

Abstract
Hypothalamic neurons expressing gonadotropin‐releasing hormone (GnRH), the “master molecule” regulating reproduction and fertility, migrate from their birthplace in the nose to their destination using a system of guidance cues, which include the semaphorins and their receptors, the neuropilins and plexins, among others. Here, we show that selectively deleting neuropilin‐1 in new GnRH neurons enhances their survival and migration, resulting in excess neurons in the hypothalamus and in their unusual accumulation in the accessory olfactory bulb, as well as an acceleration of mature patterns of activity. In female mice, these alterations result in early prepubertal weight gain, premature attraction to male odors, and precocious puberty. Our findings suggest that rather than being influenced by peripheral energy state, GnRH neurons themselves, through neuropilin–semaphorin signaling, might engineer the timing of puberty by regulating peripheral adiposity and behavioral switches, thus acting as a bridge between the reproductive and metabolic axes.
Keywords: chemotropic factors, energy homeostasis, hypothalamus, puberty onset, sexual behavior
Subject Categories: Neuroscience
A specific semaphorin guidance cue receptor, neuropilin‐1, promotes survival and migration of hypothalamic neurons expressing gonadotropin‐releasing hormone, affecting prepubertal body growth, onset of puberty, and sensitivity to male olfactory stimuli in female mice.
Introduction
Two of the major imperatives of living beings are the maintenance of energy homeostasis and the transmission of genetic material, i.e. the production of young in animals with sexual reproduction. In mammals, both processes are controlled at the level of the brain, albeit by different hypothalamic circuits.
Fertility, under the control of the hypothalamic–pituitary–gonadal (HPG) axis, is orchestrated by a small population of neuroendocrine neurons producing gonadotropin‐releasing hormone (GnRH). The release of GnRH into the hypothalamic–pituitary portal blood circulation drives gonadotropin secretion by the pituitary. The gonadotropins, luteinizing hormone (LH), and follicle stimulating hormone (FSH), in turn, act on the gonads to regulate sex steroid synthesis, gametogenesis, and the onset of puberty in both sexes (Boehm et al, 2015; Prevot, 2015; Howard & Dunkel, 2019).
While puberty consists of the permanent activation of the HPG axis and the commencement of adult reproductive function, the process is controlled by a complex array of genetic and environmental determinants, among which the acceleration of growth during the prepubertal period is thought to be a key permissive factor (Parent et al, 2003; Abreu & Kaiser, 2016; Howard & Dunkel, 2019). In patients with hypogonadotropic hypogonadism, a deficit in GnRH neuronal migration and function leads to an absence of puberty onset (Boehm et al, 2015). However, at the other end of the pathophysiological spectrum, little is known regarding the cellular and molecular mechanisms underlying precocious puberty.
GnRH‐secreting neurons originate from both the nasal placode and the neural crest during embryonic development and migrate to the forebrain and hypothalamus along olfactory/terminal nerves (Schwanzel‐Fukuda & Pfaff, 1989; Wray et al, 1989; Taroc et al, 2017). The complex developmental events leading to correct GnRH neuronal migration and secretion are tightly regulated by the specific spatiotemporal expression patterns of growth factors, adhesion molecules, and diffusible guidance cues that are either attractive or repulsive (Giacobini, 2015). The semaphorins constitute one of the largest families of phylogenetically conserved guidance cues, known to regulate multiple processes crucial for neuronal network formation (Tamagnone & Comoglio, 2004; Van Battum et al, 2015). The repulsive guidance cue semaphorin 3A (Sema3A) and its receptor, neuropilin‐1 (Nrp1), are expressed in a complementary manner in olfactory sensory neurons and are involved in the spatial encoding of sensory information in the olfactory bulb (Imai et al, 2009). Olfactory axons expressing Nrp1 also form the scaffold along which GnRH neurons migrate into the brain (Cariboni et al, 2011b; Hanchate et al, 2012). In keeping with their sites of expression, recent studies by us and others have shown that, in humans, mutations in the Sema3A:Nrp1 signaling pathway lead to dysfunctions in olfaction and fertility (Hanchate et al, 2012; Marcos et al, 2017).
Here, we studied the role of Nrp1 expression in GnRH neurons in mice. We demonstrate that Nrp1 expression in GnRH neurons is required to control the size of the GnRH neuronal population generated during embryogenesis and their migration to their proper destinations in the brain in the correct proportions. We also show that the knockout of Nrp1 specifically in GnRH neurons results in the accumulation of an abnormally high number of neurons in the accessory olfactory bulb (AOB) and the forebrain/hypothalamus, early attraction to odors of the opposite sex, precocious puberty onset, and, surprisingly, overweight, suggesting an involvement of the GnRH system in energy metabolism. Our data raise the intriguing possibility that, in patients with central precocious puberty, rather than body weight influencing the timing of puberty, GnRH neurons may themselves play a pivotal role in coordinating both peripubertal weight gain and the activation of the HPG axis, in addition to early sexual behavior.
Results
Selective deletion of Nrp1 expression in GnRH cells in mice in vivo
To investigate the role of Nrp1 receptor signaling in GnRH neurons in the mouse, we specifically knocked out Nrp1 expression in GnRH neurons, by crossing Nrp1 loxP/loxP mice (Gu et al, 2003) with a Gnrh::cre line (Yoon et al, 2005) expressing the Cre recombinase under the control of the Gnrh promoter. The resulting Gnrh::cre; Nrp1 loxP/loxP (hereafter termed “mutant”) and Nrp1 loxP/loxP (hereafter termed “control”) mice were viable and born at Mendelian frequencies. Triple transgenic mice were also produced by crossing them with Gnrh::gfp mice (Spergel et al, 1999), which express green fluorescent protein (GFP) under the control of the Gnrh promoter (Fig 1A), to visualize and isolate these neurons. We first investigated the expression of Nrp1 and the plexins, preferential co‐receptors of Nrp1, and the signal‐transducing subunits in Sema3A signaling (Takahashi et al, 1999), in GFP‐identified GnRH neurons isolated by fluorescence‐activated cell sorting (FACS) from the preoptic region of mutant and control mice at P0 (Fig 1B and C). FACS‐sorted GFP‐positive cells were 1,000–4,000 times enriched in the Gnrh transcript as compared to GFP‐negative cells (Mann–Whitney U‐test, U = 0, P = 0.03, n = 4; Fig 1C). Real‐time quantitative PCR analyses showed a 74.7% decrease in Nrp1 mRNA levels in GnRH neurons from Gnrh::gfp mutant mice as compared to Gnrh::gfp control mice (unpaired t‐test, t (9) = 3.851, P = 0.004, n = 5–6), but no significant change in Nrp2 (Mann–Whitney U‐test, U = 6, P = 0.13, n = 5–6; Fig 1D). This was associated with a 66.7% decrease in plexin‐A1 mRNA levels (unpaired t‐test, t (9) = 3.09, P = 0.01, n = 5–6), but no change in plexin‐A4 expression (unpaired t‐test, t (9) = 0.40, P = 0.70, n = 5–6) (Fig 1D). Nrp1 protein expression was also investigated during embryonic development. GnRH‐immunoreactive neurons in control embryos expressed Nrp1 protein (red immunolabeling) by embryonic day 14.5 (E14.5) (Fig 1E). In contrast, littermates with a GnRH neuron‐specific Nrp1 knockout did not show any detectable Nrp1 immunolabeling in GnRH neurons, but displayed Nrp1 in GnRH‐negative fibers, such as the vomeronasal/terminal nerves that support GnRH neurons in their migration (Fig 1E). These observations, together with the fact that in the specific Gnrh::cre mouse model we used, the expression of the transgene is very limited in ectopic neuronal populations during embryogenesis (Hoffmann et al, 2019), confirm the genetic deletion of Nrp1 in GnRH neurons in mutant mice. In addition, Nrp1 expression remains intact in the non‐GnRH hypophysiotropic systems of mutant mice (Giacobini et al, 2014), indicating that any neuroendocrine actions of Nrp1 deletion are mediated by GnRH neurons exclusively.
Figure 1. Neuropilin‐1 expression is selectively suppressed in GnRH neurons in Gnrh::cre; Nrp1 loxP/loxP mice in vivo .
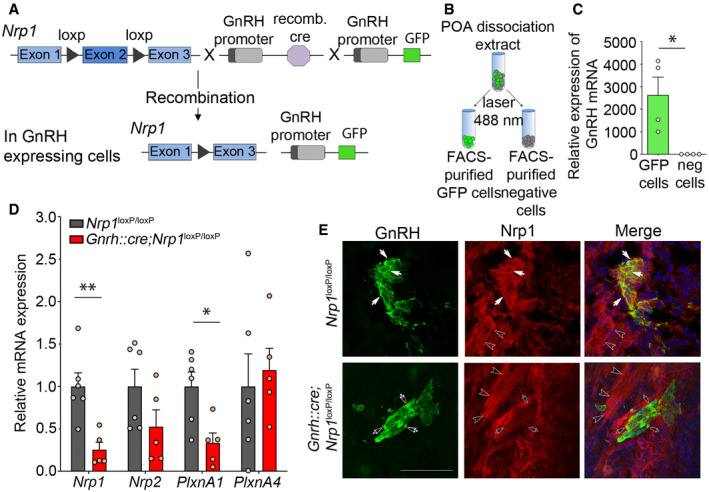
- Genetic strategy to selectively knock out Nrp1 in GnRH‐expressing cells, using Nrp1 loxP/loxP mice crossed with Gnrh::cre mice. These mice were also crossed with Gnrh::gfp mice expressing green fluorescent protein under the control of the GnRH promoter in order to generate triple transgenic mice.
- Isolation of GFP‐positive cells by FACS: schematic diagram showing the selection of the GFP‐positive population (green).
- Relative mRNA expression from real‐time PCR analysis of the GnRH transcript in GFP‐positive cells, in comparison with GFP‐negative cells. The GnRH transcript appears to be selectively expressed in GFP‐positive cells. Mann–Whitney U‐test, n = 4 mice from 2 litters.
- Relative mRNA expression from real‐time PCR analysis of neuropilin‐1 (Nrp1), Nrp2, plexin‐A1 (PlxnA1), and PlxnA4 transcripts in FACS‐isolated GFP‐positive GnRH neurons from control (Nrp1 loxP/loxP; gray) and mutant (Gnrh::cre; Nrp1 loxP/loxP; red) P0 mice. Unpaired t‐test, n = 5–6 mice per group from 3 litters.
- Representative immunofluorescence images showing GnRH neurons migrating along Nrp1‐immunoreactive vomeronasal/terminal nerve fibers (empty arrowheads) at the nose–brain junction in sagittal slices from E14.5 control and mutant embryos. GnRH neurons (green) themselves express Nrp1 (red, white arrows) in control Nrp1 loxP/loxP mice, whereas Nrp1 is undetectable in GnRH neurons from mutant Gnrh::cre; Nrp1 loxP/loxP littermates (black arrows). Scale bar: 50 μm.
Selective deletion of Nrp1 in GnRH neurons alters their number and distribution
To investigate the role of Nrp1 expression in GnRH neuron ontogenesis, we used GnRH immunofluorescence to characterize GnRH cell distribution at key stages during embryonic development. In contrast to our previous observations in mutant mice with Nrp1 deletion throughout the body (Hanchate et al, 2012), no alteration of the olfactory sensory or terminal tracts was detected in mutant embryos at any age. GnRH neurons were quantified along the migratory path from the nose to brain at three stages: E12.5, when the majority of GnRH neurons, which become detectable in the vomeronasal epithelium at E11.5, are still located in the nose; E14.5, when around half the GnRH neurons have reached the brain whereas the other half is still located in the nose; and E18.5, when most GnRH neurons have completed their migration and reached their final destination in the ventral forebrain (VFB) (Schwanzel‐Fukuda & Pfaff, 1989; Wray et al, 1989). While the total number of GnRH neurons was equivalent in mutant and control embryos at E12.5 (two‐way ANOVA, developmental period, F (2,22) = 13.31, P = 0.0002; genotype, F (1,22) = 5.63, P = 0.03; interaction, F (2,22) = 2.18, P = 0.14; Fisher's LSD multiple‐comparison test, 1078 ± 201.6 neurons in controls versus 1020 ± 96.72 neurons in mutants, t (22) = 0.36, P = 0.72, n = 4 per group), it was increased by 25.3% in mutant embryos at E14.5 (control: 1353 ± 70 neurons; mutant: 1695 ± 62 neurons, t (22) = 2.51, P = 0.02, n = 5–6) and by 23.8% at E18.5 (control: 1,386 ± 81.35 neurons; mutant: 1,716 ± 103 neurons, t (22) = 2.18, P = 0.04, n = 4–5) (Fig 2A). At E14.5, while the total number of neurons in the nose did not change (two‐way ANOVA, anatomical region, F (1,9) = 309.3, P < 0.001; genotype, F (1,9) = 12.7, P = 0.006; interaction, F (1,9) = 7.5, P = 0.02; Fisher's LSD multiple‐comparison test, control: 325 ± 36 versus mutant: 368 ± 45, t (18) = 0.63; P = 0.78, n = 6 and 5), the number of neurons reaching the brain markedly increased in mutant embryos at the same developmental stage (control: 1,027 ± 53 versus mutant: 1,327 ± 56, t (18) = 4.46; P < 0.001, n = 6 and 5). These excess cells in the brain were located in the ventral forebrain where their number was found to be significantly increased by 41% (431 ± 50 neurons in controls versus 608.80 ± 69.24 neurons in mutants, Fisher's LSD multiple‐comparison test, VFB t (27) = 2.70, P = 0.01; OB; t (27) = 1.87; P = 0.07; two‐way ANOVA, anatomical region, F (2,27) = 22.4, P < 0.0001; genotype, F (1,27) = 9.07, P = 0.006; interaction, F (2,27) = 1.07, P = 0.36, n = 6 and 5) (Fig 2B and C), suggesting that a higher proportion of cells had already migrated further toward their destinations at this embryonic stage in mutants than in control littermates.
