.html |
| EWCE package | Skene and Grant, 2016 | https://github.com/NathanSkene/EWCE |
| MicroglialDepletion package | This paper | https://github.com/NathanSkene/MicroglialDepletion |
| Other | ||
| Plain Plunger Head For PTFE Tissue Grinder | Fisherbrand | 10709382 |
| Glass Vessel for PTFE Tissue Grinder | Fisherbrand | 10075911 |
| EASYstrainer 70 μM, for 50 ML tubes, for tubes 227XXX/210XXX, blue, sterile, single packed | Greiner Bio-one | 542070 |
| OPTIMA XPN – 90 | Beckman Coulter | A94468 |
| SW 41 Ti Swinging-Bucket Rotor | Beckman Coulter | 331362 |
| 13.2 mL, Open-Top Thinwall Ultra-Clear Tube, 14 × 89mm - 50Pk | Beckman Coulter | 344059 |
| Pasteur pipette | VWR | 612-1681 |
| Falcon 5 mL Round Bottom Polystyrene Test Tube, with Cell Strainer Snap Cap, 25/Pack, 500/Case | Corning | 352235 |
| LUNA Cell Counting Slides | Westburg | LB L12001 |
| LUNA-FL Automated Fluorescence Cell Counter | Westburg | LB L20001 |
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Mark Fiers (mark.fiers@kuleuven.vib.be).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Sequencing data from single microglial cells and nuclei are available at GEO: GSE137444 for single cells, and GEO: GSE153807 for single nuclei).
Analysis of previous datasets (see Figure 1C) was performed using the EWCE package (Skene and Grant, 2016) and the MicroglialDepletion package (https://github.com/NathanSkene/MicroglialDepletion).
Experimental Model and Subject Details
Resected tissue samples were obtained from the lateral temporal neocortex of 4 epilepsy patients during neurosurgery (amygdalohippocampectomy for medial temporal lobe seizures). Patients were aged 7 (male), and 20,24, and 50 (female). Patient metadata (age, gender and epilepsy diagnosis) are presented in Table S1. Biological samples and personal data were collected in full compliance with the ethical principles of free donation, informed consent and protection of privacy according to the General Data Protection Regulation (GDPR) 2016/679 (Section 5.1.3). All procedures followed protocols approved by the local Ethical Committee at UZ Leuven (protocol number S61186).
Method Details
Isolation of human primary microglial cells
Human primary microglial cells from the Mancuso et al. (2019) study were used. Fresh brain tissue was collected at the time of surgery and immediately prepared for sequencing. Microglia were FAC-sorted from the tissue. The full protocol is described in the original study. Sequencing was performed as described for the nuclei.
Isolation of nuclei from human subjects
Nuclei from frozen biopsy tissue were isolated as follows: brain tissue was sliced on dry ice, then homogenized using a glass dounce tissue grinder (15 gentle strokes) in 1mL of ice-cold Homogenization Buffer (HB; Sucrose (320mM), Calcium chloride (5mM), Magnesium Acetate (3mM), Tris (10mM), Ethylenediaminetetraacetic acid (0.1mM), Igepal (0.1%), Phenylmethylsulfonylfluoride (0.1mM), 2-Mercaptoethanol (1mM), UltraPure water) with 5μL RNasin Plus. The homogenate was strained with a 70μm strainer and washed with 1.65mL HB to a final volume of 2.65mL. 2.65mL of Gradient medium (Calcium chloride (5mM), Optiprep (50%), Magnesium Acetate (3mM), Tris (10mM), Phenylmethylsulfonylfluoride (0.1mM), 2-Mercaptoethanol (1mM)) was added (Vf = 5.3mL). To isolate the nuclei, first an Optiprep Diluent medium (ODM; Potassium chloride (150mM), Magnesium chloride (30mM), Tris (60mM), Sucrose (250mM)) was prepared. The sample was added to a 4mL 29% cushion (Optiprep (29%), ODM) using a P1000 pipette, and the weight adjusted with HB. The sample was centrifuged in a SW41Ti rotor at 7,700 rpm for 30 minutes at 4°C. The supernatant was removed with a plastic Pasteur pipette, followed by removal of the lower supernatant with P200 pipette. Nuclei were resuspended in 200μL of resuspension buffer (10xPBS (1x), BSA (1%), RNasin Plus (0.2U μl-1)), transferred to a new tube, washed again with 100-200μL resuspension buffer, and pooled with the previous solution. Clumps were disrupted by pipetting with P200 pipette, then filtered through a Falcon tube with 0.35μm strainer. 9μL of sample was mixed with 1μL of propidium iodide (PI) stain, loaded onto a LUNA-FL slide and allowed to settle for 30 s. We viewed nuclei with the LUNA-FL Automated cell counter to check numbers and shape.
