

Abstract
Platelet-derived growth factor receptor (PDGFR)-α plays roles in cell survival, proliferation, and differentiation; however, its function in chronic liver injury sequelae, such as fibrosis, is unknown. Hepatic stellate cells (HSCs), the primary mediators of fibrosis, undergo activation, which entails differentiation to myofibroblasts, proliferation, migration, and collagen deposition, partially in response to PDGFs. To examine the role of PDGFR-α in HSCs, Lrat–Cre recombinase and Pdgfra-floxed mice were bred to generate Lrat-CrePdgfra−/− (knockout) animals, which were subjected to chronic liver injury through carbon tetrachloride treatment, bile duct ligation, and 0.1% 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Although no major difference was observed after other types of liver injury, PDGFR-α loss in HSCs led to a significant albeit transient reduction in fibrosis after carbon tetrachloride injury, associated with increased HSC death and reduced migration. There was continued alleviation of hepatocellular injury in knockout mice despite ongoing carbon tetrachloride insult, associated with increased numbers of CD68 and F480 macrophages and increased clearance of damaged hepatocytes. Altogether our findings support a profibrotic role of PDGFR-α in HSCs during chronic liver injury in vivo via regulation of HSC survival and migration and affect the immune microenvironment, especially macrophages in clearing dying hepatocytes. Thus, our study provides a preclinical foundation for the future testing of therapeutic PDGFR-α inhibition in hepatic fibrosis, especially in combination with other therapies.
Hepatic fibrosis, a manifestation of chronic liver disease, is a wound-healing response that results in excessive, dysregulated collagen deposition from activated hepatic stellate cells (HSCs) and their differentiated myofibroblast counterparts. After recurrent injury, inflammation and release of numerous paracrine and autocrine growth factors and inflammatory chemokines from injured hepatocytes, resident macrophages, infiltrating inflammatory cells, and HSCs drive a perpetual cycle of tissue destruction and subsequent tissue remodeling to form a new fibrotic matrix.
Platelet-derived growth factors (PDGFs) are cysteine knot–type growth factors that have been identified as 4 different disulfide-bonded polypeptide chains (A, B, C, and D), which form multiple dimer configurations.1 Dimeric PDGF ligands can bind differentially to PDGF receptor (PDGFR) tyrosine kinases that exist as α or β monomers in the membrane to form αα, αβ, and ββ receptor dimers. Ligand receptor–binding triggers reciprocal tyrosine phosphorylation of specific residues of each receptor to induce downstream signaling.2
PDGFR-β has been identified as the primary PDGFR isoform to mediate the activation and profibrogenic transdifferentiation of HSCs into myofibroblasts during hepatic fibrosis.3 Recently, increasing evidence has indicated that PDGFR-α also possesses distinct signaling functions in HS and /myofibroblasts—a finding that carries important implications for prospective targeted PDGFR inhibitor therapies.4 It was previously found that PDGFR-α is expressed specifically in the HSC and myofibroblast population of the murine liver and that it is functionally important to mitogenesis and cell migration in human primary HSCs.5 More recently, it has been found that PDGF-BB–treated HSCs released PDGFR-α–enriched extracellular vesicles and that extracellular vesicles released by PDGFR-α–overexpressing cells promoted HSC migration in vitro as well as in vivo murine hepatic fibrosis.6
To date, cell-specific studies of PDGFR-α designed to delineate the function of this receptor in individual cell populations of the liver in vivo are lacking. To determine whether loss of PDGFR-α in HSCs in vivo affected the progression of chronic liver injury, we generated a novel murine model of Cre-lox recombination using the promoter for lecithin retinyl acyl-transferase (Lrat) to drive Cre expression in floxed Pdgfra mice in an HSC-specific manner. By subjecting resulting Lrat-Cre Pdgfra−/− [knockout (KO)] animals and their wild-type (WT) littermate controls to multiple models of chronic liver injury, we sought to test the hypothesis that PDGFR-α expression in HSCs contributes to the progression of hepatic fibrosis.
This study found that HSC-specific PDGFR-α loss in vivo results in early reduction of fibrosis and HSC migration in a model of hepatotoxic liver injury. This finding was associated with a subsequent increase in HSC and myofibroblast cell death and clearance of dead hepatocytes survival, which was characterized by improved serum transaminase profiles because of increased restorative macrophage response in these animals. Thus, PDGFR-α loss in HSCs not only led to their overall decreased survival but also accelerated immune-mediated clearance of damaged cells, improving overall hepatic health, despite ongoing injury.
Materials and Methods
Patients
All liver tissues were collected under an institutional review board–approved protocol (PRO08010372) by the Biospecimen Repository and Processing Core at the Pittsburgh Liver Research Center. Frozen banked liver tissue from explanted livers from deidentified patients with nonalcoholic steatohepatitis (NASH) (n = 5) was obtained from the Biospecimen Repository and Processing Core. All 5 patients had advanced fibrosis and cirrhosis composed of dense fibrosis bands along with the presence of regenerating nodules. As controls, 3 background livers from the nonfibrotic, nonsteatotic healthy margin were resected from patients with fibrolamellar hepatocellular carcinoma (n = 2) and hepatocellular adenoma (n = 1). These tissues were used for protein isolation and assessed for PDGFR-α.
Animals
All animal experiments were performed under the guidelines of the NIH and the Institutional Animal Use and Care Committee at the University of Pittsburgh. The studies performed in the current report were approved by the Institutional Animal Use and Care Committee at the University of Pittsburgh. For the generation of Pdgfra KO strains, homozygous Pdgfra floxed (exons 1 to 4) were crossed with Foxa3-Cre or Lrat-Cre mice for the creation of an F1 generation with mice heterozygous for floxed Pdgfra and Cre allele. These mice were subsequently backcrossed with homozygous Pdgfra floxed mice to create Cre-positive homozygous floxed Pdgfra animals at a mendelian ratio of one-fourth. Lrat-Cre mice of a mixed background strain were provided by Dr. Robert Schwabe (Columbia University, New York, NY). Male mice were used exclusively for all experiments because of limitations of Lrat-Cre transgenic mice, which require that females with the cre transgene be used for all breeding because the males display mosaic germline transmission. Hence, all females were used for breeding and males for the experiments. Homozygous Pdgfra floxed (exons 1 to 4) were obtained from Jackson Laboratories (Bar Harbor, ME). All liver function tests were performed by the Pittsburgh Liver Research Center's Biospecimen Repository and Processing Core at the University of Pittsburgh and University of Pittsburgh Medical Center.
Carbon Tetrachloride
Mice were injected intraperitoneally twice weekly with carbon tetrachloride (1:3 dilution in corn oil) at 0.5 μL/g body weight for 4 weeks (n = 8 WT mice and 7 KO mice) or 8 weeks (n = 6 WT mice and 8 KO mice) or corn oil only for 8 weeks. Animals were sacrificed 48 hours after last injection for liver and serum harvesting.
BDL Surgery
In bile duct ligation (BDL), the peritoneal cavity is opened with the mouse under anesthesia to expose the common bile duct and is cut between two 5-0 silk ligatures. Directly after surgery and at 24 and 48 hours after surgery, mice were administered a subcutaneous injection of 5 mg/kg of ketoprofen for analgesia. Livers and serum were collected at 5 days (n = 3 WT mice and 3 KO mice) or 14 days (n = 4WT mice and 4 KO mice) after BDL. These two time points were chosen to reflect the differential contributions of portal fibroblasts and HSCs to the activated myofibroblast population at each of these times.7
DDC Diet
Animals were fed a 0.1% 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet ad libitum for 16 days (n = 7 WT mice and 5 KO mice). Livers and serum were subsequently collected for analysis.
IHC