Figure 2. Mice lacking neuropilin‐1 expression in GnRH neurons show an increased number of migrating GnRH cells and abnormal GnRH neuronal migration in the accessory olfactory bulb.
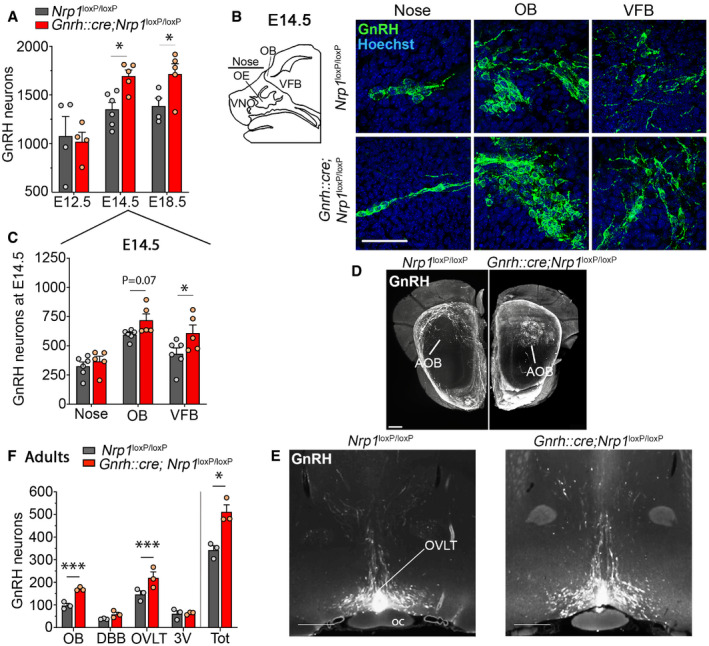
- Quantification of total number of GnRH neurons during embryogenesis, at E12.5, E14.5, and E18.5. More GnRH neurons were found in E14.5 and E18.5 mutant embryos. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 4–6 mouse embryos from at least 3 litters.
- Schematic representation of the areas containing GnRH migrating cells during embryogenesis at E14.5 and representative immunofluorescence showing migrating GnRH neurons at the level of the nose, the olfactory bulb (OB), and the ventral forebrain (VFB) in control and mutant embryos. Scale bar: 50 μm. VNO, vomeronasal organ; OE, olfactory epithelium; OB, olfactory bulbs; VFB, ventral forebrain.
- Quantitative analysis showing distribution of GnRH neurons in the nose, OB, and VFB of mutant and control embryos. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 5 and 6 mouse embryos for mutants and controls, respectively, from at least 3 litters.
- Representative image of cleared brains and immunolabeling for GnRH at the level of the olfactory bulbs, in a control (left panel) and a mutant (right panel) postnatal brain. Knockout animals show an accumulation of GnRH neurons in the accessory olfactory bulb (circle formed by the GnRH labeling, right panel). AOB, accessory olfactory bulb. Scale bar: 300 μm.
- Representative image of cleared brains and immunolabeling for GnRH at the level of the organum vasculosum of the lamina terminalis (OVLT) in the median preoptic area, from a control (left) and a mutant (right) female postnatal brain. Scale bar: 500 μm. Oc, optic chiasma.
- Quantitative analysis of the distribution of GnRH neurons in cleared brains of mutant and control littermates. OB, olfactory bulb; DBB, diagonal band of Broca; 3V, periventricular region of the preoptic area; Tot, total. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 3 adult mice from 3 litters.
We next investigated the distribution of GnRH neurons in the postnatal brain of mutant mice and their control littermates. We used an organic solvent‐based clearing methods adapted from iDISCO (Casoni et al, 2016; Belle et al, 2017) coupled with GnRH immunohistofluorescence on brain sections from adult mice to visualize the distribution of GnRH neurons in the brain. Remarkably, while GnRH neurons with abundant fibers were typically localized in the medial and basal parts of the glomerular layer of the olfactory bulbs in controls (Casoni et al, 2016), GnRH neurons in mutant mice clearly accumulated in the accessory olfactory bulb (AOB) (Fig 2D, Movies EV1 and EV2). In parallel with this abnormally high migration of GnRH neurons into the AOB, the number of GnRH neurons that had reached the forebrain/hypothalamus was higher by > 18% in mutant mice, as shown by cell counts both in 3D rendering (Fig 2E and F) and in conventional immunofluorescence (control both sex: 404.10 ± 13.35 neurons, n = 13; mutant both sex: 472.3 ± 23.81 neurons, n = 13, unpaired t‐test t (24) = 2.50, P = 0.02) (Fig EV1A). This increase in the GnRH neuronal population was largely restricted to the organum vasculosum of the lamina terminalis (transparentized brain: two‐way ANOVA, brain region, F (3,16) = 49.01, P < 0.0001; genotype, F (1,16) = 21.79, P = 0.0003; interaction, F (3,16) = 4.02, P = 0.03; Fisher's LSD multiple‐comparison test, OVLT t (16) = 4.03, P = 0.001, n = 3 per group, Fig 2E; conventional immunofluorescence: two‐way ANOVA, brain region, F (2,72) = 229.7, P < 0.0001; genotype, F (1,72) = 5.54, P = 0.02; interaction, F (2,72) = 3.15 P = 0.05; Fisher's LSD multiple‐comparison test, OVLT, control: 261.4 ± 16.26 neurons; mutant: 316.9 ± 20.89 neurons, t (72) = 3.317, P = 0.001, n = 13 per group, 9 females and 4 males per group, Fig EV1A and B). Similar differences were observed when data from female brains were analyzed alone (two‐way ANOVA, brain region, F (2,48) = 158.9, P < 0.0001; genotype, F (1,48) = 4.04 P = 0.05; interaction, F (2,48) = 2.26, P = 0.12; OVLT, control: 263.1 ± 18.87 neurons; mutant: 321.3 ± 26.55 neurons, t (48) = 2.86, P = 0.006, n = 9 per group). The distribution of GnRH neurons in the forebrain/hypothalamus of adult males (n = 4) did not appear to differ markedly from that in females (Fig EV1A). Together, these results show that alterations of GnRH neuronal number and migration during embryonic development in mutant mice have clear repercussions on their distribution in the postnatal brain.
Figure EV1. Migration of supernumerary GnRH neurons in adulthood in the hypothalamus of mice lacking neuropilin‐1 expression in GnRH neurons.
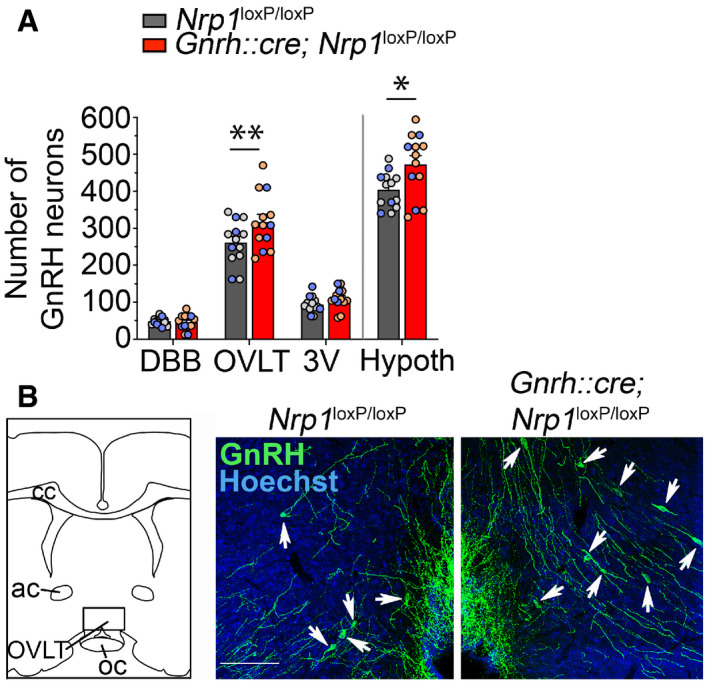
- Total number of GnRH neurons in the forebrain and the hypothalamus (Hypoth) and their regional distribution (DBB: diagonal band of Broca; OVLT: organum vasculosum of the lamina terminalis; 3V: periventricular area of the median preoptic) in Nrp1 loxP/loxP and Gnrh::cre; Nrp1 loxP/loxP littermates using conventional immunofluorescence. Individual males and females used for this analysis are represented by blue and gray/light‐red dots, respectively. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 13 (nine females and four males).
- Representative immunohistofluorescence for GnRH (green) and Hoechst staining (blue) of sections showing the OVLT region (area framed in the schematic) from control (left) and mutant (right) adult mice. More GnRH cells are observed in OVLT sections of mutant mice (white arrow). Ac, anterior commissure; cc, corpus callosum; Scale bar: 100 μm.
Knocking out Nrp1 in GnRH neurons increases their migration and survival
To understand the origin of these supernumerary GnRH neurons in mutant embryos and adult mice, we first took advantage of immortalized GnRH neurons (the GN11 line) (Radovick et al, 1991), which possess the migratory properties of GnRH neurons (Maggi et al, 2000), in addition to expressing both GnRH and Nrp1 (Cariboni et al, 2007). Semaphorin 3A (Sema3A) is a secreted guidance molecule with known Nrp1‐mediated chemorepellent properties, which plays important roles in shaping neural circuits during mammalian embryonic development (Pasterkamp, 2012). In keeping with a chemorepellent action of Sema3A, Transwell assays demonstrated that 100 ng/ml or 250 ng/ml of Sema3A, but not 50 ng/ml, inhibited cell migration by ~50% (Kruskal–Wallis test, SFM versus Sema3A 100 ng/ml, Z = 2.96, P = 0.01, n = 5 and 6; SFM versus Sema3A 250 ng/ml, Z = 2.62, P = 0.035, n = 5 and 6) (Fig 3A and B), an effect that was abolished by pretreatment with neutralizing antibodies to Nrp1 (1/200; SFM versus Sema3A 100 ng/ml + Nrp1Ab, Z = 0.24, P > 0.999, n = 5 and 6 and Sema3A 100 ng/ml versus Sema3A 100 ng/ml + Nrp1Ab, Z = 2.85, P = 0.02, n = 6 per group) (Fig 3A and B). In parallel, two‐well removable inserts were used to assess the motility of GN11 cells in response to Sema3A in the presence or absence of Nrp1‐neutralizing antibodies (Nrp1Ab) in serum‐free culture medium (SFM). Six hours after removing the insert, the area invaded by GN11 cells was significantly lower in the presence of Sema3A at 100 ng/ml (SFM 12.28 ± 0.75 pixels versus Sema3A 100 9.14 ± 0.64 pixels, n = 5 per group; two‐way ANOVA, treatment, F (4,66) = 2.78, P = 0.034; time, F (2,66) = 338.60, P < 0.0001; interaction, F (8,66) = 1.49, P = 0.18, Fisher's LSD multiple‐comparison test, t (66) = 3.33, P = 0.001) and at 250 ng/ml (9.18 ± 1.23 pixels, n = 6, SFM versus Sema3A 250, t (66) = 3.43, P = 0.001). This effect was abolished by the addition of Nrp1Ab (1/200) to the medium (10.91 ± 0.53 pixels, n = 6, Sema3A 100 ng/ml versus Sema3A 100 ng/ml+Nrp1Ab, t (66) = 1.96, P = 0.05). These in vitro results suggest that Nrp1, acting as the receptor for guidance cues, regulates GnRH cell migration.
Figure 3. Neuropilin‐1 signaling in GnRH neurons controls their survival and migration.
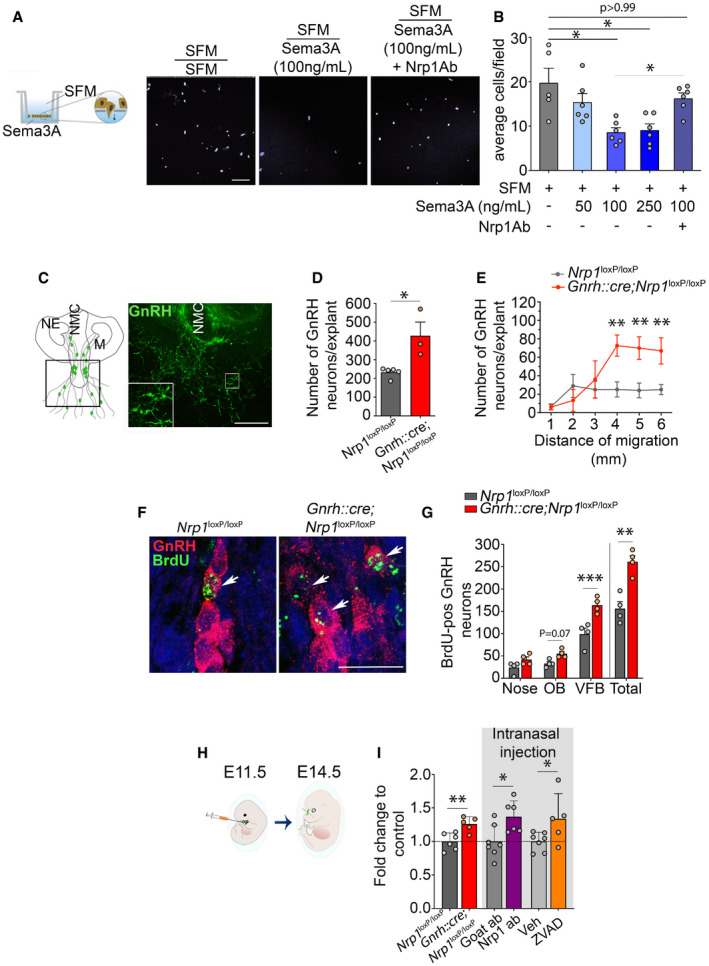
- Schematic representation of the Transwell assay used to assess the effect of Sema3A, which was placed in the lower chamber, on GN11 cell migration and representative microphotograph of cell nuclei stained with Hoechst (gray) in the lower part of the Transwell membrane after 12 h incubation in SFM (left panel), SFM with 100 ng/ml Sema3A (middle panel), and SFM with 100 ng/ml Sema3A + 1 μg Nrp1Ab (right panel). Scale bar: 100 μm.