Single nucleus sequencing
RNA sequencing was performed using the 10x Genomics Single Cell 3` Reagent Kit (v2) according to manufacturer protocols. cDNA libraries from fresh-frozen nuclei were sequenced on an Illumina HiSeq platform 4000. Table S1 provides sequencing information per sample (for cells and nuclei).
Quantification and Statistical Analysis
Single nucleus analysis
Alignment. Cellranger v2.1.1 was used to demultiplex and align sequencing output to a human reference genome (assembly hg38 build 95). We used a “pre-mRNA” database to align sequencing data to exons as well as introns (10x Genomics). Following alignment, nuclei from one patient sample (RM101.1) were removed due to poor quality (low read and gene count). See Table S1 for sample information. Unfiltered count matrices were used for downstream analysis.
Extraction of microglial nuclei. Data was processed using the Seurat v3.0.2 package (Butler et al., 2018; Stuart et al., 2019) in R v3.6.1. For each patient, the count matrix was filtered to exclude nuclei with fewer than 100 genes. Counts were normalized for library size, scaled by 10,000 and log-transformed using the natural log. FindVariableFeatures was run using a variance-stabilizing transformation (“vst”) to identify the 2,000 most variable genes in each sample. Data from the 4 patients was then integrated using Seurat’s FindIntegrationAnchors with default parameters, and IntegrateData using 40 principal components (PCs). The dataset consisted of 37,060 nuclei, with a mean UMI count of 4,305 counts per nucleus, and 1,791 genes per nucleus. Integrated data was scaled (default Seurat parameters). We ran a Principal Components Analysis (PCA), then calculated Uniform Manifold Approximation and Projection (UMAP) embeddings using 40 PCs. We identified clusters using Seurat’s FindNeighbors and FindClusters functions, again using 40 PCs. Based on abundance of known celltype markers, we assigned each cluster to a cell type. We identified 7 main cell types in 37,060 nuclei: oligodendrocytes (ODC, 34.0%), excitatory neurons (27.0%), interneurons (11.3%), oligodendrocyte precursors (OPC 9,4%), microglia (11.3%), astrocytes (6.0%), and endothelial cells (1.1%). Figures S1A and S1B show UMAP embeddings for all nuclei, colored by cell type, and selected markers for each cell type, respectively.
Figure S1C, highlighting known microglial markers (P2RY12, CSF1R, DOCK8 and CX3CR1) as well as markers for other celltypes (MBP for oligodendrocytes, GRIA1 for neurons, AQP4 for astrocytes and CLDN5 for endothelial nuclei), confirms that we extracted only microglial clusters from the original data.
Microglial nuclei were then isolated and reclustered. We identified 3,721 microglia (expressing MEF2A, P2RY12, CX3CR1, CSF1R), a macrophage cluster (enriched for CD163 and MRC1, 67 nuclei), a neutrophil cluster (72 nuclei), and a cluster containing microglial as well as astrocytic markers (marked by GFAP, 68 nuclei). The neutrophil and ambiguous clusters were discarded, leaving only microglia and brain macrophages for downstream analysis (Figure S1D). Cluster markers are provided in Table S1.
