

Key Points
Question
What is the efficacy and safety of 4 mg and 2 mg of baricitinib in combination with background topical corticosteroid (TCS) therapy in adults with moderate to severe atopic dermatitis (AD)?
Findings
In this randomized clinical trial of 329 adults with moderate to severe AD, at week 16, a validated Investigator Global Assessment for Atopic Dermatitis score of 0 (clear) or 1 (almost clear) was achieved by 31% of patients receiving 4 mg of baricitinib with TCS therapy and 24% of patients receiving 2 mg of baricitinib with TCS therapy compared with 15% receiving placebo with TCS therapy.
Meaning
This study found a clinical benefit of Janus kinase inhibition in combination with TCS therapy, the mainstay treatment for AD.
Abstract
Importance
Baricitinib, an oral selective Janus kinase 1 and 2 inhibitor, effectively reduced disease severity in moderate to severe atopic dermatitis (AD) in 2 phase 3 monotherapy studies.
Objective
To assess the efficacy and safety of 4 mg and 2 mg of baricitinib in combination with background topical corticosteroid (TCS) therapy in adults with moderate to severe AD who previously had an inadequate response to TCS therapy.
Design, Setting, and Participants
This double-blind, placebo-controlled, phase 3 randomized clinical trial, BREEZE-AD7 (Study of Baricitinib [LY3009104] in Combination With Topical Corticosteroids in Adults With Moderate to Severe Atopic Dermatitis) was conducted from November 16, 2018, to August 22, 2019, at 68 centers across 10 countries in Asia, Australia, Europe, and South America. Patients 18 years or older with moderate to severe AD and an inadequate response to TCSs were included. After completing the study, patients were followed up for up to 4 weeks or enrolled in a long-term extension study.
Interventions
Patients were randomly assigned (1:1:1) to receive 2 mg of baricitinib once daily (n = 109), 4 mg of baricitinib once daily (n = 111), or placebo (n = 109) for 16 weeks. The use of low-to-moderate potency TCSs was allowed.
Main Outcomes and Measures
The primary end point was the proportion of patients achieving a validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) score of 0 (clear) or 1 (almost clear), with a 2-point or greater improvement from baseline at week 16.
Results
Among 329 patients (mean [SD] age, 33.8 [12.4] years; 216 [66%] male), at week 16, a vIGA-AD score of 0 (clear) or 1 (almost clear) was achieved by 34 patients (31%) receiving 4 mg of baricitinib and 26 (24%) receiving 2 mg of baricitinib compared with 16 (15%) receiving placebo (odds ratio vs placebo, 2.8 [95% CI, 1.4-5.6]; P = .004 for the 4-mg group; 1.9 [95% CI, 0.9-3.9]; P = .08 for the 2-mg group). Treatment-emergent adverse events were reported in 64 of 111 patients (58%) in the 4-mg group, 61 of 109 patients (56%) in the 2-mg group, and 41 of 108 patients (38%) in the placebo group. Serious adverse events were reported in 4 patients (4%) in the 4-mg group, 2 (2%) in the 2-mg group, and 4 (4%) in the placebo group. The most common adverse events were nasopharyngitis, upper respiratory tract infections, and folliculitis.
Conclusions and Relevance
A dose of 4 mg of baricitinib in combination with background TCS therapy significantly improved the signs and symptoms of moderate to severe AD, with a safety profile consistent with previous studies of baricitinib in AD.
Trial Registration
ClinicalTrials.gov Identifier: NCT03733301
This randomized clinical trial assesses the efficacy and safety of 4 mg and 2 mg of baricitinib in combination with background topical corticosteroid therapy in adults with moderate to severe AD who previously had an inadequate response to topical corticosteroid therapy.
Introduction
Atopic dermatitis (AD) is a common, chronic, relapsing inflammatory skin disease that affects up to 25% of children and 2% to 7% of adults globally.1,2,3 Symptoms of AD include intense itch, sleep disturbance, and skin pain, leading to a significant effect on quality of life.4,5 Emollients and topical corticosteroids (TCSs) are the mainstay of treatment.6,7,8 For patients with moderate to severe AD for whom topical therapy is insufficient, the addition of phototherapy and/or systemic treatment is recommended.8,9,10 Several advances have recently been made regarding our understanding of the molecular pathogenesis of AD, and as a result, more targeted therapies are being developed. One of these, dupilumab, a human anti–interleukin 4 receptor α (IL-4Rα) antibody, has been approved in the US and European Union for the treatment of patients with moderate to severe AD.11,12 Given the heterogenous nature of AD, ongoing and future clinical studies are needed to continue to identify novel treatments that target the underlying disease pathologic features, providing additional therapeutic options for patients.