- Quantitative analyses of the mean number of GN11 cells that migrated from the upper chamber (SFM) through the perforated membrane in the presence or absence of the indicated doses of Sema3A and Nrp1Ab in the lower chamber. Kruskal–Wallis test, n = 5–6 wells in two independent experiments.
- Schematic illustration and representative photomicrograph of an organotypic culture of a nasal explant from E11.5 mouse embryos after 8 days in vitro. NE, nasal epithelium; NMC, nasal midline cartilage. Scale bar 500 μm (150 μm in inset).
- Total number of GnRH neurons that migrated out of nasal explants isolated from control and mutant embryos, quantified after 8 days in culture. Mann–Whitney U‐test, n = 3–5 embryos from at least 3 litters.
- Distance travelled by GnRH neurons from the olfactory epithelia; cells were quantified within a radius of 1–6 mm. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 3–5 explants.
- Representative immunofluorescence for GnRH (red)‐ and BrdU (green)‐positive cells in control and mutant E14.5 embryos arising from progenitors born between E9.5 and E10.5. BrdU‐positive GnRH neurons (white arrowheads) were more abundant in mutant than in control embryos. Scale bar: 20 μm.
- Number of BrdU‐positive GnRH neurons in the nose, olfactory bulb (OB), and ventral forebrain (VFB) at E14.5 and total number of cells in control and mutant embryos. Double‐labeled cells were more numerous in the mutant VFB. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 4 embryos from 2 litters.
- Schematic diagram of intra‐embryonic nasal injection in wild‐type E11.5 embryos and collection of embryos at E14.5.
- Number of GnRH neurons in E14.5 embryos injected with neutralizing antibodies to neuropilin‐1 (Nrp1Ab) compared to an isotype goat antibody, or the antiapoptotic compound ZVAD compared to its vehicle, together with fold change in GnRH neuron number as well as mutant and control embryos at the same embryonic age. Note that the data are represented as fold change, as raw values are not comparable with transgenic animals that did not undergo surgery. Unpaired t‐test, n = 5–7 embryos from at least 2 litters.
We next assessed whether cell motility is also affected in primary GnRH neurons in which Nrp1 signaling is silenced. GnRH neurons maintained in vitro in organotypic nasal placode explants exhibit many of the characteristics of migrating GnRH neurons in vivo (Wray, 2010). We cultured explants of the nasal placode from E11.5 mutant and control littermates (Fig 3C). Unexpectedly, the total number of GnRH neurons was roughly twice as high in the periphery of the nasal explants from mutant embryos compared to their controls after 8 days of culture (232.8 ± 11.96 cells in controls, 427.7 ± 72.92 cells in mutants, Mann–Whitney U‐test, U = 0, P = 0.04, n = 3–5) (Fig 3D). The distribution of GnRH cells was similar in nasal placodes from mutant and control embryos, but the number of cells found beyond a radius of 4 millimeters in mutant nasal placodes was higher by ~300% (two‐way ANOVA, distance of migration, F (5,36) = 5.60, P < 0.001; genotype, F (1,36) = 13.30, P < 0.001; interaction, F (5,36) = 3.44, P = 0.01; Fisher's LSD multiple‐comparison test, > 4 mm t (36) = 3.26, P = 0.003; > 5 mm t (36) = 3.14, > 6 mm P = 0.003; t (36) = 2.88, P = 0.007 n = 3–5) (Fig 3E). These ex vivo results suggest that Nrp1 expression in GnRH neurons, in addition to regulating neuronal migration, may also regulate GnRH cell survival.
To further investigate this possibility, we injected BrdU into pregnant females at E9.5 and E10.5, a time window in embryonic development during which the proliferation of GnRH progenitors is at its highest but the Gnrh promoter is not yet active in newly generated neurons (Wray et al, 1989), i.e., before the Cre:LoxP‐mediated knockout of Nrp1 occurs. The quantification of BrdU‐positive cells expressing GnRH at E14.5 showed a 67.3% increase in the total number of double‐labeled cells in mutants (156 ± 15.66 neurons in controls, 261 ± 13.6 neurons in mutants, unpaired t‐test, t (6) = 5.06, P = 0.002, n = 4 per group) (Fig 3F and G), primarily accounted for by an increase in their numbers in the VFB (two‐way ANOVA, anatomical region, F (2,18) = 92.57, P < 0.0001; genotype, F (1,18) = 29.19, P < 0.0001; interaction, F (2,18) = 5.39, P = 0.015; Fisher's LSD multiple‐comparison test, nose, t (18) = 1.60 P = 0.13; OB, t (18) = 1.96 P = 0.07; VFB, 99 ± 13.40 neurons in controls, 164 ± 8.79 neurons in mutants, t (18) = 5.79 P < 0.0001, n = 4 per group) (Fig 3G). These results suggest that GnRH neurons born of E9‐E10 progenitors survive in greater numbers at E14.5 in mutants than in control littermates, as the absence of GnRH expression and thus of Cre:LoxP‐mediated Nrp1 deletion at the time of their birth eliminates a potential effect at the level of proliferation. In contrast, when BrdU was injected at E11.5, when the Gnrh promoter becomes active in the nasal placode, no BrdU‐positive GnRH cells could be detected 24 h later (Fig EV2). In agreement with the current dogma (Forni & Wray, 2015), these data show that upon their differentiation into GnRH‐expressing neurons, cells of the GnRH lineage stop proliferating. This therefore suggests that Nrp1 signaling in differentiated GnRH neurons controls their own survival. To test this hypothesis, we induced a transient and site‐specific inhibition of Nrp1 signaling by infusing neutralizing antibodies to Nrp1 (1/200, 2 μl/embryo) locally into the nasal region of wild‐type mouse embryos on E11.5 (Fig 3H). In contrast to the control IgG‐injected group, blunting Nrp1 signaling in the nose at E11.5 resulted in a 34% increase in the total number of GnRH neurons at E14.5 (unpaired t‐test, t (10) = 2.22, P = 0.05, n = 5–7, Fig 3I) thus phenocopying mutant mice (26% increase; t (10) = 2.22, P = 0.006, n = 5–6). Of note, the treatment of E11.5 embryos with Nrp1‐neutralizing antibodies is likely to disrupt the vomeronasal/terminal nerve projections used by GnRH neurons to migrate from the nose to the brain (Hanchate et al, 2012), in addition to GnRH neurons themselves; because of this confounding factor, we do not report the distribution of GnRH neurons at E14.5 in the group of experiments represented in Fig 3I. This increase was also mimicked by the intranasal injection of 0.3 μg/μl ZVAD (2 μl/embryo), an inhibitor of apoptosis (inhibiting caspase‐1 and caspase‐3) (Li et al, 2000; Israel et al, 2014), compared to vehicle‐injected mice (37% increase; t (11) = 2.76, P = 0.02, n = 6–7; Fig 3I). These in vivo results thus indicate that the size of the GnRH neuronal population is regulated by apoptosis at the post‐mitotic stage and that Nrp1 signaling in newborn GnRH neurons may be involved in this process. Together with our in vitro and ex vivo results, these in vivo findings suggest that, in addition to acting as the receptor for guidance cues and thus regulating cell migration, Nrp1 regulates the survival of newly born GnRH neurons.
Figure EV2. Knocking out neuropilin‐1 in GnRH‐expressing cells does not alter cell cycle exit.
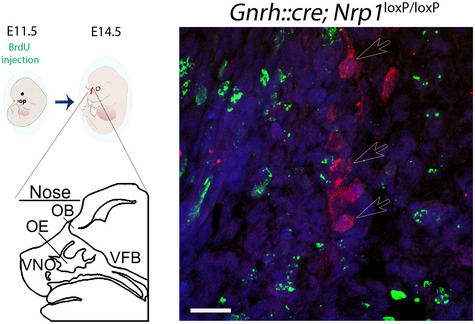
Left panels. Schematic diagram illustrating BrdU treatment of dams carrying mutant and control mouse embryos at E11.5 and their collection 3 days later (E14.5), and schematic representation of key anatomical landmarks in the head of an E14.5 embryo (VNO, vomeronasal organ; OE, olfactory epithelium, OB, olfactory bulb; VFB, ventral forebrain). Right panel. As in Nrp1 loxP/loxP control embryos, no BrdU immunofluorescence (green) was detected in GnRH‐positive cells (red) in Gnrh::cre;Nrp1 loxp/loxp mutant embryos at E14.5 (n = 3 per genotype from two independent litters). Empty arrows show GnRH‐immunoreactive neuronal cell bodies, which all lack BrdU labeling. Scale bar: 20 μm.
Female mice lacking Nrp1 expression in GnRH neurons show precocious attraction to male odors
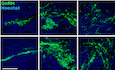
Because the AOB is a brain structure known to be involved in the perception of social odors (Brennan & Zufall, 2006; Hendrickson et al, 2008), we next investigated whether the ectopic accumulation of GnRH neurons in the AOB of mutant mice affects the detection of olfactory or pheromonal cues, by testing females for olfactory sex preference. Prepubertal and adult female littermates were exposed to urine collected from either sexually experienced males or estrous females. Adult mutant females showed a preference for male bedding with a score similar to that of control littermates (unpaired t‐test, t (9) = 0.57, P = 0.58, n = 5–6; Fig 4A). However, at P20, although control juvenile females showed a preference for female bedding, 4 out of 8 mutant females presented marked preference for male odors (one‐way ANOVA, F (2,11) = 30.79, P < 0.001; Fisher's LSD multiple‐comparison test, t (11) = 5.75, P = 0.0003, n = 4 controls and 6 mutants), while the other half showed an even more pronounced attraction for female odors than controls (t (11) = 2.60, P = 0.025, n = 4 controls and 6 mutants) (Fig 4A). Importantly, all juvenile mice (one‐way ANOVA, F (4,20) = 0.48, P = 0.75, n = 6 and 4) and adult control and mutant littermates (unpaired t‐test, t (9) = 0.79, P = 0.45, n = 5 and 6) showed equal interest toward all odors introduced into the cage (Fig 4B). These results suggest that the abnormal accumulation of GnRH neurons in the AOB of Gnrh::cre; Nrp1 loxP/loxP mice can cause a precocious switch in sex preference behavior that normally only occurs after puberty in wild‐type mice (Oboti et al, 2017).
Figure 4. Mice lacking neuropilin‐1 signaling in GnRH neurons show precocious attraction for odors of the opposite sex and early maturation of GnRH neuronal activity.
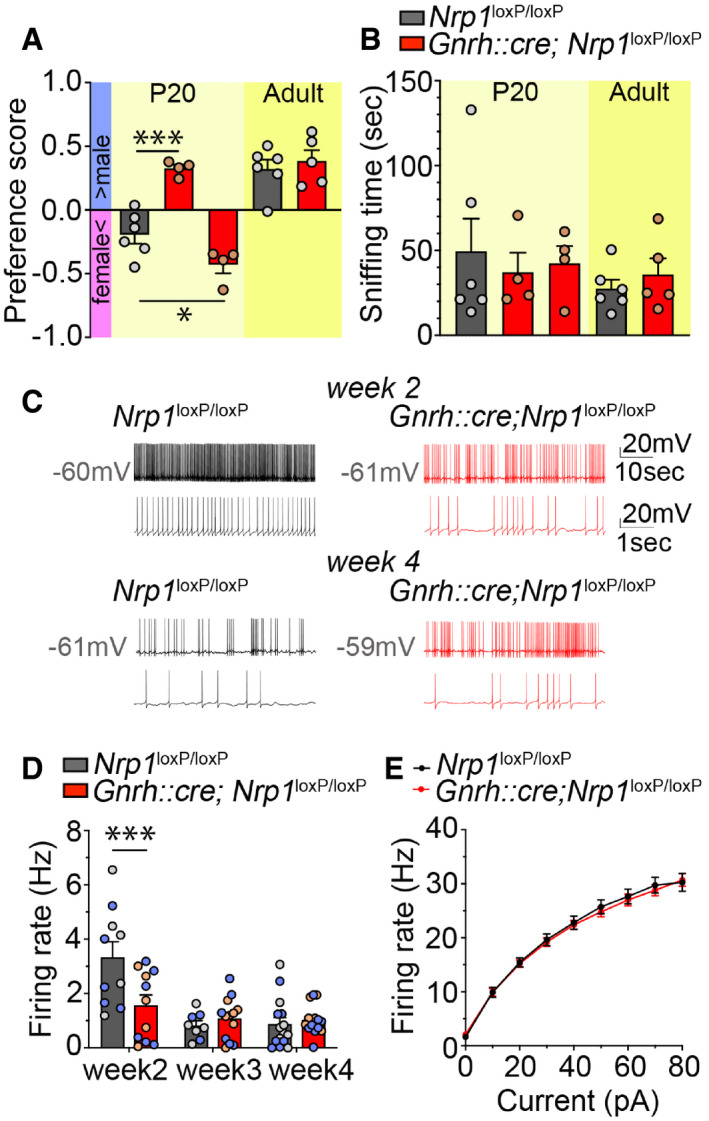
- Olfactory sex preference test in P20 (left panel) and adult (right panel) control and mutant female mice. Half of the mutant females at P20 showed a preference for males, similar to adults of both genotypes, whereas the other half showed an even more pronounced preference for females than their control littermates at P20. One‐way ANOVA, Fisher's LSD multiple‐comparison test for P20 mice and unpaired t‐test for adult mice; n = 4–6 from at least 3 litters.