Liver tissue fixed for 48 hours in 10% formalin solution was embedded in paraffin and cut into 4-μm sections for immunohistochemistry (IHC). Slides were deparaffinized in xylene and rehydrated in graded alcohol washes (100%, 95%, and 70%) before washing in distilled water. Heat-induced antigen retrieval was performed on sections by microwaving them for 12 minutes in citrate buffer (10 mmol/L sodium citrate and 0.05% Tween 20, pH 6.0) [proliferating cell nuclear antigen (PCNA)] or pressure cooking sections for 20 minutes in EDTA buffer (1 mmol/L EDTA and 0.05% Tween 20, pH 9.0) [desmin, α-smooth muscle actin (α-SMA), and CD45 IHC]. For F4/80 IHC, proteinase K buffer (catalog number 21627; Millipore, Burlington, MA) was used for antigen retrieval. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) was performed using an ApopTag peroxidase kit (Millipore). IHC for F4/80 and CD45 was quantified from representative images using ImageJ software version 1.48 (NIH, Bethesda, MD; http://imagej.nih.gov/ij). TUNEL quantification was performed by counting numbers of TUNEL-positive hepatocytes or nonparenchymal cells with spindle-shaped morphologic findings in representative high-power fields. Statistical analysis and graphs were performed in Prism version 7.0a (GraphPad Software, San Diego, CA) as described below. Primary antibodies used in IHC studies are listed in Table 1.
Table 1.
Information on Primary Antibodies Used in the Study
| Target | Host species | Size, kDa | Western blot dilution |
IF/IHC dilution |
Diluent | Company | Catalog no. |
|---|---|---|---|---|---|---|---|
| α-SMA | Mouse | 42 | 1:100 (IF) | 0.5% BSA | Sigma-Aldrich (St. Louis, MO) | C6198 | |
| Caspase-3 | Rabbit | 17, 19, 35 | 1:1000 | 5% w/v milk | Cell Signaling Technology (Beverly, MA) | 9662 | |
| CD11b | Rat | 170 | 1:100 | PBS and 0.5% BSA | BioLegend (San Diego, CA) | 101201 | |
| Cleaved caspase-3 | Rabbit | 17, 19 | 1:1000 | 5% w/v milk | Cell Signaling Technology | 9664 | |
| CD45 | Rat | 220 | 1:100 | PBS | Santa Cruz Biotechnology, Dallas, TX | sc-53665 | |
| CD68 | Rabbit | 35 | 1:250 | PBS and 0.5% BSA | Abcam (Cambridge, UK) | Ab125212 | |
| Collagen I | Rabbit | 115,120 | 1:200 | PBS and 0.5% BSA | Abcam | Ab21286 | |
| Desmin | Rabbit | 52 | 1:200 (IHC) | 1% BSA and 1% Triton X-100 | Novus Bio (Littleton, CO) | NB120–15200 | |
| Desmin | Rabbit | 52 | 1:200 (IF) | 0.5% BSA | Thermo Fisher Scientific (Waltham, MA) | RB-9014 | |
| E-Cadherin | Rat | 35 | 1:500 (IF) | 0.5% BSA | BD Biosciences (Franklin Lakes, NJ) | BD610182 | |
| F4/80 | Rat | 160 | 1:200 (IHC) | PBS | BioRad | MCA497GA | |
| PDGF-A | Mouse | 31 | 1:200 | 5% w/v milk | Santa Cruz Biotechnology | sc-9974 | |
| PDGF-C | Goat | 30 | 1:200 | 3% BSA | R&D Systems (Minneapolis, MN) | AF1560 | |
| PDGFR-α | Rabbit | 190 | 1:1000 | 5% BSA | Cell Signaling Technology | 3174 | |
| PDGFR-α | Goat | 190 | 1:40 (IF) | 0.5% BSA | R&D Systems | AF1062 | |
| PDGFR-ß | Rabbit | 190 | 1:1000 | 5% BSA | Cell Signaling Technology | 3169 | |
| P-Y572/574-PDGFR-α/ß | Rabbit | 190 | 1:1000 | 5% BSA | Invitrogen (Carlsbad, CA) | 44-1000G | |
| P-Y742-PDGFR-α | Rabbit | 190 | 1:1000 | 5% BSA | Invitrogen | 44-1006 |
BSA, bovine serum albumin; CST, cell signaling technology; IF, immunofluorescence; IHC, immunohistochemistry; PBS, phosphate-buffered saline; PDGFR, platelet-derived growth factor receptor; α-SMA, α-smooth muscle actin.
Immunofluorescence
Liver tissue was fixed in 2% formaldehyde for 2 hours before 24 incubation in 30% sucrose solution. After flash freezing in OCT compound, tissue was cut in 6-μm–thick sections and stored at −20°C. For immunofluorescence staining, tissue was rehydrated with phosphate-buffered saline (PBS) followed by 10-minute detergent permeabilization using 0.1% Triton X-100 in PBS. Next slides were incubated in distilled water for 30 minutes at 37°C followed by blocking with 2% bovine serum albumin in PBS for 45 minutes. Slides were incubated in primary antibody solution overnight at designated concentrations (Table 1) in 0.5% bovine serum albumin in PBS (PBB). The following day, slides were washed with PBB before secondary antibody detection using species-specific antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 555 diluted 1:500 in PBB for 1 hour. Slides were then washed sequentially with PBB and PBS followed by 30 seconds of Hoeschst counterstain and slide covering. Epifluorescence images were obtained using a Nikon Eclipse Ti microscope and NIS Elements version 4.40. For confocal imaging, serially stacked, 1-μm–thick images were obtained using an Olympus FluoView 1000 microscope and FV1000 ASW version 4.2 software. Primary antibodies used in immunofluorescence studies are listed in Table 1. Representative immunofluorescence images for CD11b and CD68 were quantified using NIS Elements version 4.51 (Nikon Inc., Tokyo, Japan). Statistical analysis and graphs were performed in Prism version 7.0a as described below.
Fibrosis Quantification
Polarized light images of Picrosirius Red–stained liver sections were taken at ×50 magnification using an Olympus Provis microscope and MagnaFire software version 2.1B. These images were quantified using NIS Elements version 4.51. Statistical analysis and graphs were performed in Prism version 7.0a. Hydroxyproline content was also quantitated colorimetrically from liver samples using the chloramine T method as described previously.8 Statistical analysis and graphs were performed in Prism version 7.0a.
Whole Cell Lysate Preparation in Liver Tissue
At time of harvest, mice were anesthetized by isoflurane inhalation and subsequently sacrificed by cervical dislocation. After sacrifice, the livers were extracted and washed in PBS, and then the tissue was flash frozen in liquid nitrogen and stored at −80°C until use. Both human and murine livers were homogenized using the same method. Briefly, tissue was homogenized at the time of use in radioimmunoprecipitation assay with a protease and phosphatase inhibitor manually via glass mortar and pestle on ice. Lysates were removed to a fresh 1.5-mL tube and centrifuged at 14,000 rpm for 5 minutes at 4°C to remove clear supernatant to a new 1.5-mL tube while disposing of the pellet. Samples were then used for protein applications or stored at −80 °C until use.
Cell Culture
Cell culture of human HSCs (catalog number 5300) from ScienCell Research Laboratories (Carlsbad, CA) and olaratumab (LY3012207, IMC-3G3, Eli-Lilly, Indianapolis, IN) or human IgG (Equitech, Kerrville, TX) treatment were performed as previously described.5
Western Blot Analysis
Western blots were performed on liver and cell culture lysates homogenized in radioimmunoprecipitation assay buffer with protease and phosphatase inhibitors as previously described.5 All Western blots were performed with at least three biological replicates and repeated twice to confirm the validity of the results. Primary antibodies used in Western blot studies are listed in Table 1.
Statistical Analysis
All experiments were performed with at least 3 biological replicated and repeated at least twice. Statistical analysis was performed using Prism version 7.0a using nonparametric t-tests or analysis of variance for multiple column comparisons. P < 0.05 was considered significant. All graphs from various results and analysis were also generated in Prism version 7.0a.
Results
Increased PDGFR-α Levels in Animal Models of Fibrosis and Patients with Hepatic Fibrosis
The relevance of PDGF signaling in chronic liver injury was examined using two well-characterized models of chronic liver injury: repeated carbon tetrachloride injections and BDL. After 4-week carbon tetrachloride liver injury, distinct increases in PDGFR-α and PDGF ligands, including PDGF-C and the PDGFR-α–specific ligand PDGF-A, were observed in whole liver lysates (Figure 1A). This expression was further pronounced at the 8-week carbon tetrachloride liver injury time point, which is consistent with increased numbers of activated HSCs and myofibroblasts associated with advanced fibrotic liver injury. Similarly, a corresponding up-regulation of PDGFR-β, another well-known marker of HSCs, was also noted. Indeed, densitometry found a significant increase in PDGFR-α and PDGFR-β levels at 4 and 8 weeks after carbon tetrachloride liver injury when compared with baseline (Figure 1A). To determine whether PDGFR-α was also altered in cholestatic liver injury, its expression in murine livers 5 days and 14 days after BDL was examined. Compared with control animals undergoing sham operations, BDL murine livers did not have a consistent increase in total PDGFR-α levels by Western blot analysis using whole cell lysates (Figure 1B).
Figure 1.