Pre-processing of microglial nuclei per patient. Microglia from each patient sample were analyzed individually as described for all cell types above, with the following modifications: raw counts were filtered to remove genes and counts that were ± 3 standard deviations away from the median value. After normalization, doublets were identified using DoubletFinder v2.0.2 (McGinnis et al., 2019) using 40 PCs, assuming a 7.5% doublet rate. Following removal of doublets, filtering and Seurat normalization were performed again. Data from patients was then integrated and clusters were identified as above. We discarded small clusters than contained markers for microglia as well as other cell types. After pre-processing, 3,927 nuclei remained, with a mean UMI count of 1,328 and a mean gene count of 879 genes per nucleus.
Single cell analysis
Full details of single cell processing are available in Mancuso et al. (2019). Count data was obtained by aligning raw Cellranger data to both introns and exons using a pre-mRNA database, as for the nuclei. Only cells from the four patients included in the single nucleus study were used here.
Comparisons of single cells and single nuclei
Cluster analysis
In order to identify microglial cell states in the nuclei data we calculated gene markers for each cluster using Seurat’s FindMarkers function, selecting only markers with a positive fold change. Gene markers for cell clusters were extracted from the original Mancuso et al. (2019) study. Markers for nuclei and cells are available in Table S1. For the analysis, we kept the top 40 significant markers (padj < 0.05) based on log fold change for the nuclear clusters and cellular clusters. For each nucleus, we calculated the mean abundance levels of each cell cluster marker set against the aggregated abundance of random control gene sets, using Seurat’s AddModuleScore function. This gave us the MS40 score for each cell marker set (Figure S1E). We performed two-sided Fisher’s Exact tests with Benjamini Hochberg corrections to determine the overlap of cell cluster markers with nuclear cluster markers (selecting the top 40 markers for each set), using the union of all genes in the cell and nuclei datasets as a background (padj < 0.05 was considered significant). Our nuclei were able to recover a cytokine response cluster (CRM), marked by CCL3, CCL4, and an activation-like cluster, equivalent to the “in vitro microglia” identified in the original study (original markers included APOC1, GPNMB, SPP1, APOE). Homeostatic markers appeared ubiquitously through-out the nuclei dataset, and we were not able to distinguish a reduction of these markers in the activation-like response cluster, as we would expect from transcriptomic profiling of microglia in mice (Keren-Shaul et al., 2017; Sala Frigerio et al., 2019). Finally, the CAM (macrophage) cluster (CD163, MRC1), separated out from the bulk of the microglia, and was easily-recognizable based on its MS40 score. Cluster markers are provided in Table S1.
In order to quantify the differences between cells and nuclei in more detail, we examined the overlap of the top 40 markers between nuclei clusters and cell clusters (Figure S1F). The cell macrophage (CAM) and cell cytokine (CRM) clusters showed the largest overlaps with Nuc1 and Nuc5 (27 and 24 of 40 markers, respectively). Other clusters only showed overlaps of between 1 and 5 genes. Cluster Nuc3 showed similar overlaps between “in vitro 1” and “in vitro 2” (5 genes). Cluster Nuc0 showed an overlap of 5 genes with “in vivo HM,” and cluster Nuc2 showed an overlap of 2 genes with “in vivo HM.” Cluster Nuc4 showed similarities with the “in vitro 2” cluster, suggesting it could be a cluster of activation, however all 5 overlapping genes were mitochondrial genes. Cluster Nuc3 markers RPS12, TPT1, FTL, RPS18 and EEF1A1 also appeared as markers of “in vitro 2.”
We performed similar analyses using more markers, however we found that introducing more markers resulted in nuclei markers overlapping with more than one cell cluster. We also noticed that introducing more markers resulted in overlaps between markers of the cellular clusters with each other. Selecting 40 markers allowed us to align cellular and nuclear clusters in an almost one-to-one fashion (see Figure S1F).