- Time spent sniffing after the introduction of both male and female odors into the cage of control and mutant littermates. One‐way ANOVA, Fisher's LSD multiple‐comparison test for P20 mice and unpaired t‐test for adult mice; n = 4–6 from at least 3 litters.
- Representative traces of spontaneous firing in GnRH neurons from control (left, black) and mutant (right, red) animals at Week 2 (upper panels) and Week 4 (lower panels); lower traces show an enlargement of the upper trace.
- Average spontaneous firing frequency in GnRH neurons from control and mutant mice at Weeks 2, 3, and 4. Individual males and females used for this analysis are represented by blue and gray/light‐red dots, respectively. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 8–14 neurons from at least three animals from different litters. Note that individual neurons from male (n = 3–8) and female mice (n = 5–8) used for this analysis are represented by blue and gray dots, respectively.
- Quantification of evoked firing in GnRH neurons from control and mutant mice in response to current steps. Two‐way repeated‐measures ANOVA, n = 10–11 neurons from at least three mice from different litters.
GnRH neurons lacking Nrp1 show an early postnatal decrease in firing frequency
Sexual maturation has recently been shown to be accompanied by changes in the firing frequency of GnRH neurons during postnatal development (Dulka & Moenter, 2017). To determine whether the alterations in the number and distribution of GnRH neurons in the absence of Nrp1 signaling are also associated with alterations in the postnatal maturation of GnRH neuronal activity, we next studied the firing properties of GnRH neurons 2, 3, and 4 weeks after birth in control and mutant littermates. Patch‐clamp electrical recordings were performed in GFP‐identified GnRH neurons in the preoptic region from mutant Gnrh::Gfp; Gnrh::cre; Nrp1 loxP/loxP and control Gnrh::Gfp; Nrp1 loxP/loxP littermates. Spontaneous firing was measured in whole‐cell current‐clamp mode in GnRH neurons held at a resting potential of −60.25 ± 2.74 mV. In cells from control mice, firing activity was high at postnatal Week 2, i.e., during the infantile period, before declining to juvenile levels (Fig 4C). In contrast, cells from mutant infantile mice displayed a 53% decrease in the mean firing rate at Week 2 compared to those from control littermates (two‐way ANOVA, age, F (2,65) = 15.16, P < 0.001; genotype, F (1,65) = 3.24, P = 0.08; interaction, F (2,65) = 6.53, P = 0.003, Fisher's LSD multiple‐comparison test, t (65) = 3.89, P = 0.0002, n = 10–11) both in males (t (29) = 2.18, P = 0.038, n = 5–6) and in females (t (30) = 3.14, P = 0.004, n = 5 per group) (Fig 4C and D). However, firing frequency was not different between the two groups after weaning (both sexes, Week 3, t (65) = 0.55, P = 0.59; Week 4, t (65) = 0.39 P = 0.70; Fig 4C). To understand whether the lower firing rate at Week 2 was due to a difference in cell excitability, we compared the action potential output of these cells in response to current injection (0‐80 pA, 10 pA increments, 1s). Measurement of evoked action potential firing in response to ± 10 pA current steps showed no difference between the groups (two‐way repeated‐measures ANOVA, current step, F (7,133) = 865.80, P < 0.0001; genotype, F (1,19) = 0.09, P = 0.76; interaction, F(7,133) = 0.54, P = 0.80, Fig 4E). The input resistance of GnRH neurons was also comparable between the two groups (control, 1358 ± 204.8 MOhm; mutant, 1291 ± 124.9 MOhm, unpaired t‐test, t (20) = 0.28, P = 0.78). Altogether, these data suggest that Nrp1 signaling does not influence the intrinsic excitability of GnRH neurons and that differences in spontaneous activity may therefore be the result of their altered connectivity.
Genetic deletion of Nrp1 in GnRH neurons causes weight gain and central precocious puberty in female mice
During postnatal development, male mutant and control littermates showed no significant difference in their body weight curves (two‐way ANOVA, age, F (5,42) = 8.73, P < 0.001; genotype, F (1,42) = 3.39, P = 0.07; interaction, F (5,42) = 0.23, P = 0.95; Fisher's LSD multiple‐comparison test at P60, t (42) = 0.80, P = 0.42, n = 5; at P80 t (42) = 1.3, P = 0.20 n = 4) (Fig EV3A) or age at the onset of sexual maturation, as assessed by balanopreputial separation, which occurred at ~P30 (Mann–Whitney U‐test, U = 6.5, P = 0.13, n = 5–6) (Fig EV3B). In contrast, the body weight of female mice lacking Nrp1 signaling in GnRH neurons was significantly higher than in controls (two‐way ANOVA, age, F (8,216) = 1144, P < 0.0001; genotype, F (1,216) = 30.79, P < 0.0001; interaction, F (8,216) = 2, P = 0.047), as early as P21, i.e., the late juvenile period (Fisher's LSD multiple‐comparison test, t (216) = 2.20, P = 0.03 n = 11–17) (Fig 5A). This overweight (~1 g) persisted into adulthood (P60, t (216) = 3.43, P = 0.0007, n = 12 and 16; P100, t (216) = 3.28, P = 0.001, n = 7 and 8) (Fig 5A) and was associated specifically with a 33% increase in the volume of white adipose tissue (unpaired t‐test, t (14) = 2.20, P = 0.045, n = 8, Fig 5B), with lean tissue, as measured by CT scan, remaining unchanged (Mann–Whitney U‐test, U = 30, P = 0.88, n = 8, Fig 5C). No difference was noted between mutant and control littermates in the amplitude of the minipubertal FSH surge, which triggers ovarian steroidogenesis and an infantile increase in circulating estrogens levels (Kuiri‐Hanninen et al, 2014; Prevot, 2015; Francois et al, 2017) (control: 5.47 ± ± 0.62 ng/ml versus mutant: 5.87 ± 0.96 ng/ml, unpaired t‐test, t (20) = 0.35, P = 0.73, n = 11 per group), or the timing of vaginal opening, an external marker of the initiation of sexual maturation (control: 34.46 ± 0.75 days versus mutant: 33.2 ± 0.7 days, Mann–Whitney U‐test, U = 60, P = 0.21, n = 13 per group) (Fig 5D). However, the first ovulation, i.e., puberty, was significantly advanced in mutant mice (45.15 ± 0.67 postnatal days, n = 13) as compared to their control littermates (50.85 ± 1.25 postnatal days, n = 13; unpaired t‐test, t (24) = 4.02, P = 0.0005, n = 13, Fig 5E). Indeed, 80% of mutant mice had already reached puberty at P46 in contrast to only 15% for control females; 80% of control females were seen to reach puberty only at P55, i.e., 9 days later. Consistent with these findings, while the body weight at puberty was comparable between control mice (16.46 ± 0.38 g, n = 10) and mutant littermates (16.84 ± 0.19 g, n = 11, unpaired t‐test, t (19) = 0.95, P = 0.36), mutant female mice reached this mean pubertal weight more than 7 days earlier than their control littermates (Fig 5A) and weighed about 1 g more at P45, the average age at which mutant mice reach puberty (unpaired t‐test, body weight at P45 control versus mutant, t (21) = 3.25, P = 0.004, n = 9–14; Fig 5F). Similarly, plasma LH levels (Mann–Whitney U‐test, U = 3.5, P = 0.03, n = 5–6, Fig 5G) and the ratio of uterine weight to body weight (Mann–Whitney U‐test, U = 6, P = 0.008, n = 6–10, Fig 5H), a marker of circulating estrogen levels, were also significantly higher in mutant female mice than in control littermates at P45, indicating increased central GnRH release.
Figure EV3. Male mice lacking neuropilin expression in GnRH neurons show no alteration in body weight evolution and initiation of sexual maturation.
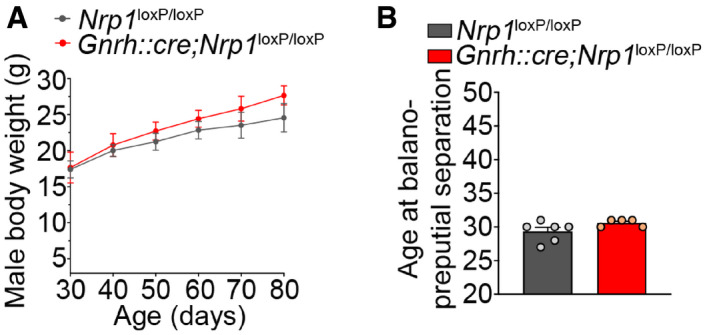
- Evolution of body weight in control and mutant male littermates from 30 to 80 days of age. Two‐way ANOVA, n = 3–5 mice.
- Age at balanopreputial separation in males. Mann–Whitney U‐test, n = 5–6 mice.
Figure 5. Selectively knocking out neuropilin‐1 expression in GnRH neurons leads to central precocious puberty and overweight in female mice.
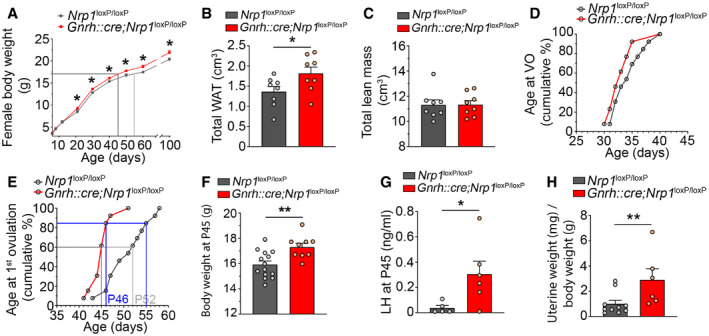
- Evolution of body weight in control and mutant female littermates from 6 to 100 days of age. Mutant females show a significantly higher body weight from P21. The gray lines mark body weight at puberty onset. Two‐way ANOVA, Fisher's LSD multiple‐comparison test, n = 7–17 mice from at least 4 litters.
- Fat mass or volume of white adipose tissue (WAT). Adult mutant females showed higher WAT volumes. Unpaired t‐test, n = 8 mice.
- Volume of lean tissue. Mann–Whitney U‐test, n = 8 mice.
- Cumulative percentage of the age at vaginal opening, n = 13 mice.
- Cumulative percentage of animals undergoing first ovulation as assessed by the occurrence of first estrus at different ages, n = 13 mice.
- Bar graph showing body weight at P45, which is the mean age at puberty in mutants. Unpaired t‐test, n = 9–14 mice.
- Plasma LH levels at P45 in control and mutant mice. Mann–Whitney U‐test, n = 5–6 mice.
- Mean uterine weight normalized to body weight (mg/g BW). Unpaired t‐test, n = 6–10 mice.
Adult female mice lacking Nrp1 expression in GnRH neurons display no marked alteration in adult reproductive physiology
In contrast to the changes observable in the pre‐ and peripubertal period, mutant female mice did not show any apparent defect in adult reproductive function. There were no differences between control and mutant female mice in estrous cyclicity (Fig EV4A and B), ovarian cytology (as evaluated by the stage of ovarian follicles present; two‐way ANOVA, follicular stage, F (4,45) = 37.4, P < 0.0001; genotype, F (1,45) = 0.10, P = 0.75; interaction, F (4,45) = 1, P = 0.43, Fig EV4C), or circulating LH levels either in diestrus (Mann–Whitney U‐test, U = 60, P > 0.99, n = 11 per group; Fig EV4D) or in females exposed to male odors (male‐induced LH surge, unpaired t‐test, t (15) = 0.29, P = 0.78, n = 7–10; Fig EV4E).
Figure EV4. Female mice lacking neuropilin‐1 expression in GnRH neurons show no alteration of adult reproductive physiology.
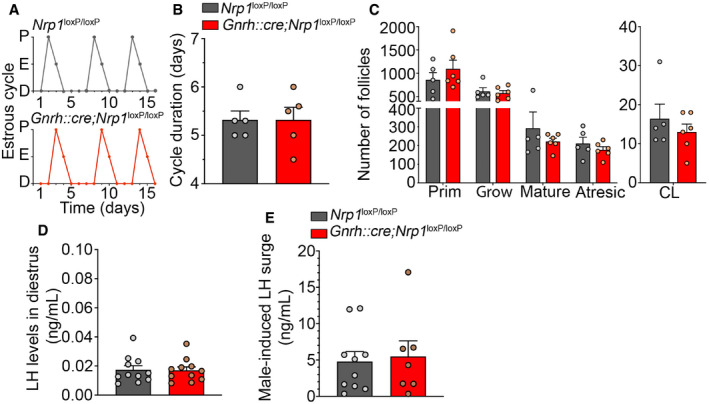
- Representative estrous cycles from an Nrp1 loxP/loxP control female (top) and a Gnrh::cre;Nrp1 loxP/loxP mutant female (bottom). D = diestrus, E = estrus, P = proestrus.
- Estrous cycle duration. Unpaired t‐test, n = 5.
- Number of follicles in both ovaries in control and mutant females. Prim, primordial; Grow, growing; CL, corpora lutea. Two‐way ANOVA, n = 5–6.
- Nadir LH levels in diestrus. Mann–Whitney U‐test, n = 11.
- Surge levels of LH induced by male odors. Unpaired t‐test, n = 7–10.
Discussion
In this study, we identified Nrp1 expression in GnRH neurons as a potential mechanism coordinating prepubertal body growth, the onset of puberty, and sensitivity to male olfactory stimuli in female mice. Sema3A:Nrp1 signaling has been shown to play a key role in the establishment of the scaffold along which GnRH neurons migrate from the nose to the brain during embryogenesis (Hanchate et al, 2012). Our data suggest that Nrp1 signaling in developing GnRH neurons contributes not only to the migration but to the survival of these neurons during embryogenesis and, surprisingly, to the regulation of body weight during postnatal development. In addition, we show that the genetic deletion of Nrp1 in GnRH neurons results in central precocious puberty and a marked advancement of sexual receptivity in some females. Overall, our results suggest that Nrp1‐mediated signaling in GnRH neurons could be a linchpin that holds together various neuroanatomical, physiological, and behavioral adaptations involved in triggering puberty and achieving adult reproductive function.