Increased platelet-derived growth factor receptor (PDGFR)-α levels in animal models of fibrosis and patients with hepatic fibrosis. A: Representative Western blots showing increased expression of PDGFR-α (arrow) in whole liver lysates of mice subjected to a 4-week or 8-week course of carbon tetrachloride injections compared with corn oil–injected control mice (top left panel). A similar increase in PDGFR-β (arrow), platelet-derived growth factor (PDGF)-A, and PDGF-C is shown. Both PDGFR-α and PDGFR-β levels are significantly increased after carbon tetrachloride treatment compared with corn oil treatment as shown by densitometry on representative Western blots (top right and bottom panels). B: Western blots showing no notable differences in PDGFR-α (arrow) levels in murine livers after bile duct ligation (BDL) compared with mice undergoing sham operations. C: Confocal co-immunofluorescence imaging showing increased expression of PDGFR-α (green) co-localizing with α-smooth muscle actin (α-SMA)–positive (red) myofibroblasts in mice subjected to 8 weeks of carbon tetrachloride (middle panel) or 14 days after BDL (right panel) compared with normal liver (left panel). D: Increased PDGFR-α (arrow) levels in whole liver lysates from patients with nonalcoholic steatohepatitis (NASH) compared with normal liver tissue control. PDGFR-α levels are significantly increased in NASH cirrhotic livers compared with control livers as shown by densitometry. ∗P < 0.05, ∗∗P < 0.01 versus 8 week corn oil (non-parametric t-tests); †P < 0.05 versus normal (non-parametric t-tests). Scale bars = 100 μm. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
This study then addressed which cell types in the liver may show PDGFR-α localization after carbon tetrachloride and BDL injury. Double immunofluorescence for PDGFR-α and various cell-specific markers was performed. In the carbon tetrachloride model, there was a notable localization of PDGFR-α to activated myofibroblasts, as seen by co-localization with α-SMA (Figure 1C), a very small number of hepatocytes (subset of E-cadherin–positive cell), and no co-localization with endothelial cells [lymphatic vessel endothelial receptor (Lyve)-1] (Supplemental Figure S1A). In the BDL model, a notable localization of PDGFR-α was found only to activated myofibroblasts, as seen by co-localization with α-SMA (Supplemental Figure S1C) but none in hepatocytes (E-cadherin), cholangiocytes (epithelial cell adhesion molecule), or endothelial cells (Lyve-1) (Supplemental Figure S1B).
Next, lysates from explanted livers from patients with cirrhosis secondary to NASH (n = 5) were analyzed. Compared with normal nonfibrotic and nonsteatotic livers, a statistically significant increase was found in the levels of total PDGFR-α in NASH cirrhosis (Figure 1D).
Collectively, these findings indicate an increased expression of PDGFR-α and its ligands in hepatic fibrosis, consistent with a previous study that found increased PDGFR-α expression in activated HSCs and myofibroblasts in the carbon tetrachloride and BDL models and in advanced fibrosis associated with NASH in patients.5
Lrat-CrePdgfra−/− (KO) Mice Have Reduced Expression of PDGFR-α in HSCs after Chronic Liver Injury
To address the role of PDGFR-α in HSCs, we generated an HSC-specific Pdgfra null mice through breeding of Lrat promoter–driven Cre-expressing animals with Pdgfra floxed mice. The resultant Lrat-Cre Pdgfra−/− mice were confirmed through genotyping (Figure 2A). To confirm loss of PDGFR-α expression in KO mice, whole liver lysates through Western blot were examined, which showed that PDGFR-α expression was lost or reduced in KO mouse livers at baseline (Figure 2B). The mice were born at a normal mendelian ratio and had no differences in the liver weight to body weight ratio or any other overt phenotype as indicated by normal histologic and serum biochemistry results (data not shown).
Figure 2.

Lrat–CrePdgfra−/− [knockout (KO)] mice have reduced expression of platelet-derived growth factor receptor (PDGFR)-α in hepatic stellate cells (HSCs) after chronic liver injury. A: Genotyping of KO mice showing expression of floxed Pdgfra allele and Cre. B and C: Representative Western blots showing reduction of total PDGFR-α expression in whole liver lysates of Lrat-CrePdgfra−/− and wild-type (WT) littermate controls at baseline (B) and after 4 weeks of carbon tetrachloride, 16 days of 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), or 14 days after bile duct ligation (BDL) (C). D: Representative confocal immunofluorescence microscopy showing reduction in PDGFR-α expression in desmin-positive HSCs of KO mice after 4-week carbon tetrachloride, 16-day DDC, or 14-day BDL chronic liver injury. Scale bars = 100 μm. CCl4, carbon tetrachloride; Het, heterozygous; NT, no treatment.
These mice were next subjected to carbon tetrachloride treatment for 4 or 8 weeks, BDL for 5 days or 14 days, and DDC treatment for 16 days. Carbon tetrachloride is a model of hepatocellular injury–driven hepatic fibrosis, whereas BDL and DDC lead to cholestatic injury and ensuing fibrosis. As indicated in representative samples from carbon tetrachloride, BDL, and DDC treatments, PDGFR-α levels remained low or absent in KO mice compared with their littermate WT controls (Figure 2C). The loss of nearly all PDGFR-α expression in KO livers supports the finding that PDGFR-α is primarily expressed in HSCs and myofibroblasts. To specifically confirm loss of PDGFR-α expression in activated HSCs in KO mice, co-localization immunofluorescence confocal microscopy of PDGFR-α with desmin in carbon tetrachloride–, DDC-, and BDL-injured livers of KO and WT mice (Figure 2D) was performed. In each model of liver injury, a clear loss of PDGFR-α in desmin-positive HSCs in KO animals compared with WT controls was observed.
Transient Reduction in Hepatic Fibrosis in KO Mice in Hepatotoxic Liver Injury
To determine whether loss of PDGFR-α in KO mice affected the progression of hepatic fibrosis in hepatotoxic liver injury, KO mice after 4 weeks or 8 weeks of carbon tetrachloride–induced liver injury were evaluated. After 4 weeks of carbon tetrachloride–induced liver injury, fibrosis was significantly reduced in KO mice compared with WT controls as seen by Picrosirius Red staining, which was clearly notable at a higher magnification (100×) (Figure 3, A and B). However, at 8 weeks of continued carbon tetrachloride treatment, comparable fibrosis was observed by Sirius Red staining in WT and KO mice (Figure 3A). To verify these differences, the Sirius Red staining images were quantified using NIS Elements as described in Materials and Methods, which confirmed a modest but statistically significant decrease only at 4 weeks (Figure 3C). To confirm these changes, Masson Trichrome staining was also used. Representative sections show a decrease in collagen staining at 4 weeks in KO compared with WT mice but comparable staining between the two groups at 8 weeks (Figure 3D). The difference in Trichrome staining at 4 weeks after carbon tetrachloridewas more clearly visible at a higher magnification (200×) (Figure 3E)
Figure 3.