Differential Abundance
We discarded all non-microglial clusters (brain macrophages, neutrophils), leaving 3,721 nuclei and 14,435 cells. Differential abundance analysis was performed with the Seurat package, using a two-sided Wilcoxon rank sum test, with a Bonferroni correction for multiple testing. Genes with padj < 0.05 and fold change > |2| were considered significant. As Seurat applies a pseudocount of +1 to data before calculating natural log fold changes, a fold change of 2 corresponds to a log fold change of 0.63. Log fold changes calculated by Seurat were used for further analysis in gene set enrichment analysis.
Scatterplots
We calculated the mean of the normalized abundance levels for cells and for nuclei, and log-transformed these values.
Assessment of nuclear-enriched or cell-enriched gene sets in public scRNA-Seq and snRNA-Seq datasets
We followed the methodology described in Skene et al. (2018): genes that were significantly more abundant in nuclei or more abundant in cells (see Differential abundance methodology above) were used, creating two gene sets. 8 public datasets (see Public datasets below) were reduced to contain six major cell types: pyramidal neurons, interneurons, astrocytes, interneurons, microglia and oligodendrocyte precursors. Within each dataset, for each gene in our gene sets, we calculated a celltype specificity score using the EWCE R package (Github version committed July 29, 2019; Skene and Grant, 2016). For each pair of datasets, X and Y, we subtracted the mean microglial specificity score of Y from X. We then calculated the same scores for 10,000 random gene sets: the probability and z-score for the difference in specificity for the dendritic genes is calculated using these. Finally, the depletion z-score for each gene set was equal to: (mean subtracted microglial specificity score – bootstrapped mean) / (bootstrapped standard deviation). A large positive z-score thus indicated that the gene set was depleted in microglia of dataset Y relative to dataset X. Benjamini-Hochberg multiple testing corrections were applied.
Public datasets
For the Karolinska Institutet (KI) dataset (Skene et al., 2018), we used S1 pyramidal neurons. For the Zeisel 2018 dataset (Zeisel et al., 2018) we used all ACTE∗ cells as astrocytes, TEGLU∗ as pyramidal neurons, TEINH∗ as interneurons, OPC as oligodendrocyte precursors and MGL∗ as microglia. For the Saunders dataset (Saunders et al., 2018), we used all Neuron.Slc17a7 celltypes from the frontal cortex (FC), hippocampus (HC) or posterior cortex (PC) as pyramidal neurons; all Neuron.Gad1Gad2 cell types from FC, HC or PC as interneurons; Polydendrocye as OPCs; Astrocyte as astrocytes, and Microglia as microglia. The Lake datasets both came from a single publication (Lake et al., 2018) which had data from frontal cortex, visual cortex and cerebellum. The cerebellum data was not used here. Data from frontal and visual cortices were analyzed separately. All other datasets - Dronc Human (Habib et al., 2017), Dronc Mouse (Habib et al., 2017), Allen Institute for Brain Science (AIBS) (Hodge et al., 2019), Tasic (Tasic et al., 2016) and Habib (Habib et al., 2016) – were used as described previously (Skene et al., 2018). Table S1 lists all public datasets used. An R package is available for the analysis at https://github.com/NathanSkene/MicroglialDepletion.
Functional analysis
We performed Gene Set Enrichment Analysis (GSEA) using the R package fgsea v1.8.0 (Sergushichev, 2016), using default parameters. Gene sets were mapped against a list of genes ranked according to fold change between cellular abundance and nuclear abundance. Gene ontology (GO) sets were obtained from MSigDB (Liberzon et al., 2011; Subramanian et al., 2005). Other gene sets were obtained from previous studies (see Table S1). padj < 0.05 (Benjamini-Hochberg correction) was considered significant.