The genetic deletion of Nrp1 in GnRH neurons in our model was associated with the downregulation of the expression of one of its major co‐receptors, plexin‐A1, but had no effect on plexin‐A4 or Nrp2 transcripts. Interestingly, rare heterozygous variants for these genes, all involved in Sema3 signaling, have recently been found both in patients with GnRH deficiency (Marcos et al, 2017) and in some with severe obesity (van der Klaauw et al, 2019). However, in contrast to patients with hypogonadotropic hypogonadism carrying Nrp1 and plexin‐A1 variants and to knockout mice for these genes (Hanchate et al, 2012; Marcos et al, 2017; Oleari et al, 2019), animals lacking Nrp1 signaling in GnRH neurons do not present any GnRH deficiency, but, on the contrary, display an early maturation of the HPG axis and precocious puberty. This apparent discrepancy could be due to the fact that whole‐body Nrp1 deficiency in both humans and mice alters the projection of the vomeronasal and terminal nerve fibers in the frontonasal region and thus disrupts the migration of GnRH neurons into the brain (Hanchate et al, 2012; Marcos et al, 2017), whereas the impairment of Nrp1 signaling selectively in GnRH neurons increases their survival and therefore their numbers, accelerates their migration, and potentially alters their integration into the hypothalamic network controlling their activity after birth. It has been shown that Nrp1 signaling, besides playing a decisive role in neuronal guidance and axonal growth (Gu et al, 2003; Imai et al, 2009), also induces programmed cell death in neurons during development, both in vitro (Bagnard et al, 2001) and in vivo (Wehner et al, 2016). In agreement with this proapoptotic role of Nrp1 signaling, but in contrast to previously reported findings by others in whole‐body Nrp1‐null mice (Cariboni et al, 2011a), our results demonstrate that selectively knocking out Nrp1 in GnRH neurons markedly increases their numbers during embryogenesis. Notably, a similar phenomenon is produced by injecting Nrp1‐neutralizing antibodies or the antiapoptotic drug ZVAD into the nose of wild‐type mouse embryos. The antiapoptotic effect of Nrp1 deletion in GnRH neurons results in the migration of supernumerary neurons into the brain, including to the preoptic region of the hypothalamus where most GnRH cell bodies normally reside.
In the present study, the lack of Nrp1 signaling also appears to affect the neurophysiology of post‐migratory GnRH neurons, as seen by the early acquisition of a mature firing pattern by GnRH neurons during postnatal development. Because the absence of Nrp1 signaling in GnRH neurons does not affect intrinsic GnRH neuronal excitability, our findings suggest that these differences in spontaneous GnRH neuronal activity are likely the result of differential network inputs to the neurons (or alternatively, inadequate inputs given the large increase in GnRH neuron numbers). This inference is supported by recent studies showing that Sema‐mediated signaling can affect the stabilization of synaptic transmission in the postnatal brain (Orr et al, 2017). While the firing pattern of GnRH neurons lacking Nrp1 signaling in our mice does not affect the occurrence of the minipubertal surge of FSH in the infantile period (Prevot et al, 2003; Messina et al, 2016), i.e., the first postnatal activation of the HPG axis promoting gonadal steroidogenesis (Kuiri‐Hanninen et al, 2014; Prevot, 2015; Francois et al, 2017), it is associated with precocious puberty. GnRH release at the median eminence has indeed been shown to be dependent on action potentials (Glanowska & Moenter, 2015), and the decreased frequency of GnRH pulsatile release during the infantile–juvenile transition period has been associated with precocious puberty in a rat model of early‐life exposure to high concentrations of endocrine disruptors (Franssen et al, 2016). However, recent human genetic studies show that mutations in IGSF10, which cause a dysregulation of GnRH neuronal migration, are associated with delayed puberty onset (Howard et al, 2016), suggesting that the correct timing of puberty could be influenced by the number of GnRH neurons arriving at their destination. It is thus tempting to speculate that early puberty onset in Gnrh::cre; Nrp1 loxP/loxP mice may also be, at least in part, triggered by the increased number of GnRH neurons reaching the hypothalamus. Whether the observed sexual dimorphism with regard to precocious puberty is a real phenomenon cannot be deduced at present. While we used both male and female mice in our experiments, it is not possible to pinpoint the onset of puberty in males (i.e., presence of spermatozoa in the cauda epididymis–vas deferens junction), unlike females, in a minimally invasive manner (McKinney & Desjardins, 1973). In addition, the duration of spermatozoal maturation following HPG axis activation in the infantile period is fairly incompressible (Hess & Renato de Franca, 2008), greatly reducing the margin for any acceleration of puberty (McGee & Narayan, 2013; Prevot, 2015). However, given the changes we see in GnRH neuron numbers and their aberrant migration and activation in both sexes, it is likely that some changes, even if statistically insignificant, do occur in the timing of the maturation of the GnRH system in males also.
The phenotype of our model, in particular the association of overweight with the prepubertal alteration of GnRH neuron firing, is an interesting one. While it is partly reminiscent of a preclinical mouse model of polycystic ovary syndrome [PCOS; a condition often associated with obesity in humans (Barber et al, 2019)], it differs from PCOS in the absence of any alterations of estrous cyclicity or ovarian histology. However, it does evoke the phenotype of obese prepubertal girls without PCOS (McCartney et al, 2009). Indeed, it has been shown in age‐matched obese and non‐obese prepubertal girls that while there is no difference in FSH release during the prepubertal period, LH pulse frequency—an indicator of GnRH pulse frequency—is reduced in obese girls when compared with controls, at least until puberty (McCartney et al, 2009). Nevertheless, although the acceleration in growth during the peripubertal period, which is accompanied by an increase in fat mass in girls and lean mass in boys (Ahmed et al, 1999), is one of the earliest manifestations of puberty (Parent et al, 2003; Howard & Dunkel, 2019), and is usually thought to be a permissive component of pubertal maturation (Prevot, 2015; Tena‐Sempere, 2015), our observations on weight gain in female mice lacking Nrp1 in GnRH neurons suggest that the opposite is true. In other words, weight gain in the peripubertal period might actually be tightly controlled by the central activation of the HPG axis and of GnRH neurons themselves during the juvenile period. Premature increases in fat mass and higher body mass index could thus be harbingers of the early activation of GnRH neurons and of central precocious puberty (He & Karlberg, 2001; Howard & Dunkel, 2019).
In humans, while the age of onset of pubertal development is primarily thought to be driven by genetic factors (Parent et al, 2003; Canton et al, 2019; Howard & Dunkel, 2019), the genetic determinants of this timing and, in particular, the factors leading to central precocious puberty are largely unknown. To date, the most frequently mutated gene in pedigrees with central precocious puberty is MKRN3 (Abreu et al, 2013), whose hypothalamic expression decreases during postnatal development both in primates and rodents (Abreu et al, 2020), and whose early repression causes precocious puberty in female rats (Heras et al, 2019). Other such genes are DLK1 (patients who display overweight and obesity as additional clinical features) (Dauber et al, 2017) and two genes also mutated in patients with hypogonadotropic hypogonadism (Boehm et al, 2015), KISS1 (Silveira et al, 2010) and GPR54 (Teles et al, 2008). The fact that the genetic deletion of Nrp1 in GnRH neurons leads to central precocious puberty in our mice raises the possibility that some rare variants of NRP1 and, more generally, of genes in the signaling pathway of SEMA3A could also account for this condition in the human population. Variants in SEMA3A have recently been reported in patients with a severe early‐onset form of obesity (van der Klaauw et al, 2019), a phenotype that has been suggested to be due, at least in part, to defective melanocortin circuit development, since the disruption of Sema3 signaling in POMC‐expressing anorexigenic neurons decreases their ability to establish proper connections with their target brain regions during postnatal development and causes moderate weight gain (van der Klaauw et al, 2019). However, this metabolic feature is closely mimicked by our mice, which selectively lack Nrp1, the receptor of Sema3A, in GnRH neurons, unmasking a new explanation for this phenotype and a hitherto unappreciated role for the GnRH system in the control of energy homeostasis. Whether patients with SEMA3A‐associated early‐onset obesity also display central precocious puberty has not been documented, but warrants investigation in light of our current findings.
Another factor that could contribute to central precocious puberty in mutant mice is the abnormally high accumulation of GnRH neurons in the AOB, a brain region in which they are usually more sparsely distributed in both rodents and humans (Witkin & Silverman, 1983; Casoni et al, 2016), possibly because of the repulsive effect of high Sema3A expression in this region during embryogenesis (Giger et al, 1996). Changes in GnRH levels within the AOB have been associated with LH release in the vole upon pheromonal stimulation (Dluzen et al, 1981). Signals triggered by pheromones from both odor and pheromonal relays in mice have been shown to be transmitted to GnRH neurons, which can themselves influence both odor and pheromonal processing via feedback loops (Boehm et al, 2005). Furthermore, a recent study has demonstrated that the expression of GnRH is required to mediate proper male‐directed mate preference in adult female mice (Hellier et al, 2018). Our data show that 50% of juvenile female mice lacking Nrp1 expression in GnRH neurons are precociously attracted by male urine, although this switch in female‐to‐male sexual odor preference usually only occurs after puberty, i.e., when the neuroendocrine and behavioral aspects of sexual maturation match to respond effectively to physiological demands (Mucignat‐Caretta et al, 1998; Oboti et al, 2017). Our results suggest that ectopic GnRH release in the AOB may itself be responsible for these changes in sexual receptivity during postnatal development. Whether similar changes in the perception of social odors also occur in juvenile male mutants remains to be explored. Altogether, these findings not only provide the neuroanatomical and neurochemical basis to suggest that the neuroendocrine system controlling reproduction can also regulate sexual behavior, but also the framework for understanding how the alteration of its postnatal development can create maladaptive behaviors. This is of particular relevance with regard to studies examining psychosocial risks in children with precocious puberty, which affects mostly girls (60–80% of patients) (Golub et al, 2008). Children experiencing early puberty are indeed at risk for sexual abuse or early sexual debut (Golub et al, 2008). Interestingly, human imaging studies have recently shown that the treatment of individuals with kisspeptin, a peptide known to activate the release of GnRH, both enhances responses to sexual stimuli and reduces sexual aversion (Comninos et al, 2017, 2018) and that treatment with a GnRH antagonist diminishes the risk of sexual offense in men with pedophilic disorder (Landgren et al, 2020). In addition to evoking behavioral disorders, the early maturation of the HPG axis in juvenile females may also alter the expression of chemical signals known to block sexual approaches by males in mice (Ferrero et al, 2013) and sexual arousal in men (Gelstein et al, 2011).
Altogether, the intriguing concept that the activity of GnRH neurons during postnatal development could constitute the missing link between weight gain, the onset of puberty, and sexual behavior not only paves the way to a clearer understanding of the mechanism of sexual maturation and the involvement of energy homeostasis in this process, but could hold therapeutic implications for the prognosis and management of altered puberty in children.
Materials and Methods
Animals
All C57Bl/6J mice were housed under specific pathogen‐free conditions in a temperature‐controlled room (21–22°C) with a 12‐h light/dark cycle and ad libitum access to food and water. Gnrh::cre (Tg(Gnrh1::Cre)1Dlc) and Gnrh::gfp mice were a generous gift from Dr. Catherine Dulac (Howard Hughes Medical Institute, Cambridge MA) (Yoon et al, 2005) and Dr. Daniel J. Spergel (Section of Endocrinology, Department of Medicine, University of Chicago, IL) (Spergel et al, 1999), respectively. Nrp1 loxp/loxp (B6.129(SJL)‐Nrptm2Ddg/J (Stock No 005247) mice were purchased from Jackson Laboratory, Maine, USA (Gu et al, 2003). Genotyping was released by polymerase chain reaction (PCR) and the following primers: Nrp1‐sense: 5′‐AGGTTAGGCTTCAGGCCAAT‐3′, Nrp1‐antisense: 5′‐GGTACCCTGGGTTTTCGATT‐3′; Gnrh::cre‐sense: 5′‐CTGGTGTAGCTGATGATCCG‐3′, Gnrh::cre‐antisense: 5′‐ATGGCTAATCGCCATCTTCC‐3′; and Gnrh::gfp‐sense: 5′‐GAAGTACTCAACCTACAACGGAAG‐3′, Gnrh::gfp‐antisense 5′‐GCCATCCAGTTCCACAGAATTGG‐3′. Importantly, unexpected germline recombination has recently been reported to occur in distinct cre driver lines (Luo et al, 2020). Because recombination in the Gnrh::cre mouse line can occur in some oocytes (Hoffmann et al, 2019), only male Gnrh::Cre mice are used to generate bigenic mice (i.e., Gnrh::Cre (het); Nrp1 loxp/loxp males are crossed with Gnrh::Cre (wt);Nrp1 loxp/loxp females to generate about 50% mutants and 50% controls in the same litter). Animal studies were performed with the approval of the Institutional Ethics Committees for the Care and Use of Experimental Animals of the University of Lille and the French Ministry of National Education, Higher Education and Research (APAFIS#2617‐2015110517317420 v5 and APAFIS#13387‐2017122712209790 v9), and under the guidelines defined by the European Union Council Directive of September 22, 2010 (2010/63/EU).
Isolation of hypothalamic GnRH neurons using fluorescence‐activated cell sorting
The preoptic regions of hypothalamus from Gnrh::cre; Nrp1 loxp/loxp; Gnrh::gfp and Nrp1 loxp/loxp; Gnrh::gfp mice were microdissected and enzymatically dissociated using a Papain Dissociation System (Worthington, Lakewood, NJ) to obtain single‐cell suspensions. FACS was performed using an EPICS ALTRA Cell Sorter Cytometer Device (BD Bioscience) as described previously (Messina et al, 2016). The sort decision was based on measurements of GFP fluorescence (excitation: 488 nm, 50 mW; detection: GFP bandpass 530/30 nm, autofluorescence bandpass 695/40 nm) by comparing cell suspensions from Gnrh::Gfp and wild‐type animals. For each animal, about 400 GFP‐positive cells were sorted directly into 10 μl extraction buffer: 0.1% Triton® X‐100 (Sigma‐Aldrich) and 0.4 U/μl RNaseOUT™ (Life Technologies).