Transient reduction in hepatic fibrosis in knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) in hepatotoxic liver injury. A: KO mice have reduced fibrosis as assessed by Sirius Red staining at 4 weeks of carbon tetrachloride–induced liver injury compared with wild-type (WT) controls. No change in hepatic fibrosis is seen in KO mice at the 8-week carbon tetrachloride time point compared with WT controls. B: Higher-magnification representative image confirms decrease in Sirius Red staining at the 4-week time point. C: Picrosirius Red staining was quantified using polarized light microscopy and verified significant decrease in collagen is observed in KO mice at the 4-week time point only. D: Representative Mason trichrome staining images show decreased collagen in KO mice at 4 weeks of carbon tetrachloride treatment compared with WT mice but comparable staining between the 2 groups at 8 weeks. E: Higher-magnification images also show decreased trichrome staining in KO mice at 4 weeks of carbon tetrachloride treatment compared with WT mice. n = 7 KO mice at 4 weeks (A); n = 8 WT mice at 4 weeks and KO mice at 8 weeks (A); n = 6 WT controls at 8 weeks (A). ∗∗∗P < 0.001. Original magnification: ×50 (A); ×100 (B and D); ×200 (E)
IHC for desmin and α-SMA showed the presence of HSCs in areas of injury and fibrosis in both the WT and KO mice, albeit a notable decrease in desmin-positive cells was evident in KO mice at the 4-week time point (Figure 4A). Comparable numbers of desmin- and α-SMA–positive HSCs were present at the site of injury and scar formation in WT and KO mice at the 8-week time point (Figure 4B).
Figure 4.

Decreased fibrosis at 4 weeks of carbon tetrachloride treatment coincides with fewer stellate cells in hepatic parenchyma. A: Representative liver sections from knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) show fewer desmin-positive cells by immunohistochemistry (IHC) after 4 weeks of carbon tetrachloride injury compared with wild-type (WT) controls. No change in α-smooth muscle actin (α-SMA) IHC is observed at this time point. B: No change in desmin or α-SMA IHC is evident in KO versus WT controls at the 8-week carbon tetrachloride time point. C: Hydroxyproline assay shows comparable content hydroxyproline (micrograms) content per gram of hepatic tissue at 4 and 8 weeks of carbon tetrachloride treatment in WT versus KO mice. n = 7 KO mice at 4 weeks (A); n = 8 WT mice at 4 weeks (A) and KO mice at 8 weeks (B); n = 6 WT controls at 8 weeks (B). Original magnification, ×50 (A and B).
A hydroxyproline assay was performed to examine any differences in collagen content in the KO versus WT mice at both 4 and 8 weeks of carbon tetrachloride treatment. Intriguingly, no significant differences were evident between the two groups at either time point, suggesting a discrepancy between staining for intact collagen and content of hydroxyproline, reflective of both intact and degraded collagen (Figure 4C). In a previous study of the effect of PDGFR-α inhibition in human hepatic stellate cells in vitro, PDGFR-α inhibition with olaratumab decreased HSC proliferation and migration but had no significant effect on the overall expression of fibrosis-associated gene expression.5 The relative expression of fibrosis-associated genes in KO and WT cohorts at the 4-week carbon tetrachloride time point was examined and no significant change in the expression of these targets, including Acta2, Tgfb1, Tgfb2, Fn1, Mmp9, Mmp13, Timp1, or Pdgfrb (Supplemental Figure S2) was observed.
To extend these findings to other models in which liver fibrosis is secondary to cholestasis, the overall fibrosis through Sirius Red staining in the BDL and DDC models was examined. Intriguingly, no differences were observed in Sirius Red, desmin, or α-SMA in WT and KO animals in these cholestatic liver injury models (Supplemental Figure S3). On the basis of these findings, we conclude that HSC-specific PDGFR-α primarily promotes hepatocellular injury–driven but not biliary injury–driven hepatic fibrosis.
Altered Morphologic Features and Reduced MidZonal Distribution of HSCs in KO Mice after Chronic Liver Injury
Migration is an important function of activated HSCs and facilitates their ability to reach areas of liver injury and initiate the deposition of extracellular matrix associated with hepatic fibrosis. PDGFR-α signaling contributes toward HSC and myofibroblast migration in vitro.5,6 To address such changes in vivo, the presence of HSCs in midzonal regions of the hepatic lobule in the three injury models was assessed. A clear decrease in the numbers of midzonal HSCs was evident after 4 week of carbon tetrachloride treatment in the KO versus WT animals (Figure 5A). Despite the absence of significant changes in overall levels of fibrosis in BDL- or DDC-induced liver injury models, closer analysis of HSC distribution in these models also revealed a distinct decrease in midzonal HSC distribution in KO livers as observed by desmin IHC (Supplemental Figure S4). Three-dimensional reconstruction of consecutive confocal slices of representative liver sections after 4 weeks of desmin staining also verified overall fewer midzonal HSCs present in KO livers, which also found a distinct reduction in spindle-shaped morphologic features compared with the WT littermates (Figure 5B).
Figure 5.

Altered morphologic findings and reduced midzonal distribution of hepatic stellate cells (HSCs) in knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) after chronic liver injury. A: Representative lower-magnification and higher magnification images of desmin immunohistochemistry-stained liver sections highlight reduced distribution of HSCs in midzonal areas in KO livers compared with wild-type (WT) controls after 4-week carbon tetrachloride–mediated liver injury. Boxed areas are shown at higher magnification in the bottom row.B: A 3-dimensional reconstruction of confocal image slices of a representative midzonal area in carbon tetrachloride–mediated liver injury shows reduced size and decreased spindle morphologic features of desmin-positive HSCs in KO liver compared with WT control. Original magnification: ×50 (A, top row); ×200 (A, bottom row, and B).
Transient reduction of fibrosis in KO mice is followed by improvement in liver function after carbon tetrachloride–induced hepatocellular liver injury. Given the transient, although significant, reduction in fibrosis in KO mice after carbon tetrachloride–induced hepatocellular injury, it was investigated if loss of PDGFR-α from HSCs contributed to any difference in overall hepatobiliary injury. Next, serum liver function tests in KO and WT animals at 4 and 8 weeks of carbon tetrachloride–induced liver injury were analyzed as a clinical surrogate of hepatocellular and hepatobiliary injury. Although comparable levels of serum transminases were observed at 4 weeks after carbon tetrachloride injury, the alanine transaminase and aspartate transaminase levels continued to elevate in WT animals from 4 weeks to 8 weeks of carbon tetrachloride, whereas these values plateaued in KO mice (Figure 6A, Supplemental Figure S5). Thus, although the profibrotic effect of HSC-specific PDGFR appears to be transient, KO mice benefit from a subsequent decrease in overall hepatocellular injury in carbon tetrachloride injury.
Figure 6.