Clustering of gene ontology terms
GSEA of GO terms resulted in many functional categories with overlapping genes. In order to reduce this redundancy, the top and bottom 100 GO terms according to normalized enrichment score (with padj < 0.05) were clustered as follows: a Jaccard index (the size of the intersection of the two datasets, divided by the size of the union of the two datasets, multiplied by 100) of the overlapping genes was calculated between each significant GO set. The resulting similarity matrix was converted to a dissimilarity matrix, and hierarchical clustering was performed on the matrix. We selected a k value of 16 to group the GO terms based on the hierarchical clustering (see Table S1). Gene sets were merged, and each new “super” GO was assigned an annotation manually. GSEA analysis was performed on these super-GO gene sets as described above.
Gene sets from previous studies
We extracted gene sets from previous studies for this analysis. A full list of gene sets is available in Table S1. Where data was selected from mouse datasets, we converted the mouse gene to its human ortholog using R’s BioMaRt package v2.40.5 (Drost and Paszkowski, 2017), selecting only orthologs that displayed 1-to-1 orthology. For the ARM gene set we selected the top 200 ARM genes based on log fold change (Sala Frigerio et al., 2019). For the Gerrits human gene set, we took the union of all genes that showed significant differential abundance between cells and nuclei (microglia) from donor 1 and donor 2 (Gerrits et al., 2019). For the LPS gene set, we took the union of all genes significantly upregulated in LPS in cells and in nuclei (microglia) from the Gerrits study (Gerrits et al., 2019). For GSEA of coding sequence (CDS) length we took the 200 genes with the longest CDS and the 200 genes with the shortest CDS from Ensembl (Zerbino et al., 2018). For GSEA of GC content, we took the top 200 genes with the highest GC content and the top 200 genes with the lowest GC content from Ensembl (Zerbino et al., 2018). Ensembl was accessed using BioMartR v0.92 (Drost and Paszkowski, 2017).
Downsampling of cell counts
To match cell sequencing depth to nucleus sequencing depth (see Figure S2B), we sampled without replacement the number of reads in the cells by a proportion of 0.32, using the downsampleMatrix function of the DropletUtils R package v1.4.3 (Griffiths et al., 2018; Lun et al., 2019). This resulted in 1,304 mean reads per cell, compared with the original count of 3,979 reads per cell.
Downsampling of cells
To ensure the high number of cells relative to nuclei did not affect the result (Figure S2C), we randomly sampled 3,927 cells from the count matrix.
Definition of ambient RNA profile in nuclei
We extracted nuclei with less than 700 counts from the original unfiltered raw count matrix of all cell types (resulting in 2,414 nuclei with a mean read depth of 590), and summed the gene counts, under the assumption that these were empty drops rather than nuclei. We took the top 300 genes to represent the ambient RNA profile.
GSEA analysis of ARM gene length
From the Sala Frigerio et al. (2019) study, we calculated the log fold change between ARM cells and homeostatic cells for each gene (Seurat’s FindMarkers function). For each gene, we extracted the CDS length from Ensembl, using BioMartR as described above. We took the 200 genes with the longest CDS and the 200 genes with the shortest CDS as input into a GSEA analysis against the ARM log fold change.
Analysis of Gerrits data
We downloaded the human data from GEO (accession number GSE135618). Each sample (fresh nuclei, frozen nuclei, and cells, each from 2 donors) was processed to remove genes that were found in less than 10 cells, and cells that contained less that 100 genes or more than 2,000 counts (nuclei) or more than 3,500 counts (cells), and cells that contained more than 20% mitochondrial reads. Counts were normalized for library size, scaled by 10,000 and log-transformed. Data from the 2 patients was then merged to create a frozen nuclei dataset, a fresh nuclei dataset, and a cellular dataset. FindVariableFeatures was run using a variance-stabilizing transformation (“vst”) to identify the 2,000 most variable genes in each dataset. We ran a Principal Components Analysis (PCA), then calculated Uniform Manifold Approximation and Projection (UMAP) embeddings using 12 PCs. We identified clusters using Seurat’s FindNeighbors and FindClusters functions, again using 12 PCs. Based on abundance of known celltype markers, we assigned each cluster to a cell type. In the frozen nuclei dataset, microglial clusters (clusters 3,5 and 6 in Figures S3A and S3B) were identified using known markers including P2RY12, CSF1R, CX3CR1 (resulting in 2,659 microglial nuclei), and extracted for downstream analysis. In the fresh nuclei and cells, examination of these markers in the clustering confirmed that all cells were microglial. Scatterplots were then then generated from the normalized data of these datasets.