Quantitative RT–PCR analyses
For gene expression analyses, mRNAs obtained from FACS‐sorted GnRH neurons were reverse‐transcribed using SuperScript® III Reverse Transcriptase (Life Technologies) and a linear preamplification step was performed using the TaqMan® PreAmp Master Mix Kit Protocol (P/N 4366128, Applied Biosystems) and described previously (Messina et al, 2016). Real‐time PCR was carried out on Applied Biosystems 7900HT Fast Real‐Time PCR System using exon‐boundary‐specific TaqMan® Gene Expression Assays (Applied Biosystems): Gnrh1 (Gnrh1‐Mm01315605_m1), Nrp1 (Nrp1‐Mm01253208_m1), Nrp2 (Nrp2‐Mm00803099_m1), PlexinA1 (PlexinA1‐Mm00501110_m1), and PlexinA4 (PlexinA4‐Mm00558881_m1). Control housekeeping genes: r18S (18S‐Hs99999901_s1); ACTB (Actb‐Mm00607939_s1).
BrdU injections
5‐Bromo 2‐deoxyuridine (Sigma) was injected intraperitoneally at the concentration of 3 mg/kg to timed‐pregnant mice at 9.5 and 10.5 or at 11.5 days of gestation.
Intra‐embryonic nasal injections
Timed‐pregnant mice carrying E11.5 embryos were anesthetized under isoflurane, and the uterine horns were gently placed outside the abdominal cavity and constantly hydrated with 35°C sterile saline. Using a NanoFil syringe and a 35‐G needle attachment (World Precision Instruments), 0.2/0.4 μg of ZVAD (Sigma‐Aldrich), diluted in saline 1% DMSO, or goat anti‐Nrp1‐neutralizing antibody 1/200 in saline was injected in utero in the olfactory placode of the embryos as described previously (Croizier et al, 2016; Malone et al, 2019). The respective controls were injected with either saline 1% DMSO or goat IgG. The uteri were put back and the mothers were sutured, and the animals were monitored for few days. Embryos were collected at embryonic day 14.5 (E14.5) for GnRH neuron quantification.
Tissue preparation
For immunohistochemical analysis, adult female mice (3–5 months old) were anesthetized with 50–100 mg/kg of ketamine‐HCl and 5–10 mg/kg xylazine‐HCl and perfused transcardially with 10 ml of saline, followed by 50–100 ml of 4% paraformaldehyde (PFA) in PBS, pH 7.4. Brains were collected, post‐fixed in the same fixative for 2–4 h at 4°C, cryoprotected with PBS 20% sucrose, embedded in OCT embedding medium (Tissue‐Tek), frozen in isopentane cooled with nitrogen, and stored at −80°C until coronal cryosectioning. Embryos were removed from uterus of pregnant females, after sacrifice, at E11.5, E12.5, E14.5, and E18.5, or were sacrificed at postnatal day 0 (P0). The entire head was dissected and post‐fixed in 4% PFA from 3 to 4 h to overnight according to the age (overnight for P0), at 4°C, then cryoprotected with PBS 20% sucrose, embedded in OCT, frozen in isopentane cooled with nitrogen, and stored at 80°C until sagittal cryosectioning.
Nasal explants
Embryos were obtained from timed‐pregnant animals. Nasal pits of E11.5 were isolated under aseptic conditions in Gey's balanced salt solution (Invitrogen) enriched with glucose (Sigma‐Aldrich) and plated. Explants were placed onto glass coverslips coated with 10 μl of chicken plasma (Cocalico Biologicals). 10 μl of thrombin (Sigma‐Aldrich) was then added to adhere (thrombin/plasma clot) the explant to the coverslip. Explant was maintained in defined serum‐free medium (SFM) (Fueshko & Wray, 1994; Giacobini et al, 2008) containing 2.5 mg/ml Fungizone (Sigma‐Aldrich) at 37°C with 5% CO2 for 7 days in vitro. From culture day 3, fresh medium containing fluorodeoxyuridine (8 × 10−5 M; Sigma‐Aldrich) was provided to inhibit the proliferation of dividing olfactory neurons and fibroblasts. The medium was replaced with fresh SFM every 3 days for 8 days and then fixed with 4% PFA for processing immunohistofluorescence.
Immunohistofluorescence
Tissues were cryosectioned (Leica cryostat) at 35 μm (free‐floating sections, postnatal brains, and coronal sections) or 16 μm (glass sections, embryos and P0, and sagittal sections). Immunohistochemistry was performed as previously reported (Messina et al, 2011; Hanchate et al, 2012) using Alexa Fluor 488 (1:500), Alexa Fluor 568 (1:500), and Alexa Fluor 650 (1:500) secondary antibodies (Invitrogen), and Hoechst 33258 (pentahydrate bis‐benzimide, 1 μg/ml, Invitrogen Cat#H3570). Fluorescent specimens were mounted using MOWIOL (Calbiochem). The primary antisera used were the rabbit anti‐GnRH 1:3,000 (gift from G. Tramu, CNRS URA 339, Université de Bordeaux) (Beauvillain & Tramu, 1980; Prevot et al, 1999) or guinea pig anti‐GnRH (1:10,000) (Hrabovszky et al, 2011), goat anti‐Nrp1 (1:400; R&D Cat#AF566), and rat anti‐BrdU (1:300; Bio‐Rad Cat# OBT0030G). Analyses of the total GnRH cell number have been performed as described previously (Hanchate et al, 2012). In adult females, cell counts were performed on 35‐μm coronal sections in an antero‐posterior distribution into the hypothalamus and were divided into different regions of interest. For each animal, counts were performed on 1 serial series out of 2 and multiplied by 2. In embryos, cell counts were performed on 16‐μm sagittal sections according to their anatomical location (nose includes all the nasal regions, olfactory bulbs (OB) include the junction between the nose and brain, and brain includes the ventral forebrain (VFB)). For each animal, counts were performed on 2 series out of 4 and multiplied by 2. GnRH neuron counts were performed by eye directly using the microscope to account for the deepness of the tissue.
Whole‐mount clearing and imaging
Experiments were performed as previously described (Casoni et al, 2016; Belle et al, 2017) and detailed below.
Sample pretreatment with methanol
Samples were washed in PBS (twice for 1 h), followed by 50% methanol in PBS (once for 1 h), 80% methanol (once for 1 h), and 100% methanol (twice for 1 h). Next, samples were bleached in 5% H2O2 in 20% DMSO/methanol (twice for 1 h), 80% methanol (once for 1 h), 50% methanol (once for 1 h), PBS (twice for 1 h), and finally PBS/0.2% Triton X‐100 (twice for 1 h) before proceeding to the labeling procedures.
Whole‐mount immunolabeling
Samples were incubated at 37°C on an adjustable rotator in 10 ml of a blocking solution (PBSGNaT) of 1× PBS containing 0.2% gelatin (Sigma), 0.5% Triton X‐100 (Sigma‐Aldrich), and 0.01% NaAzide for 3 nights. Samples were transferred to 10 ml of PBSGNaT containing primary antibody rabbit anti‐GnRH 1:3,000 (gift from G. Tramu, CNRS URA 339, Université de Bordeaux) and placed at 37°C in rotation for 7 days. This was followed by six washes of 30 min in PBSGT at RT and a final wash in PBSGT overnight at 4°C. Next, samples were incubated in secondary antibody (1:400, Goat anti‐rabbit Alexa 647) diluted in 10 ml PBSGNaT for 2 days at 37°C in a rotating tube. After 6 × 30‐min washes in PBS at room temperature, the samples were stored in PBS at 4°C in the dark until clearing.
Tissue clearing
All incubation steps were performed at RT in a fume hood, on a tube rotator at 14 rpm, protected from light. Samples were dehydrated in a graded series of 1‐h washes in 20, 40, 60, 80, and 100% methanol in PBS before 66% DCM and 33% methanol for 1 h. It was followed by a delipidation step of 30–40 min in 100% dichloromethane (DCM; Sigma‐Aldrich). Samples were cleared in dibenzylether (DBE; Sigma‐Aldrich) for 2 h. Finally, samples were moved into fresh DBE and stored in glass tubes at RT until imaging. We could image samples, as described below, without significant fluorescent loss for up to 6 months. 3D Imaging was performed as previously described (Casoni et al, 2016; Belle et al, 2017). An ultramicroscope (LaVision BioTec) using ImSpector Pro software (LaVision BioTec) was used. The light sheet was generated by a laser (wavelength 647, Coherent Sapphire Laser, LaVision BioTec) and 2 cylindrical lenses. A binocular stereomicroscope (MXV10, Olympus) with a 4× objective (LaVision BioTec.; LVMI‐Fluor 4x/0.3 WD6) was used with either 1× or 2× magnification. Samples were placed in an imaging reservoir made of 100% quartz (LaVision BioTec) filled with DBE and illuminated from the side by the laser light. A PCO Edge SCMOS CCS camera (2.560 × 2.160 pixel size, LaVision BioTec) was used to acquire images. The step size between each image was fixed at 2 μm.
Electrophysiological recordings
Mice were anesthetized with isoflurane, and, after decapitation, the brain was rapidly removed and put into ice‐cold oxygenated (O2 95%/CO2 5%) artificial cerebrospinal fluid (ACSF) containing the following (in mM): 120 NaCl, 3.2 KCl, 1 NaH2PO4, 26 NaHCO3, 1 MgCl2, 2 CaCl2, and 10 glucose, pH 7.4 (with O2 95%/CO2 5%). After removal of the cerebellum, the brain was glued and coronal slices (200 μm thickness) were cut throughout the septum and preoptic area using a vibratome (VT1200S; Leica). Before recording, slices were incubated at 34°C to recover for 1 h. After recovery, slices were placed in a submerged recording chamber (32.8°C; Warner Instruments) and continuously superfused (2 ml/min) with oxygenated ACSF. Gfp‐positive GnRH neurons in the hypothalamic preoptic area at the level of the OVLT were visually identified with a 40× objective magnification and an upright Leica DM LFSA microscope under an FITC filter and their cell bodies observed by using IR‐differential interference contrast. Whole‐cell patch‐clamp recordings were performed in current clamp with bridge mode by using a MultiClamp 700B amplifier (Molecular Devices). Data were filtered at 1 kHz and sampled at 5 kHz with Digidata 1440A interface and pCLAMP‐10 software (Molecular Devices). Pipettes (from borosilicate capillaries, World Precision Instruments) had a resistance of 5–7 MΩ when filled with an internal solution containing the following (in mM): 140K‐gluconate, 10 KCl, 1 EGTA, 2 Mg‐ATP, and 10 HEPES, pH 7.3 (with KOH). Bridge balance was adjusted to compensate for pipette resistance. Data from cells showing series resistance higher than 20 MOhm were discarded. All recordings were analyzed with Clampfit 10 (Molecular Devices). Junction potential (15 mV) was determined to allow correction of membrane potential values and was applied post hoc by adding the value to the resting membrane potential. Data from cells showing a resting membrane potential more depolarized than −55 mV or more hyperpolarized than −65 mV were discarded; the average resting membrane potential was 60.25 ± 2.74 mV. Electrical membrane properties were measured in current‐clamp mode by applying a series of current pulses from −60 to +80 pA (1s, 10pA increments). Input resistance (Rin) was determined by measuring the slope of the linear portion of the current–voltage (I‐V) curve. Cells showing Rin lower than 500 MOhm were not used, and data from cells showing more than 10% change in Rin between the beginning and the end of the protocol were excluded. Spontaneous firing frequency was averaged over a period of 5 min after measuring membrane properties.
Cell culture
GN11 cells (Zakaria et al, 1996) were grown in a monolayer at 37°C in a 5% CO2 atmosphere, in DMEM (Life Technologies, Inc.), containing 110 mg/l sodium pyruvate and 4,500 mg/l glucose, supplemented with 1% l‐glutamine (200 mM, Life Technologies, Inc.), 1% penicillin–streptomycin (10,000 U/ml Life Technologies, Inc.), and 10% FBS (Life Technologies, Inc.). The medium was replaced at 2‐ to 3‐day intervals. Subconfluent cells were routinely harvested by trypsinization and seeded onto 50‐cm2 dishes (100,000 cells). For all experiments, only cells within six passages were used.
Transwell assay
Transwell chambers were used according to manufacturer's instructions (Falcon). In brief, GN11 cells grown in complete medium until subconfluence were harvested and re‐suspended at a density of 1 × 105 cells/μl in serum‐free medium (SFM). Cell were seeded on the upper side of an 8‐μm pore membranes and incubated for 12 h with SFM, at 37°C in a 5% CO2 atmosphere. GN11 cells were incubated in the presence of SFM, recombinant human Sema3A/Fc chimera (1250‐S3, R&D Systems) at different concentrations (50 ng/ml, 100 ng/ml, or 250 ng/ml) or Sema3A and Nrp1‐neutralizing antibody (Goat anti‐Nrp1, R&D AF566, 1:200), placed on the lower chamber. Cells on the upper side of the membranes were mechanically removed and cells on the lower side fixed with 4% PFA for 30 min before nuclei labeling with Hoechst. Two non‐overlapping regions were imaged per membrane, with nuclei counted.