Improved serum transaminase profile in knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) in the absence of hepatocellular proliferative advantage after carbon tetrachloride–mediated liver injury. A: KO and wild-type (WT) mice have similar alanine transaminase (ALT) and aspartate transaminase (AST) values at the 4-week carbon tetrachloride time point, whereas ALT and AST values in KO mice (red lines) are markedly lower than in WT mice (blue lines) at the 8-week carbon tetrachloride time point. B: Proliferating cell nuclear antigen immunohistochemistry shows no increase in hepatocellular proliferation in KO animals at the 4-week or 8-week carbon tetrachloride time points compared with WT controls. ∗P < 0.05. n = 4 KO mice (A) and WT mice (A and B); n = 5 KO mice (B). Original magnification, ×100.
Consistent with a lack of difference in hepatic fibrosis between KO and WT mice after BDL, no differences were observed in markers of hepatobiliary injury after 2 weeks of BDL (Supplemental Figure S6A) or at any other point during BDL (Supplemental Figure S6B).
Improved serum transaminase profile in KO mice after carbon tetrachloride injury is associated with increased HSC and myofibroblast cell death and reduced numbers of dying hepatocytes. To further investigate the potential mechanism for the improvement in serum transaminase profile in KO animals, hepatocyte regeneration by PCNA IHC were first examined and relative levels of necrosis and apoptosis assessed by TUNEL staining of liver sections in KO and WT animals after 4 and 8 weeks of carbon tetrachloride injury. No increase in hepatocyte proliferation as assessed by PCNA at 4 or 8 weeks after carbon tetrachloride injury in KO animals compared with WT controls were observed, which could indicate enhanced regeneration as a mechanism of alleviation of hepatocyte injury at the 8-week time point (Figure 6B). However, an intriguing inverse relationship between the TUNEL staining of hepatocytes and TUNEL staining of spindle-shaped HSCs was noted, which differed between the KO and WT groups (Figure 7, A–C). Specifically, KO livers had a statistically significant reduction in the number of TUNEL-positive hepatocytes compared with WT control livers, especially in the pericentral zone (Figure 7B). Conversely, KO livers had a statistically significant increase in TUNEL-positive, spindle-shaped HSCs after 8 weeks of carbon tetrachloride injury (Figure 7C).
Figure 7.

Reduced hepatocellular death and hepatic stellate cell (HSC) and myofibroblast survival in knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) after long-term carbon tetrachloride–induced injury. A: Representative terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining of KO liver sections after 8 weeks of carbon tetrachloride–induced liver injury shows increased numbers of TUNEL-positive HSCs and myofibroblasts (arrows) while lacking the many TUNEL-positive pericentral hepatocytes (arrowheads) seen in wild-type (WT) littermate controls. B: Quantification of TUNEL-positive hepatocytes (arrowheads) in WT versus KO mice at 8 weeks of hepatocellular injury shows a significant decrease in cell death in KO mice. C: Quantification of TUNEL-positive HSCs and myofibroblasts in WT versus KO mice at 8 weeks of hepatocellular injury shows a significant increase in cell death in KO mice. D: Platelet-derived growth factor-α–-specific monoclonal antibody inhibitor olaratumab induces activation of caspase-3 in human hepatic stellate cells in vitro. n = 4 WT mice (A); n = 5 KO mice (A). ∗P < 0.05. Original magnification, ×100 (A).
PDGFR-α Inhibition in Vitro in Human Hepatic Stellate Cells Lead to Increased Cell Death
The presence of increased numbers of TUNEL-positive HSCs in the KO mice after carbon tetrachloride injury suggests that PDGFR-α may promote survival of activated myofibroblasts in advanced hepatic fibrosis. To directly investigate this, the effect of PDGFR-α–specific inhibition in human hepatic stellate cells using the monoclonal PDGFR-α inhibitor olaratumab (Lartruvo) was examined. Olaratumab exposure, but not control IgG, induced an increase in cleaved caspase-3 after 6-hour exposure in culture (Figure 7D). Thus, PDGFR-α signaling seems to be an important prosurvival signal in HSCs, the absence of which may indirectly contribute to decreased hepatocellular injury through mechanisms that are discussed further below.
Increased Restorative Hepatic Macrophage Response in KO Mice after Long-Term Carbon Tetrachloride–Induced Injury
To address how HSC-specific loss of PDGFR-α in KO animals led to decreased TUNEL-positive hepatocytes and decreased serum hepatocellular injury markers after 8-week carbon tetrachloride–induced liver injury, any differences in inflammatory cell response in KO and WT mice were next assessed. IHC was performed for CD45 (leukocyte common antigen), a general marker of inflammation, and for F4/80, a broad macrophage marker at the 4-week and 8-week carbon tetrachloride time points. No differences were observed in CD45 at the 4-week or 8-week carbon tetrachloride time points (Figure 8A). In contrast, there was a statistically significant increase in the number of F4/80-positive macrophages present in KO livers compared with WT controls at the 8-week time point (Figure 8, B and C).
Figure 8.