Acknowledgments
Work in the De Strooper laboratory was supported by the European Union (ERC 834682, CELLPHASE_AD), the Fonds voor Wetenschappelijk Onderzoek (FWO), KU Leuven, VIB, UK-DRI (Medical Research Council, ARUK, and Alzheimer Society), a Methusalem grant from KU Leuven and the Flemish Government, the “Geneeskundige Stichting Koningin Elisabeth,” Opening the Future campaign of the Leuven Universitair Fonds (LUF), the Belgian Alzheimer Research Foundation (SAO-FRA), and the Alzheimer's Association USA. B.d.S. is holder of the Bax-Vanluffelen Chair for Alzheimer’s Disease. Cell sorting was performed at the KU Leuven FACS core facility, and sequencing was carried out by the VIB Nucleomics Core. R.M. is the recipient of a postdoctoral fellowship from the Alzheimer's Association, USA. P.M.M. acknowledges generous support from the Edmond J. Safra Foundation and Lily Safra, the NIHR Biomedical Research Centre at Imperial College, and the UK Dementia Research Institute. C.S.F. and N.G.S. acknowledge support from the UK Dementia Research Institute. N.F. is recipient of a PhD fellowship from Fonds voor Wetenschappelijk Onderzoek (FWO). T.T. is a senior clinical investigator of FWO Flanders (FWO 1830717N).
Author Contributions
Conceptualization: B.d.S., M.F., C.S.F., and R.M.; surgery and extraction of patient tissue samples: T.T.; investigation: R.M., L.W., and S.P.; formal analysis: N.T., Y.F., N.F., M.F., N.G.S., and P.M.M.; writing – original draft: N.T., C.S.F., and M.F.; writing – review & editing: B.d.S., R.M., N.G.S., and P.M.M.; supervision: M.F. and B.d.S.
Declaration of Interests
P.M.M. has been reimbursed for service on a Scientific Advisory Board to Ipsen Pharmaceuticals. He has received consultancy fees from Roche, Adelphi Communications, Celgene, Neurodiem, and Medscape. He has received honoraria or speakers’ fees from Novartis and Biogen and has received research or educational funds from Biogen, Novartis, and GlaxoSmithKline.
Published: September 29, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108189.
Contributor Information
Bart de Strooper, Email: bart.destrooper@kuleuven.vib.be.
Mark Fiers, Email: mark.fiers@kuleuven.vib.be.