Image analysis
For confocal observations and analyses, an inverted laser scanning Axio Observer microscope (LSM 710, Zeiss, Oberkochen, Germany) with EC Plan NeoFluar 10×/0.3 NA, 20×/0.5 NA, and 40×/1.3 NA (Zeiss) objectives was used (Imaging Core Facility of IFR114 of the University of Lille 2, France). Images, 3D volumes, and movies were generated using Imaris x64 software (version 7.6.1 Bitplane). Stack images were first converted to Imaris files (.ims) using ImarisFileConverter, and 3D reconstruction was performed using “volume rendering”. Optical slices of samples were obtained using the “orthoslicer” tools. The surface of the samples was created using the “surface” tool by creating a mask around each volume. 3D pictures were generated using the “snapshot” tool. Images from explant cultures and Transwell assays were taken with a Zeiss 20× objective (N.A. 0.8) mounted on an Axio Imager Z2 light microscope (Zeiss). ImageJ (National Institutes of Health, Bethesda, MD) and Photoshop CS5 (Adobe Systems, San Jose, CA) were used to process, quantify, adjust, and merge the photomontages. Figures were prepared using Adobe Photoshop and Adobe Illustrator CS6.
Physiological measurements
Puberty onset
Weaned female mice were checked daily for vaginal opening. After vaginal opening, vaginal smears were performed daily and analyzed under an inverted microscope to identify the specific day of the estrous cycle. The first estrus was designed as the day of puberty onset. Weight was recorded between P6 and P100.
Olfactory sex preference test
Subjects were coded so that the investigator was blind to the phenotype of each animal. The day before the experiment, the housing cage was changed to clean cage. The tests were conducted in the animal's home cage to minimize both manipulation and exposure to external stimuli. Prepubertal (P20) or adult (P60) control and mutant females were first exposed to urine from adult C57BL6/J wt sexually experienced male and not experienced estrus female, respectively, for 30 min. Urine samples (approximately 50 μl) were delivered on a piece of filter paper placed within a Petri dish (enabling direct contact with the source). Samples were removed for 30 min. For the testing paradigm, male and female urine samples were placed on opposite sides of the cage, equidistant from the cage walls. Trials lasted 10 min during which mouse behavior directed toward the two urine sources was recorded. The amount of time spent sniffing the Petri dish containing the sample was used as an indication of the mouse's interest in gaining further information from the scent source. The duration of sniffing behavior was recorded and quantified using the EthoVision software (Noldus). A preference score was calculated by dividing the time spent investigating the male sample minus the time spent investigating the female sample by the total time spent investigating both compartments. A positive value for the preference score indicates olfactory preference directed toward the male stimulus, whereas negative value indicates olfactory preference directed toward female stimulus.
LH measurements
Blood samples (5 μl) were taken from the tail and diluted in PBS‐Tween and immediately frozen. LH was assayed in duplicate using a protocol previously described by others (Steyn et al, 2013). A 96‐well high‐affinity binding microplate (9018; Corning) was coated with 50 μl of capture antibody (monoclonal antibody, anti‐bovine LH beta subunit, 518B7; University of California) at a final dilution of 1:1,000 (in NaHCO3/NaH2CO3 solution, pH = 9.6) and incubated overnight at 4°C. Then, wells were incubated with 200 μl of blocking buffer [5% (w/v) skim milk powder in 1× PBS‐T (1× PBS with 0.05% Tween 20)] for 2 h at room temperature (RT). A standard curve was generated using a twofold serial dilution of mLH (reference preparation, AFP‐5306A; National Institute of DIABETES and Digestive and Kidney Diseases−National Hormone and Pituitary Program [NIDDK‐NHPP]) in 1% (w/v) BSA−1× PBS‐T, and 25 μl serum was incubated in duplicates for 1.5 h. After 3 washing steps with 1× PBS‐T, the wells were incubated with 50 μl of detection antibody (polyclonal antibody, rabbit LH antiserum, AFP240580Rb; NIDDK‐NHPP) at a final dilution of 1:10,000 for 1.5 h (at RT). Each well containing bound substrate was incubated with 50 μl of horseradish peroxidase‐conjugated antibody (polyclonal goat anti‐rabbit, PI‐1000 Vector Laboratories) at a final dilution of 1:2,000. After a 1.5‐h incubation, 100 μl of o‐phenylenediamine (002003; Invitrogen), substrate containing 0.1% H2O2 was added to each well and left at RT for 10–30 min (estimated in function of the intensity of the reaction). The reaction was stopped by addition of 50 μl of 3 M HCl, and absorbance of each well was read at a wavelength of 490 nm (Multiskan Ascent Thermo LabSystems, Ascent Software). The concentration of LH in serum samples was determined by interpolating the OD values of unknowns against a nonlinear regression of the LH standard curve.
FSH measurements
Serum was isolated from trunk blood collected from control and mutant littermates at 12 days of age (P12). FSH levels were measured using a rodent FSH ELISA Kit (ERK R7014, Endocrine Technologies). Briefly, 50 μl of standards and serum was dispensed into the precoated wells of the kit. 100 μl of enzyme conjugate was added to the wells, and the plate was incubated for 3 h. Importantly, the serum from two pups of the same genotype was pooled to obtain the required serum volume and to obtain one data point. After 5 washing steps with wash buffer, 100 μl of TMB solution was added to each well. The plate was incubated for 20 min in the dark. The reaction was stopped by adding 50 μl of 2 N HCl to each well and the absorbance was read at a wavelength of 450 nm (Multiskan Ascent Thermo LabSystems, Ascent Software). The concentration of FSH in serum samples was determined by interpolating the OD values of unknowns against a nonlinear regression of the LH standard curve.
Measurement of body composition by CT Scan
Body composition analysis of fat/lean mass was performed using a CTscan, LaTheta 100 X‐ray Computed Tomography scanner (LCT‐100A; Zinsser Analytic).
Statistics
All analyses were performed using Prism 7 (GraphPad Software) and assessed for normality (Shapiro–Wilk test) and variance, when appropriate. Sample sizes were chosen according to standard practice in the field. For normal distribution, data were compared using unpaired two‐tailed Student's t‐test and Fisher's LSD test (one‐way and two‐way ANOVA). For non‐normally distributed values, Mann–Whitney and Kruskal–Wallis (one‐way analysis) tests were used. The significance level was set at P ≤ 0.05. Data are indicated as single‐value plots and means ± SEM. The number of animals, P values, and degrees of freedom are indicated either in the main text or in the figure legends.
Author contributions
CV, PG, and VP conceived the study, and designed and interpreted the experiments. CV performed the experiments and analyzed the results. ST performed olfactory tests, and GT, ST, and SM performed 3D imaging. SS performed the electrophysiological experiments. FC performed ex vivo experiments on nasal explants. AM, SC, NKH, and PC shared essential expertise. PC, SGB, and SR were involved in experimental design, data analysis, and interpretation. VP and PG supervised the study. CV, SR, and VP wrote the manuscript with contributions from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Acknowledgements
This work was supported by the Métropole Européenne de Lille (MEL, No: Convention_2017_ESR_04 to S.S. and V.P.), the Agence Nationale de la Recherche (ANR, France: ANR‐14‐CE12‐0015 RoSes and GnRH to P.G. and ANR‐17‐CE16‐0015 GRAND to V.P. and P.C.), and EU COST Action (BM1105 to V.P. and P.G.). We thank Nathalie Jouy (cell sorting) and Meryem Tardivel (confocal microscopy) from the BioImaging Center of Lille (BiCeL), and Julien Devassine (animal core facility) of the UMS2014‐US41 for their expert technical support.
The EMBO Journal (2020) 39: e104633
Data availability
This study includes no data deposited in external repositories.
References
- Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, Cukier P, Thompson IR, Navarro VM, Gagliardi PC et al (2013) Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med 368: 2467–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu AP, Kaiser UB (2016) Pubertal development and regulation. Lancet Diabetes Endocrinol 4: 254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu AP, Toro CA, Song YB, Navarro VM, Bosch MA, Eren A, Liang JN, Carroll RS, Latronico AC, Rønnekleiv OK et al (2020) MKRN3 inhibits the reproductive axis through actions in kisspeptin‐expressing neurons. J Clin Invest 130: 4486–4500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ML, Ong KK, Morrell DJ, Cox L, Drayer N, Perry L, Preece MA, Dunger DB (1999) Longitudinal study of leptin concentrations during puberty: sex differences and relationship to changes in body composition. J Clin Endocrinol Metab 84: 899–905 [DOI] [PubMed] [Google Scholar]
- Bagnard D, Vaillant C, Khuth ST, Dufay N, Lohrum M, Puschel AW, Belin MF, Bolz J, Thomasset N (2001) Semaphorin 3A‐vascular endothelial growth factor‐165 balance mediates migration and apoptosis of neural progenitor cells by the recruitment of shared receptor. J Neurosci 21: 3332–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber TM, Hanson P, Weickert MO, Franks S (2019) Obesity and polycystic ovary syndrome: implications for pathogenesis and novel management strategies. Clin Med Insights Reprod Health 13: 1179558119874042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvillain JC, Tramu G (1980) Immunocytochemical demonstration of LH‐RH, somatostatin, and ACTH‐like peptide in osmium‐postfixed, resin‐embedded median eminence. JHistochemCytochem 28: 1014–1017 [DOI] [PubMed] [Google Scholar]
- Belle M, Godefroy D, Couly G, Malone SA, Collier F, Giacobini P, Chedotal A (2017) Tridimensional visualization and analysis of early human development. Cell 169: 161–173 [DOI] [PubMed] [Google Scholar]
- Boehm U, Zou Z, Buck LB (2005) Feedback loops link odor and pheromone signaling with reproduction. Cell 123: 683–695 [DOI] [PubMed] [Google Scholar]
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A et al (2015) Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 11: 547–564 [DOI] [PubMed] [Google Scholar]
- Brennan PA, Zufall F (2006) Pheromonal communication in vertebrates. Nature 444: 308–315 [DOI] [PubMed] [Google Scholar]
- Canton APM, Seraphim CE, Brito VN, Latronico AC (2019) Pioneering studies on monogenic central precocious puberty. Arch Endocrinol Metab 63: 438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariboni A, Hickok J, Rakic S, Andrews W, Maggi R, Tischkau S, Parnavelas JG (2007) Neuropilins and their ligands are important in the migration of gonadotropin‐releasing hormone neurons. J Neurosci 27: 2387–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariboni A, Davidson K, Dozio E, Memi F, Schwarz Q, Stossi F, Parnavelas JG, Ruhrberg C (2011a) VEGF signalling controls GnRH neuron survival via NRP1 independently of KDR and blood vessels. Development 138: 3723–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariboni A, Davidson K, Rakic S, Maggi R, Parnavelas JG, Ruhrberg C (2011b) Defective gonadotropin‐releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: implications for the aetiology of hypogonadotropic hypogonadism. Hum Mol Genet 20: 336–344 [DOI] [PubMed] [Google Scholar]
- Casoni F, Malone SA, Belle M, Luzzati F, Collier F, Allet C, Hrabovszky E, Rasika S, Prevot V, Chedotal A et al (2016) Development of the neurons controlling fertility in humans: new insights from 3D imaging and transparent fetal brains. Development 143: 3969–3981 [DOI] [PubMed] [Google Scholar]
- Comninos AN, Wall MB, Demetriou L, Shah AJ, Clarke SA, Narayanaswamy S, Nesbitt A, Izzi‐Engbeaya C, Prague JK, Abbara A et al (2017) Kisspeptin modulates sexual and emotional brain processing in humans. J Clin Invest 127: 709–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comninos AN, Demetriou L, Wall MB, Shah AJ, Clarke SA, Narayanaswamy S, Nesbitt A, Izzi‐Engbeaya C, Prague JK, Abbara A et al (2018) Modulations of human resting brain connectivity by kisspeptin enhance sexual and emotional functions. JCI Insight 3: e121958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croizier S, Prevot V, Bouret SG (2016) Leptin controls parasympathetic wiring of the pancreas during embryonic life. Cell Rep 15: 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauber A, Cunha‐Silva M, Macedo DB, Brito VN, Abreu AP, Roberts SA, Montenegro LR, Andrew M, Kirby A, Weirauch MT et al (2017) Paternally inherited DLK1 deletion associated with familial central precocious puberty. J Clin Endocrinol Metab 102: 1557–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dluzen DE, Ramirez VD, Carter CS, Getz LL (1981) Male vole urine changes luteinizing hormone‐releasing hormone and norepinephrine in female olfactory bulb. Science 212: 573–575 [DOI] [PubMed] [Google Scholar]
- Dulka EA, Moenter SM (2017) Prepubertal development of gonadotropin‐releasing hormone neuron activity is altered by sex, age, and prenatal androgen exposure. Endocrinology 158: 3943–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero DM, Moeller LM, Osakada T, Horio N, Li Q, Roy DS, Cichy A, Spehr M, Touhara K, Liberles SD (2013) A juvenile mouse pheromone inhibits sexual behaviour through the vomeronasal system. Nature 502: 368–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forni PE, Wray S (2015) GnRH, anosmia and hypogonadotropic hypogonadism ‐ where are we? Front Neuroendocrinol 36: 165–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois CM, Petit F, Giton F, Gougeon A, Ravel C, Magre S, Cohen‐Tannoudji J, Guigon CJ (2017) A novel action of follicle‐stimulating hormone in the ovary promotes estradiol production without inducing excessive follicular growth before puberty. Sci Rep 7: 46222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen D, Gerard A, Hennuy B, Donneau AF, Bourguignon JP, Parent AS (2016) Delayed neuroendocrine sexual maturation in female rats after a very low dose of bisphenol A through altered GABAergic neurotransmission and opposing effects of a high dose. Endocrinology 157: 1740–1750 [DOI] [PubMed] [Google Scholar]