Increased inflammation and hepatic macrophage response in knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) after long-term carbon tetrachloride–induced injury: A and B: CD45 (A) and F4/80 (B) immunohistochemistry of liver sections from 4-week and 8-week carbon tetrachloride–treated KO and wild-type (WT) mice showing no change in CD45-positive or F4/80-positive cells at the 4-week carbon tetrachloride time point. C: A statistically significant increase `in F4/80-positive macrophages is observed in KO mice at the 8-week time point. D and E: Immunofluorescent staining of livers from 8-week carbon tetrachloride–treated mice for macrophage subset markers CD68 (D) and CD11b (E) shows increased numbers of pericentral CD68-positive macrophages but comparable levels of CD11b-positive macrophages. n = 5 KO mice (A and B); n = 4 WT mice (A and B). ∗P < 0.05. Scale bars = 100 μm (D and E). Original magnification, ×50 (A and B).
Notably, the qualitative increase in F4/80-positive macrophages was primarily observed in fibrotic foci centered around pericentral regions. A major function of hepatic macrophages as part of the innate immune response is the clearance of cellular debris and apoptotic bodies. Previous studies of hepatic macrophage populations have identified specific macrophage subsets that are associated with this function, including CD68-positive macrophages, which represent a restorative subset characterized by high phagocytic activity.9 In contrast, CD11b-positive macrophages were identified as having a proclivity for proinflammatory cytokine secretion.8 To further investigate the hypothesis of macrophage-mediated debris clearance, levels of CD68-positive and CD11b-positive macrophages in pericentral foci of injury were stained and quantified in the 4-week and 8-week carbon tetrachloride KO and WT cohorts. Similar levels of CD11b-positive macrophage infiltration were evident in KO and WT livers at the 4-week or 8-week carbon tetrachloride time point (Figure 8E, Supplemental Figure S7B). However, significantly higher numbers of CD68-positive macrophages in KO livers compared with WT controls at the 8-week, but not the 4-week, carbon tetrachloride time point (Figure 8D, Supplemental Figure S7A) were found. Thus, a heightened CD68-positive hepatic macrophage response during advanced fibrosis may be the mechanism of reduced numbers of TUNEL-positive hepatocytes seen in these animals, allowing for reduced hepatocellular injury. Notably, levels of CD68- and CD11b-positive macrophages were unchanged between the KO and WT cohorts at the 4-week carbon tetrachloride time point (Supplemental Figure S6), which also coincided with lack of any improvement in serum biochemical profiles in the KO mice at this time.
Discussion
Studies of chronic liver injury on the mechanisms of hepatic fibrosis progression have focused on the potential for therapeutic intervention to prevent or reverse fibrosis. The ultimate goal of such intervention is to prevent the progression of hepatic fibrosis to end stage liver diseases, such as cirrhosis or hepatocellular carcinoma, which accompany devastating clinical consequences. Alternatively, intervention to reverse fibrosis after successful treatment of the underlying hepatic insult, such as hepatitis C virus, is also of increasing relevance. The diverse signaling functions of PDGFR-α in human HSC cells were previously identified in vitro, using the PDGFR-α monoclonal antibody inhibitor olaratumab (Lartruvo),5 which is currently being studied in multiple clinical trials for soft tissue sarcoma,10 pediatric solid tumors (A Study of Olaratumab Alone and in Combination With Standard Chemotherapies in Children With Cancer), and metastatic pancreatic cancer (A Study of Nab-Paclitaxel and Gemcitabine With or Without Olaratumab (LY3012207) in Participants With Metastatic Pancreatic Cancer). Our current study provides proof-of-principle that PDGFR-α loss specifically in HSCs ameliorates early carbon tetrachloride–induced fibrosis in vivo. Furthermore, these findings highlight the nuanced, dynamic relationship among fibrosis, inflammation, and hepatocellular injury.
Lrat plays an important role in the formation of retinyl ester lipid droplets in HSCs, which is necessary for the storage of retinoids (vitamin A and its metabolites) and is one of the primary characteristics of quiescent HSCs. In a seminal study of the efficiency and specificity of Lrat promoter expression, Lrat-Cre mice expressed Cre in 99% of HSCs, and through fate tracing in toxic and cholestatic liver injury models, these HSCs produced 82% to 96% of myofibroblasts.11 Our study is the first to examine the loss of PDGFR-α in an HSC-specific manner using the HSC-specific promoter for Lrat to direct Cre expression for the excision of floxed Pdgfra in a highly specific and efficient manner. The specificity of this model was validated by confocal co-localization immunofluorescence to show loss of PDGFR-α in HSCs of KO animals.
In this study, decreased collagen deposition in KO animals was observed after 4-week carbon tetrachloride–induced liver injury but not after 8 weeks of carbon tetrachloride (Figure 3). This finding was observed with both Sirius Red and Masson Trichrome staining. However, this modest but significant decrease in collagen staining was not reflected by hydroxyproline content. However, the hydroxyproline assay detects total collagen protein in the liver (including degraded protein), whereas Sirius Red and Masson Trichrome detect intact collagen fibers that have accumulated within the tissue.12 These relative differences in sensitivity and specificity of the assays therefore likely explain these results. The transient decrease in overall fibrosis through PDGFR-α suppression in stellate cells suggests not only a contributory role of PDGFR-α but also that is likely not an effective monotherapy in the setting of chronic ongoing hepatocellular insult. Furthermore, KO animals did not have a significant reduction in fibrosis in biliary fibrosis (BDL) at 5 or 14 days after surgery. A potential explanation for the discrepancy between hepatotoxic (carbon tetrachloride) and cholestatic liver injury (BDL) is the activation of a population of portal fibroblasts during biliary liver injury. Portal fibroblasts have been previously identified as a precursor that contributes to the myofibroblast population in biliary fibrosis and show PDGFR-α expression in our model.11 Mederacke et al11 found that although most myofibroblasts originated from Lrat-tdTomato-expressing cells in toxic (carbon tetrachloride and thioacetamide) and cholestatic (BDL) forms of liver injury, the overlap between tdTomato expression and myofibroblast marker α-SMA was less robust in cholestatic liver injury (82% to 85%) compared with toxic liver injury (93% to 96%) models. In our studies, nearly all PDGFR-α was lost in whole liver lysates and myofibroblasts (as observed by immunofluorescence) at 14 days after BDL. Even so, no notable effect was seen on overall fibrosis, despite reduced parenchymal presence of HSCs likely because of reduced migration. This finding may be attributable to a less important role of PDGFR-α in portal fibroblasts. Indeed, there is conflicting evidence for a major role of PDGF signaling in portal fibroblast fibrogenicity.7,13 A study of murine BDL-derived activated portal fibroblasts found that these fibroblasts were unresponsive to PDGF in contrast to HSCs,7 whereas primary rat portal fibroblasts had no mitogenic activity in response to PDGF-BB stimulation in vitro.13 Further studies will be needed to determine whether PDGFR-α is expressed specifically in the portal fibroblast subpopulation and whether potential retention of PDGFR-α in this population can help to supplement the myofibroblast supply in a compensatory manner in Lrat-Cre Pdgfra−/− animals during biliary fibrosis.
It was found that PDGFR-α contributes to proliferation and migration of human HSCs, while lacking a direct effect on the expression of fibrosis-associated genes, including ACTA2 (α-SMA).5 These findings provide a functional explanation for how loss of PDGFR-α in vivo may alter the progression of hepatic fibrosis. For example, our finding that desmin IHC is reduced in KO livers after 4-week CCl4 injury supports the notion of impaired HSC proliferation (Figure 3A). Increased HSC death at this time point is unlikely to explain the reduction in desmin positivity because KO and WT livers had similar low levels of TUNEL staining at this time point. Consistent with our previously published findings in human HSCs,5 no differences in α-SMA expression between the WT and KO cohorts were seen under any conditions, suggesting a lack of an effect of PDGFR-α on myofibroblast transdifferentiation.
Examination of the midzonal regions of KO livers after desmin IHC also suggests a possible effect of PDGFR-α loss on HSC migration. Migration of HSCs facilitates the movement of activated HSCs toward areas of injury and inflammation and is likely to contribute the formation of characteristic bridging fibrosis seen in advanced hepatic fibrosis. On the basis of these assumptions, we posit that a reduction in the midzonal distribution of HSCs in KO livers may be partially attributable to a defect in HSC migration, particularly in biliary fibrosis, where overall HSC proliferation appears to be unaffected because of similar levels of desmin positivity. These findings support previous studies that indicate that the PDGFR-α–specific monoclonal antibody inhibitor olaratumab inhibits migration in human HSCs.5 It is challenging to address HSC migration in vivo; hence, we are unable to address how much of the reduced midzonal HSC distribution in KO mice in various liver injury models is a function of defect in migration versus reduced proliferation or survival. Because reduced fibrosis and improvement in hepatocellular injury was evident in KO mice only after carbon tetrachloride and not BDL or DDC, HSC migration may not be an equal contributor to overall injury and fibrosis in all models.
Alanine transaminase and aspartate transaminase values are the most commonly used clinical surrogate measures of hepatocyte death and overall liver injury. During carbon tetrachloride–mediated liver injury, a remarkable plateau was observed in the transaminases between the 4-week and 8-week time points in KO animals. Consistent with these changes, decreased TUNEL-positive hepatocytes were observed in KO livers, representing an overall improvement in hepatic health when compared with WT controls. Of particular interest was a concomitant increase in TUNEL-positive HSCs in KO mice at this time point. Apoptosis is a known mechanism of myofibroblast reduction during regression of fibrosis.14 Although PDGFR-α is known to play a role in cell survival in mesenchymal cell types15 against apoptotic stimuli, studies thus far have not reported a contribution of PDGFRs in promoting HSC survival independent of their effect on HSC transdifferentiation to myofibroblasts during liver injury. In fact, PDGFR-β siRNA does not induce apoptosis of HSCs isolated from rats after BDL.16 In another study of activated HSCs isolated from rat livers after BDL, PDGF-AB did not reduce rat HSC apoptosis in response to serum deprivation.17 Therefore, our findings suggest a novel and distinct prosurvival function of PDGFR-α signaling in HSCs, which was evident in KO mice and in human HSCs after exogenous PDGFR-α inhibition by olaratumab. Additional studies will be needed to determine the exact mechanism PDGFR-α–mediated HSC survival. Such mechanisms may include both ligand-dependent and -independent mechanisms, the latter of which include transforming growth factor (TGF)-β–conferred antiapoptosis, which has also previously been reported in rat HSCs in vitro18,19 and could in turn result from the PDGF ligand–independent interactions of PDGFR-α and internalized TGF-β receptor I/TGF-β receptor II heterodimer complexes after binding to TGF-β and has been demonstrated in human HSCs.20
The increase in F4/80-positive cells in KO animals centered at the pericentral hepatocyte foci of injury at 8 weeks of carbon tetrachloride–induced injury suggests that hepatic macrophages may be driving an accelerated immune-mediated clearance of apoptotic and necrotic hepatocytes and cell debris in these animals. This mechanism is further supported by the presence of increased pericentral CD68-positive cells in KO animals after 8-week carbon tetrachloride–induced liver injury, which have previously been identified as a subset of hepatic macrophages characterized by phagocytic activity.9 We posit that this effect may be responsible for the overall reduction in liver injury seen at this time point. Immune-mediated clearance of dead hepatocytes is considered an important step for effective hepatocyte regeneration. Furthermore, a previous study found that hepatic macrophage depletion in rats after carbon tetrachloride–mediated injury has differential effects during different periods of injury and recovery, with macrophage depletion early in liver injury alleviating fibrosis and macrophage depletion during carbon tetrachloride recovery resulting in slower recovery and decreased fibrosis regression.21 This dual nature of hepatic macrophages during chronic liver injury may explain the discrepancy between inflammatory macrophage infiltration and hepatocellular injury in 8-week carbon tetrachloride–induced liver injury of KO mice. Although more studies are needed, increased HSC death in KO livers may indirectly promote a beneficial inflammatory response that improves clearance of hepatocyte debris and ameliorates further progression of liver injury.
Thus, PDGFR-α has an important role in HSC biology. PDGFR-α in HSCs during hepatocellular injury appears to be essential for HSC survival, migration, and proliferation at least temporally and can regulate the immune environment, especially macrophages, to perpetuate injury and fibrosis (Figure 9). Successful inhibition of PDGFR-α may thus synergize with other inhibitors of HSC activation for a more profound effect. This finding is highly relevant because of the emergence in recent years of PDGFR-α–specific inhibitors being tested for therapeutic utility. Our study is the first to assess HSC-specific PDGFR-α loss in murine chronic liver injury and highlights both cell intrinsic and cell extrinsic mechanisms by which it regulates liver fibrosis and overall liver health.
Figure 9.