Supplemental Information
References
- Bahar Halpern K., Caspi I., Lemze D., Levy M., Landen S., Elinav E., Ulitsky I., Itzkovitz S. Nuclear Retention of mRNA in Mammalian Tissues. Cell Rep. 2015;13:2653–2662. doi: 10.1016/j.celrep.2015.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken T.E., Hodge R.D., Miller J.A., Yao Z., Nguyen T.N., Aevermann B., Barkan E., Bertagnolli D., Casper T., Dee N. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 2018;13:e0209648. doi: 10.1371/journal.pone.0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-T., Lu A., Craessaerts K., Pavie B., Sala Frigerio C., Corthout N., Qian X., Laláková J., Kühnemund M., Voytyuk I. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell. 2020;182:976–991.e19. doi: 10.1016/j.cell.2020.06.038. [DOI] [PubMed] [Google Scholar]
- Del-Aguila J.L., Li Z., Dube U., Mihindukulasuriya K.A., Budde J.P., Fernandez M.V., Ibanez L., Bradley J., Wang F., Bergmann K. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res. Ther. 2019;11:71. doi: 10.1186/s13195-019-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J., Adiconis X., Simmons S.K., Kowalczyk M.S., Hession C.C., Marjanovic N.D., Hughes T.K., Wadsworth M.H., Burks T., Nguyen L.T. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020;38:737–746. doi: 10.1038/s41587-020-0465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost H.-G., Paszkowski J. Biomartr: genomic data retrieval with R. Bioinformatics. 2017;33:1216–1217. doi: 10.1093/bioinformatics/btw821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efthymiou A.G., Goate A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017;12:43. doi: 10.1186/s13024-017-0184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca M.I., Chu S.-H., Hernandez M.X., Fang M.J., Modarresi L., Selvan P., MacGregor G.R., Tenner A.J. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflammation. 2017;14:48. doi: 10.1186/s12974-017-0814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10x Genomics. Creating a Reference Package with cellranger mkref -Software -Single Cell Gene Expression -Official 10x Genomics Support. https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references#premrna.
- Gerrits E., Heng Y., Boddeke E.W.G.M., Eggen B.J.L. Transcriptional profiling of microglia; current state of the art and future perspectives. Glia. 2019;68:740–755. doi: 10.1002/glia.23767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D., Skola D., Coufal N.G., Holtman I.R., Schlachetzki J.C.M., Sajti E., Jaeger B.N., O’Connor C., Fitzpatrick C., Pasillas M.P. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017;356:eaal3222. doi: 10.1126/science.aal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths J.A., Richard A.C., Bach K., Lun A.T.L., Marioni J.C. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun. 2018;9:2667. doi: 10.1038/s41467-018-05083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A., Chew G., Ouyang J.F., Sun G., Choo X.Y., McLean C., Simmons R.K., Buckberry S., Vargas-Landin D.B., Poppe D. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019;22:2087–2097. doi: 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- Habib N., Li Y., Heidenreich M., Swiech L., Avraham-Davidi I., Trombetta J.J., Hession C., Zhang F., Regev A. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353:925–928. doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N., Avraham-Davidi I., Basu A., Burks T., Shekhar K., Hofree M., Choudhury S.R., Aguet F., Gelfand E., Ardlie K. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods. 2017;14:955–958. doi: 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A.-S., Donlin-Asp P.G., Leitch B., Herzog E., Schuman E.M. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science. 2019;364:eaau3644. doi: 10.1126/science.aau3644. [DOI] [PubMed] [Google Scholar]
- Hasselmann J., Coburn M.A., England W., Figueroa Velez D.X., Kiani Shabestari S., Tu C.H., McQuade A., Kolahdouzan M., Echeverria K., Claes C. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron. 2019;103:1016–1033.e10. doi: 10.1016/j.neuron.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge R.D., Bakken T.E., Miller J.A., Smith K.A., Barkan E.R., Graybuck L.T., Close J.L., Long B., Johansen N., Penn O. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019;573:61–68. doi: 10.1038/s41586-019-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen I.E., Savage J.E., Watanabe K., Bryois J., Williams D.M., Steinberg S., Sealock J., Karlsson I.K., Hägg S., Athanasiu L. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019;51:404–413. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O’Loughlin E., Xu Y., Fanek Z. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47:566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle B.W., Grenier-Boley B., Sims R., Bis J.C., Damotte V., Naj A.C., Boland A., Vronskaya M., van der Lee S.J., Amlie-Wolf A. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019;51:414–430. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake B.B., Ai R., Kaeser G.E., Salathia N.S., Yung Y.C., Liu R., Wildberg A., Gao D., Fung H.-L., Chen S. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake B.B., Codeluppi S., Yung Y.C., Gao D., Chun J., Kharchenko P.V., Linnarsson S., Zhang K. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 2017;7:6031. doi: 10.1038/s41598-017-04426-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake B.B., Chen S., Sos B.C., Fan J., Kaeser G.E., Yung Y.C., Duong T.E., Gao D., Chun J., Kharchenko P.V., Zhang K. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018;36:70–80. doi: 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C., DeStafano A.L., Bis J.C., Beecham G.W., Grenier-Boley B. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T.L., Riesenfeld S., Andrews T., Dao T.P., Gomes T., Marioni J.C., participants in the 1st Human Cell Atlas Jamboree EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019;20:63. doi: 10.1186/s13059-019-1662-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso R., Van Den Daele J., Fattorelli N., Wolfs L., Balusu S., Burton O., Liston A., Sierksma A., Fourne Y., Poovathingal S. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 2019;22:2111–2116. doi: 10.1038/s41593-019-0525-x. [DOI] [PubMed] [Google Scholar]
- Maniatis S., Äijö T., Vickovic S., Braine C., Kang K., Mollbrink A., Fagegaltier D., Saiz-Castro G., Cuevas M., Watters A. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89–93. doi: 10.1126/science.aav9776. [DOI] [PubMed] [Google Scholar]
- Marioni R.E., Harris S.E., Zhang Q., McRae A.F., Hagenaars S.P., Hill W.D., Davies G., Ritchie C.W., Gale C.R., Starr J.M. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry. 2018;8:99. doi: 10.1038/s41398-018-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J.Z., Menon M., He L., Abdurrob F., Jiang X. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer P.L., Itagaki S., Tago H., McGeer E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B., Zappia L., Oshlack A. Gene length and detection bias in single cell RNA sequencing protocols. F1000Res. 2017;6:595. doi: 10.12688/f1000research.11290.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C., Wolfs L., Fattorelli N., Thrupp N., Voytyuk I., Schmidt I., Mancuso R., Chen W.-T., Woodbury M.E., Srivastava G. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019;27:1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A., Macosko E.Z., Wysoker A., Goldman M., Krienen F.M., de Rivera H., Bien E., Baum M., Bortolin L., Wang S. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell. 2018;174:1015–1030.e16. doi: 10.1016/j.cell.2018.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. 2016 doi: 10.1101/060012. [DOI] [Google Scholar]
- Skene N.G., Grant S.G.N. Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front. Neurosci. 2016;10:16. doi: 10.3389/fnins.2016.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene N.G., Bryois J., Bakken T.E., Breen G., Crowley J.J., Gaspar H.A., Giusti-Rodriguez P., Hodge R.D., Miller J.A., Muñoz-Manchado A.B. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018;50:825–833. doi: 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl P.L., Salmén F., Vickovic S., Lundmark A., Navarro J.F., Magnusson J., Giacomello S., Asp M., Westholm J.O., Huss M. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B., Menon V., Nguyen T.N., Kim T.K., Jarsky T., Yao Z., Levi B., Gray L.T., Sorensen S.A., Dolbeare T. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 2016;19:335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A., Hochgerner H., Lönnerberg P., Johnsson A., Memic F., van der Zwan J., Häring M., Braun E., Borm L.E., La Manno G. Molecular Architecture of the Mouse Nervous System. Cell. 2018;174:999–1014.e22. doi: 10.1016/j.cell.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Song W.M., Andhey P.S., Swain A., Levy T., Miller K.R., Poliani P.L., Cominelli M., Grover S., Gilfillan S. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020;26:131–142. doi: 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data from single microglial cells and nuclei are available at GEO: GSE137444 for single cells, and GEO: GSE153807 for single nuclei).
Analysis of previous datasets (see Figure 1C) was performed using the EWCE package (Skene and Grant, 2016) and the MicroglialDepletion package (https://github.com/NathanSkene/MicroglialDepletion).