- Fueshko S, Wray S (1994) LHRH cells migrate on peripherin fibers in embryonic olfactory explant cultures: an in vitro model for neurophilic neuronal migration. Dev Biol 166: 331–348 [DOI] [PubMed] [Google Scholar]
- Gelstein S, Yeshurun Y, Rozenkrantz L, Shushan S, Frumin I, Roth Y, Sobel N (2011) Human tears contain a chemosignal. Science 331: 226–230 [DOI] [PubMed] [Google Scholar]
- Giacobini P, Messina A, Morello F, Ferraris N, Corso S, Penachioni J, Giordano S, Tamagnone L, Fasolo A (2008) Semaphorin 4D regulates gonadotropin hormone‐releasing hormone‐1 neuronal migration through PlexinB1‐Met complex. J Cell Biol 183: 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini P, Parkash J, Campagne C, Messina A, Casoni F, Vanacker C, Langlet F, Hobo B, Cagnoni G, Gallet S et al (2014) Brain endothelial cells control fertility through ovarian‐steroid‐dependent release of semaphorin 3A. PLoS Biol 12: e1001808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini P (2015) Shaping the reproductive system: role of semaphorins in gonadotropin‐releasing hormone development and function. Neuroendocrinology 102: 200–215 [DOI] [PubMed] [Google Scholar]
- Giger RJ, Wolfer DP, De Wit GM, Verhaagen J (1996) Anatomy of rat semaphorin III/collapsin‐1 mRNA expression and relationship to developing nerve tracts during neuroembryogenesis. J Comp Neurol 375: 378–392 [DOI] [PubMed] [Google Scholar]
- Glanowska KM, Moenter SM (2015) Differential regulation of GnRH secretion in the preoptic area (POA) and the median eminence (ME) in male mice. Endocrinology 156: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub MS, Collman GW, Foster PM, Kimmel CA, Rajpert‐De Meyts E, Reiter EO, Sharpe RM, Skakkebaek NE, Toppari J (2008) Public health implications of altered puberty timing. Pediatrics 121: S218–S230 [DOI] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD (2003) Neuropilin‐1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell 5: 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchate NK, Giacobini P, Lhuillier P, Parkash J, Espy C, Fouveaut C, Leroy C, Baron S, Campagne C, Vanacker C et al (2012) SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with kallmann syndrome. PLoS Genet 8: e1002896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Karlberg J (2001) Bmi in childhood and its association with height gain, timing of puberty, and final height. Pediatr Res 49: 244–251 [DOI] [PubMed] [Google Scholar]
- Hellier V, Brock O, Candlish M, Desroziers E, Aoki M, Mayer C, Piet R, Herbison A, Colledge WH, Prevot V et al (2018) Female sexual behavior in mice is controlled by kisspeptin neurons. Nat Commun 9: 400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson RC, Krauthamer S, Essenberg JM, Holy TE (2008) Inhibition shapes sex selectivity in the mouse accessory olfactory bulb. J Neurosci 28: 12523–12534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heras V, Sangiao‐Alvarellos S, Manfredi‐Lozano M, Sanchez‐Tapia MJ, Ruiz‐Pino F, Roa J, Lara‐Chica M, Morrugares‐Carmona R, Jouy N, Abreu AP et al (2019) Hypothalamic miR‐30 regulates puberty onset via repression of the puberty‐suppressing factor, Mkrn3. PLoS Biol 17: e3000532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess RA, Renato de Franca L (2008) Spermatogenesis and cycle of the seminiferous epithelium In Molecular mechanisms in spermatogenesis, Cheng CY. (ed), New York, NY: Landes Bioscience and Springer Science + Business Media; [DOI] [PubMed] [Google Scholar]
- Hoffmann HM, Larder R, Lee JS, Hu RJ, Trang C, Devries BM, Clark DD, Mellon PL (2019) Differential CRE expression in Lhrh‐cre and GnRH‐cre alleles and the impact on fertility in Otx2‐Flox mice. Neuroendocrinology 108: 328–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard SR, Guasti L, Ruiz‐Babot G, Mancini A, David A, Storr HL, Metherell LA, Sternberg MJ, Cabrera CP, Warren HR et al (2016) IGSF10 mutations dysregulate gonadotropin‐releasing hormone neuronal migration resulting in delayed puberty. EMBO Mol Med 8: 626–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard SR, Dunkel L (2019) Delayed puberty‐phenotypic diversity, molecular genetic mechanisms, and recent discoveries. Endocr Rev 40: 1285–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabovszky E, Molnar CS, Sipos MT, Vida B, Ciofi P, Borsay BA, Sarkadi L, Herczeg L, Bloom SR, Ghatei MA et al (2011) Sexual dimorphism of kisspeptin and neurokinin B immunoreactive neurons in the infundibular nucleus of aged men and women. Front Endocrinol 2: 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Yamazaki T, Kobayakawa R, Kobayakawa K, Abe T, Suzuki M, Sakano H (2009) Pre‐target axon sorting establishes the neural map topography. Science 325: 585–590 [DOI] [PubMed] [Google Scholar]
- Israel JM, Cabelguen JM, Le Masson G, Oliet SH, Ciofi P (2014) Neonatal testosterone suppresses a neuroendocrine pulse generator required for reproduction. Nat Commun 5: 3285 [DOI] [PubMed] [Google Scholar]
- van der Klaauw AA, Croizier S, Mendes de Oliveira E, Stadler LKJ, Park S, Kong Y, Banton MC, Tandon P, Hendricks AE, Keogh JM et al (2019) Human semaphorin 3 variants link melanocortin circuit development and energy balance. Cell 176: 729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiri‐Hanninen T, Sankilampi U, Dunkel L (2014) Activation of the hypothalamic‐pituitary‐gonadal axis in infancy: minipuberty. Horm Res Paediatr 82: 73–80 [DOI] [PubMed] [Google Scholar]
- Landgren V, Malki K, Bottai M, Arver S, Rahm C (2020) Effect of gonadotropin‐releasing hormone antagonist on risk of committing child sexual abuse in men with pedophilic disorder: a randomized clinical trial. JAMA Psychiatry 77: 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Ona VO, Guegan C, Chen M, Jackson‐Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S et al (2000) Functional role of caspase‐1 and caspase‐3 in an ALS transgenic mouse model. Science 288: 335–339 [DOI] [PubMed] [Google Scholar]
- Luo L, Ambrozkiewicz MC, Benseler F, Chen C, Dumontier E, Falkner S, Furlanis E, Gomez AM, Hoshina N, Huang WH et al (2020) Optimizing nervous system‐specific gene targeting with cre driver lines: prevalence of germline recombination and influencing factors. Neuron 106: 37–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi R, Pimpinelli F, Molteni L, Milani M, Martini L, Piva F (2000) Immortalized luteinizing hormone‐releasing hormone neurons show a different migratory activity in vitro . Endocrinology 141: 2105–2112 [DOI] [PubMed] [Google Scholar]
- Malone SA, Papadakis GE, Messina A, Mimouni NEH, Trova S, Imbernon M, Allet C, Cimino I, Acierno J, Cassatella D et al (2019) Defective AMH signaling disrupts GnRH neuron development and function and contributes to hypogonadotropic hypogonadism. Elife 8: e47198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos S, Monnier C, Rovira X, Fouveaut C, Pitteloud N, Ango F, Dode C, Hardelin JP (2017) Defective signaling through plexin‐A1 compromises the development of the peripheral olfactory system and neuroendocrine reproductive axis in mice. Hum Mol Genet 26: 2006–2017 [DOI] [PubMed] [Google Scholar]
- McCartney CR, Prendergast KA, Blank SK, Helm KD, Chhabra S, Marshall JC (2009) Maturation of luteinizing hormone (gonadotropin‐releasing hormone) secretion across puberty: evidence for altered regulation in obese peripubertal girls. J Clin Endocrinol Metab 94: 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee SR, Narayan P (2013) Precocious puberty and Leydig cell hyperplasia in male mice with a gain of function mutation in the LH receptor gene. Endocrinology 154: 3900–3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney TD, Desjardins C (1973) Postnatal development of the testis, fighting behavior, and fertility in house mice. Biol Reprod 9: 279–294 [DOI] [PubMed] [Google Scholar]
- Messina A, Ferraris N, Wray S, Cagnoni G, Donohue DE, Casoni F, Kramer PR, Derijck AA, Adolfs Y, Fasolo A et al (2011) Dysregulation of Semaphorin7A/beta1‐integrin signaling leads to defective GnRH‐1 cell migration, abnormal gonadal development and altered fertility. Hum Mol Genet 20: 4759–4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina A, Langlet F, Chachlaki K, Roa J, Rasika S, Jouy N, Gallet S, Gaytan F, Parkash J, Tena‐Sempere M et al (2016) A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat Neurosci 19: 835–844 [DOI] [PubMed] [Google Scholar]
- Mucignat‐Caretta C, Caretta A, Baldini E (1998) Protein‐bound male urinary pheromones: differential responses according to age and gender. Chem Senses 23: 67–70 [DOI] [PubMed] [Google Scholar]
- Oboti L, Trova S, Schellino R, Marraudino M, Harris NR, Abiona OM, Stampar M, Lin W, Peretto P (2017) Activity dependent modulation of granule cell survival in the accessory olfactory bulb at puberty. Front Neuroanat 11: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleari R, Caramello A, Campinoti S, Lettieri A, Ioannou E, Paganoni A, Fantin A, Cariboni A, Ruhrberg C (2019) PLXNA1 and PLXNA3 cooperate to pattern the nasal axons that guide gonadotropin‐releasing hormone neurons. Development 146: dev176461 [DOI] [PubMed] [Google Scholar]
- Orr BO, Fetter RD, Davis GW (2017) Retrograde semaphorin‐plexin signalling drives homeostatic synaptic plasticity. Nature 550: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP (2003) The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev 24: 668–693 [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ (2012) Getting neural circuits into shape with semaphorins. Nat Rev Neurosci 13: 605–618 [DOI] [PubMed] [Google Scholar]
- Prevot V, Croix D, Bouret S, Dutoit S, Tramu G, Stefano GB, Beauvillain JC (1999) Definitive evidence for the existence of morphological plasticity in the external zone of the median eminence during the rat estrous cycle: implication of neuro‐glio‐endothelial interactions in gonadotropin‐releasing hormone release. Neuroscience 94: 809–819 [DOI] [PubMed] [Google Scholar]
- Prevot V, Rio C, Cho GJ, Lomniczi A, Heger S, Neville CM, Rosenthal NA, Ojeda SR, Corfas G (2003) Normal female sexual development requires neuregulin‐erbB receptor signaling in hypothalamic astrocytes. J Neurosci 23: 230–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevot V (2015) Puberty in mice and rats In Knobil and Neill's physiology of reproduction, Plant TM, Zeleznik J. (eds), pp 1395–1439. New York: Elsevier; [Google Scholar]
- Radovick S, Wray S, Lee E, Nicols DK, Nakayama Y, Weintraub BD, Westphal H, Cutler GB Jr, Wondisford FE (1991) Migratory arrest of gonadotropin‐releasing hormone neurons in transgenic mice. Proc Natl Acad Sci USA 88: 3402–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanzel‐Fukuda M, Pfaff DW (1989) Origin of luteinizing hormone‐releasing hormone neurons. Nature 338: 161–164 [DOI] [PubMed] [Google Scholar]
- Silveira LG, Noel SD, Silveira‐Neto AP, Abreu AP, Brito VN, Santos MG, Bianco SD, Kuohung W, Xu S, Gryngarten M et al (2010) Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab 95: 2276–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spergel DJ, Kruth U, Hanley DF, Sprengel R, Seeburg PH (1999) GABA‐ and glutamate‐activated channels in green fluorescent protein‐tagged gonadotropin‐releasing hormone neurons in transgenic mice. J Neurosci 19: 2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyn FJ, Wan Y, Clarkson J, Veldhuis JD, Herbison AE, Chen C (2013) Development of a methodology for and assessment of pulsatile luteinizing hormone secretion in juvenile and adult male mice. Endocrinology 154: 4939–4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM (1999) Plexin‐neuropilin‐1 complexes form functional semaphorin‐3A receptors. Cell 99: 59–69 [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Comoglio PM (2004) To move or not to move? Semaphorin signalling in cell migration. EMBO Rep 5: 356–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taroc EZM, Prasad A, Lin JM, Forni PE (2017) The terminal nerve plays a prominent role in GnRH‐1 neuronal migration independent from proper olfactory and vomeronasal connections to the olfactory bulbs. Biol Open 6: 1552–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, Seminara SB, Mendonca BB, Kaiser UB, Latronico AC (2008) A GPR54‐activating mutation in a patient with central precocious puberty. N Engl J Med 358: 709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tena‐Sempere M (2015) Physiological mechanisms for the metabolic control of reproduction In Knobil and Neill's physiology of reproduction, Plant TM, Zeleznik J. (eds), pp 1605–1636. New York: Elsevier; [Google Scholar]
- Van Battum EY, Brignani S, Pasterkamp RJ (2015) Axon guidance proteins in neurological disorders. Lancet Neurol 14: 532–546 [DOI] [PubMed] [Google Scholar]
- Wehner AB, Abdesselem H, Dickendesher TL, Imai F, Yoshida Y, Giger RJ, Pierchala BA (2016) Semaphorin 3A is a retrograde cell death signal in developing sympathetic neurons. Development 143: 1560–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JW, Silverman AJ (1983) Luteinizing hormone‐releasing hormone (LHRH) in rat olfactory systems. J Comp Neurol 218: 426–432 [DOI] [PubMed] [Google Scholar]
- Wray S, Grant P, Gainer H (1989) Evidence that cells expressing luteinizing hormone‐releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci USA 86: 8132–8136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S (2010) From nose to brain: development of gonadotrophin‐releasing hormone‐1 neurones. J Neuroendocrinol 22: 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, Enquist LW, Dulac C (2005) Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell 123: 669–682 [DOI] [PubMed] [Google Scholar]
- Zakaria M, Dunn IC, Zhen S, Su E, Smith E, Patriquin E, Radovick S (1996) Phorbol ester regulation of the gonadotropin‐releasing hormone (GnRH) gene in GnRH‐secreting cell lines: a molecular basis for species differences. Mol Endocrinol 10: 1282–1291 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.