Proposed time course of the effect of Pdgfra loss in hepatic stellate cells (HSCs) during carbon tetrachloride–induced chronic liver injury in mice. During the early stages of chronic hepatocyte injury, Pdgfra loss from HSCs leads to modest reduction in fibrosis likely as a result of impaired HSC proliferation and migration. As chronic liver injury progresses, HSCs and myofibroblasts without Pdgfra are susceptible to cell death, which in turn leads to increased inflammatory cells and hepatic macrophages, likely because of proinflammatory cell debris and chemokines released from dying HSCs and myofibroblasts. The resulting influx of phagocytic hepatic macrophages improves clearing of necrotic hepatocytes near injury foci, ameliorating overall liver injury. ALT, alanine transaminase; AST, aspartate transaminase; PDGF, platelet-derived growth factor.
Footnotes
Supported by NIH grants 4R01DK095498 (S.P.M.) and 5F30DK107129 (A.K.); and Pittsburgh Liver Research Center for the Biospecimen Repository and Processing Core grant P30DK120531.
Some of the data presented in the study was taken from the graduate thesis work of A.K.
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2020.06.006.
Supplemental Data
Supplemental Figure S1.
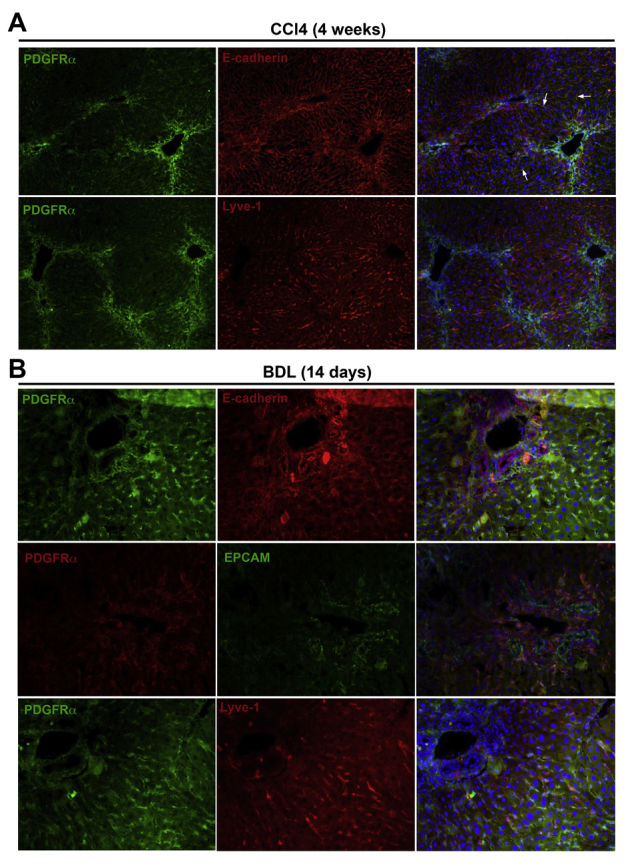
Platelet-derived growth factor receptor (PDGFR)-α expression is absent in cholangiocytes and liver sinusoidal endothelial cells and only minimally expressed in hepatocytes in murine models of chronic liver injury. A: Immunofluorescence staining for PDGFR-α and E-cadherin shows co-localization in a few hepatocytes (arrows) and PDGFR-α and lymphatic vessel endothelial receptor (Lyve)-1 shows lack of co-localization in endothelial cells in livers of 4-week carbon tetrachloride (CCl4)–treated mice. B: Immunofluorescence staining for PDGFR-α and E-cadherin, PDGFR-α and epithelial cell adhesion molecule (EPCAM), and PDGFR-α and Lyve-1 in the livers of mice 14 days after bile duct ligation (BDL) showing absence of any co-localization in hepatocytes, cholangiocytes, or endothelial cells, respectively. Original magnification: ×100 (A); ×200 (B).
Supplemental Figure S2.

Knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) have no reduction of fibrosis-associated gene expression. Real-time RT-PCR shows no significant changes in expression of Acta2, Tgfb1, Tgfb2, Fn1, Mmp9, Mmp13, Timp1, or Pdgfrb in cDNA derived from KO livers compared with wild-type (WT) controls at the 4-week carbon tetrachloride time point of liver injury. All assays were performed in triplicate, and the results are normalized to the mean gene expression of the WT cohort. n = 7 KO mice and 6 WT mice.
Supplemental Figure S3.

Knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) have no reduction in fibrosis in bile duct ligation (BDL) or 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet models of cholestatic liver injury. A and B: No change is seen at 5 days after BDL or at 14 days after BDL as assessed by Sirius Red staining, desmin, and α-smooth muscle actin (αSMA) immunohistochemistry. Collagen was quantified by quantification of Picrosirius Red images taken with polarized light microscopy (brightfield images shown). C: KO and wild-type (WT) mice fed a DDC diet for 16 days have no differences in overall hepatic fibrosis as assessed by Sirius Red, desmin, and α-SMA immunohistochemistry. Relative collagen expression was quantified using immunofluorescence staining of collagen to avoid signal interference with intraductal porphyrin plugs. n = 3 mice at 5 days and 4 mice at 14 days per genotype (A and B); n = 5 KO mice and 7 WT mice (C). Original magnification, ×50.
Supplemental Figure S4.
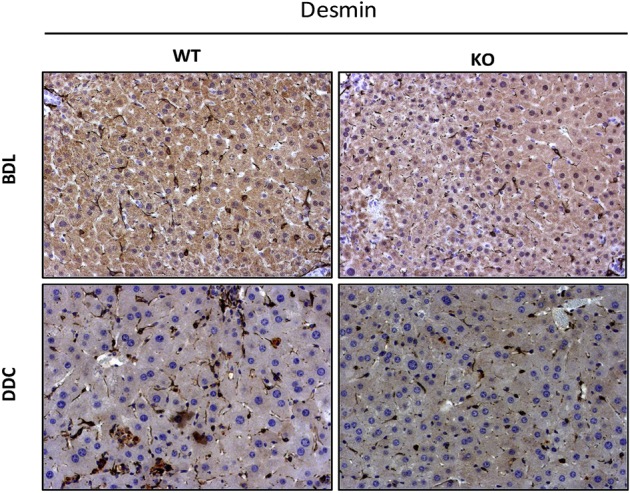
Altered morphologic features and reduced midzonal distribution of hepatic stellate cells (HSCs) in knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) after bile duct ligation (BDL) and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet–induced chronic liver injury. Representative images of desmin immunohistochemistry-stained liver sections highlight reduced distribution of HSCs in midzonal areas in KO livers compared with WT controls after BDL or DDC diet–induced chronic liver injury. Original magnification: ×100 (top row); ×200 (bottom row).
Supplemental Figure S5.

Improved serum transaminase profile in knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) after carbon tetrachloride (CCl4)–mediated liver injury. A and B: KO and wild-type (WT) mice have similar alanine transaminase (ALT) and aspartate transaminase (AST) values at the 4-week CCl4 time point (A), whereas ALT and AST values in KO mice are significantly lower than in WT mice at the 8-week CCl4 time point (B). n = 4 per cohort. ∗P < 0.05. ALP, alkaline phosphatase.
Supplemental Figure S6.

A: Knockout (KO) mice (Lrat-Cre Pdgfra−/− mice) have no reduction in serum markers of liver injury after bile duct ligation (BDL)–induced liver injury. KO and wild-type (WT) mice have no differences in alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP) total bilirubin, or direct bilirubin values at 14 days. B: Comparable longitudinal trends in serum ALT, AST, ALP, total bilirubin, or direct bilirubin values between KO and WT at 0 days, 5 days, or 14 days after BDL. n = 2 at 0 days, 3 at 5 days, and 3 at 14 days per genotype.
Supplemental Figure S7.

No difference in CD68- or CD11b-positive cells in knockout (KO) livers of 4-week carbon tetrachloride (CCl4)–treated mice (Lrat-Cre Pdgfra−/− mice) compared with wild-type (WT) controls. A and B: Immunofluorescence in 4-week CCl4-treated KO and WT mice shows no differences in the numbers of CD68-positive (A) or CD11b-positive (B) macrophages. n = 4 KO and 4 WT mice. Scale bar = 100 μm.
References
- 1.Andrae J., Gallini R., Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tallquist M., Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15:205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Kocabayoglu P., Lade A., Lee Y.A., Dragomir A.C., Sun X., Fiel M.I., Thung S., Aloman C., Soriano P., Hoshida Y., Friedman S.L. Beta-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J Hepatol. 2015;63:141–147. doi: 10.1016/j.jhep.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kikuchi A., Monga S.P. PDGFRalpha in liver pathophysiology: emerging roles in development, regeneration, fibrosis, and cancer. Gene Expression. 2015;16:109–127. doi: 10.3727/105221615X14181438356210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kikuchi A., Pradhan-Sundd T., Singh S., Nagarajan S., Loizos N., Monga S.P. Platelet-derived growth factor receptor alpha contributes to human hepatic stellate cell proliferation and migration. Am J Pathol. 2017;187:2273–2287. doi: 10.1016/j.ajpath.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kostallari E., Hirsova P., Prasnicka A., Verma V.K., Yaqoob U., Wongjarupong N., Roberts L.R., Shah V.H. Hepatic stellate cell-derived PDGFRalpha-enriched extracellular vesicles promote liver fibrosis in mice through SHP2. Hepatology. 2018;68:333–348. doi: 10.1002/hep.29803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keiko I., Chunyan J., Mingjun Z., Min C., Thomas Joseph M.-M., Tae Jun P., Xiao L., Jun X., Ping W., Yong-Han P., Fanli M., Masataka A., Lynne A.M., Alan F.H., Takashi I., Christopher K.G., David A.B., Tatiana K. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci. 2014;111:305. doi: 10.1073/pnas.1400062111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beier J.I., Kaiser J.P., Guo L., Martinez-Maldonado M., Arteel G.E. Plasminogen activator inhibitor-1 deficient mice are protected from angiotensin II-induced fibrosis. Arch Biochem Biophys. 2011;510:19–26. doi: 10.1016/j.abb.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinoshita M., Uchida T., Sato A., Nakashima M., Nakashima H., Shono S., Habu Y., Miyazaki H., Hiroi S., Seki S. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatology. 2010;53:903–910. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 10.Tap W.D., Wagner A.J., Schoffski P., Martin-Broto J., Krarup-Hansen A., Ganjoo K.N., Yen C.C., Abdul Razak A.R., Spira A., Kawai A., Le Cesne A., Van Tine B.A., Naito Y., Park S.H., Fedenko A., Papai Z., Soldatenkova V., Shahir A., Mo G., Wright J., Jones R.L., Investigators A. Effect of doxorubicin plus olaratumab vs doxorubicin plus placebo on survival in patients with advanced soft tissue sarcomas: the ANNOUNCE randomized clinical trial. JAMA. 2020;323:1266–1276. doi: 10.1001/jama.2020.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mederacke I., Hsu C.C., Troeger J.S., Huebener P., Mu X., Dapito D.H., Pradere J.P., Schwabe R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsons C.J., Bradford B.U., Pan C.Q., Cheung E., Schauer M., Knorr A., Krebs B., Kraft S., Zahn S., Brocks B., Feirt N., Mei B., Cho M.S., Ramamoorthi R., Roldan G., Ng P., Lum P., Hirth-Dietrich C., Tomkinson A., Brenner D.A. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology. 2004;40:1106–1115. doi: 10.1002/hep.20425. [DOI] [PubMed] [Google Scholar]
- 13.Wells R.G., Kruglov E., Dranoff J.A. Autocrine release of TGF-beta by portal fibroblasts regulates cell growth. FEBS Lett. 2004;559:107–110. doi: 10.1016/S0014-5793(04)00037-7. [DOI] [PubMed] [Google Scholar]
- 14.Kisseleva T., Cong M., Paik Y., Scholten D., Jiang C., Benner C., Iwaisako K., Moore-Morris T., Scott B., Tsukamoto H., Evans S.M., Dillmann W., Glass C.K., Brenner D.A. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA. 2012;109:9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonner J.C. Mesenchymal cell survival in airway and interstitial pulmonary fibrosis. Fibrogenesis Tissue Repair. 2010;3:15. doi: 10.1186/1755-1536-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S.W., Chen Y.X., Zhang X.R., Qian H., Chen W.Z., Xie W.F. Targeted inhibition of platelet-derived growth factor receptor-beta subunit in hepatic stellate cells ameliorates hepatic fibrosis in rats. Gene Ther. 2008;15:1424–1435. doi: 10.1038/gt.2008.93. [DOI] [PubMed] [Google Scholar]
- 17.Issa R., Williams E., Trim N., Kendall T., Arthur M.J., Reichen J., Benyon R.C., Iredale J.P. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut. 2001;48:548–557. doi: 10.1136/gut.48.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saile B., Matthes N., Knittel T., Ramadori G. Transforming growth factor beta and tumor necrosis factor alpha inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology. 1999;30:196–202. doi: 10.1002/hep.510300144. [DOI] [PubMed] [Google Scholar]
- 19.Saile B., Matthes N., El Armouche H., Neubauer K., Ramadori G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic stellate cells. Eur J Cell Biol. 2001;80:554–561. doi: 10.1078/0171-9335-00182. [DOI] [PubMed] [Google Scholar]
- 20.Liu C., Li J., Xiang X., Guo L., Tu K., Liu Q., Shah V.H., Kang N. PDGF receptor-alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-beta receptors. Am J Physiol Gastrointest Liver Physiol. 2014;307:G749–G759. doi: 10.1152/ajpgi.00138.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duffield J.S., Forbes S.J., Constandinou C.M., Clay S., Partolina M., Vuthoori S., Wu S., Lang R., Iredale J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clinical Investigation. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]