Baricitinib, an oral selective Janus kinase (JAK)1/JAK2 inhibitor,13 inhibits several cytokines in AD pathogenesis, including thymic stromal lymphopoietin, IL-4, IL-5, IL-13, IL-22, and IL-31.14,15,16 In the Studies of Baricitinib (LY3009104) in Patients with Moderate to Severe Atopic Dermatitis, BREEZE-AD1 and BREEZE-AD2, 2 independent, 16-week, phase 3 trials of patients with moderate to severe AD who had an inadequate response to TCSs, 2-mg and 4-mg once-daily oral baricitinib monotherapy was superior to placebo for improving several clinical signs and symptoms of AD, with an acceptable safety profile.17 The BREEZE-AD1 and BREEZE-AD2 studies validated the role of JAK1/JAK2 signaling in AD pathogenesis. This study reports the first, to our knowledge, 16-week, phase 3 trial (BREEZE-AD7) investigating baricitinib treatment in combination with background TCS therapy in patients with moderate to severe AD who previously had an inadequate response to TCS therapy, a regimen that more closely mirrors clinical practice.
Methods
Study Design
In this multicenter, double-blind, placebo-controlled, parallel-arm, 16-week, phase 3 randomized clinical trial (NCT03733301), investigators evaluated patients at 68 centers in 10 countries in Asia, Australia, Europe, and South America. The study was conducted from November 16, 2018, to August 22, 2019 (trial protocol given in Supplement 1). Eligible patients were 18 years or older and had a diagnosis of AD, as defined by the American Academy of Dermatology,18 at least 12 months before screening and a documented history of inadequate response to topical therapies within 6 months before screening. Full inclusion and exclusion criteria are given in the eAppendix in Supplement 2. The study was conducted in accordance with ethical principles of the Declaration of Helsinki19 and Good Clinical Practice guidelines. All investigation sites received approval from the appropriate authorized institutional review board or ethics committee. All patients provided written informed consent before the study-related procedures were undertaken. All data were deidentified. This study followed the Consolidated Standards of Reporting Trials (CONSORT) reporting guideline.
Patients were randomly allocated 1:1:1 by computer-generated random sequence to placebo (n = 109), 2 mg of baricitinib (n = 109), or 4 mg of baricitinib (n = 111). Patients were stratified by baseline disease severity (validated Investigator Global Assessment for Atopic Dermatitis [vIGA-AD]20 score of 3 [moderate] or 4 [severe]) and region (Europe, Japan, or other [Argentina, Australia, South Korea, and Taiwan]). Double-blind investigational product tablets were provided to patients at each visit (eMethods in Supplement 2). eFigure 1 in Supplement 2 shows the study design.
Patients discontinued topical therapy 2 weeks and systemic therapy 4 weeks before randomization. Patients applied emollients daily during the 14 days that preceded randomization and throughout the study. After an 8- to 35-day screening period, once-daily oral baricitinib or placebo was given for 16 weeks. All patients received moderate- and/or low-potency TCSs (such as 0.1% triamcinolone cream and 2.5% hydrocortisone ointment, respectively) for active lesions; topical calcineurin inhibitors and/or crisaborole, in countries where approved, could be used in place of TCSs, with guidance to limit use to areas considered inadvisable for TCSs. Rescue therapy with high- or ultrahigh-potency TCSs or systemic therapies was available for patients who experienced worsening and unacceptable AD symptoms after 2 weeks of treatment. A further description of TCS use and rescue procedures is given in the eMethods in Supplement 2. Patients who completed the 16-week treatment period were eligible to participate in the long-term extension study, BREEZE-AD3.21
Per protocol, clinical laboratory tests, vital signs, and other safety assessments were evaluated on scheduled study visits. A daily electronic diary captured patient-reported questionnaires for Itch Numeric Rating Scale (NRS), Skin Pain NRS, AD Sleep Scale (ADSS), Patient Global Impression of Severity for AD, and TCS use.
Outcomes
The primary objective was adjusted for multiplicity and tested the superiority of 4 mg of baricitinib plus TCSs or 2 mg of baricitinib plus TCSs with placebo plus TCSs by assessing the proportion of patients with AD who achieved a vIGA-AD score of 0 (clear) or 1 (almost clear) and had a 2-point or greater improvement from baseline at week 16. The vIGA-AD is a static 5-point scale with scores from 0 (clear) to 4 (severe) that measures the physician’s global assessment of the patient’s overall disease severity. Key secondary outcomes were adjusted for multiplicity and included the following: proportion of patients who achieved 75% and 90% improvement in Eczema Area and Severity Index (EASI) score (EASI75 and EASI90) at week 16, 75% improvement in the SCORing Atopic Dermatitis index (SCORAD75) at week 16, and 4-point or greater improvement on the Itch NRS among patients with baseline score ≥4 on day 2 and weeks 1, 2, 4, and 16; percent change from baseline (CFB) in total EASI score at week 16; and mean CFB on the Skin Pain NRS at week 16 and on the Item 2 score of the ADSS (number of nighttime awakenings) on weeks 1 and 16. The Itch NRS and Skin Pain NRS are 11-point scales (0 indicating no itch or pain and 10 indicating worst itch or pain imaginable); patients rated the worst severity in the previous 24 hours in the daily diaries. Other secondary outcomes not adjusted for multiplicity and post hoc end points are described in the eMethods in Supplement 2. Safety assessments included adverse events (AEs), laboratory studies, and vital signs (detailed in the eMethods in Supplement 2).
Statistical Analysis
Randomization of approximately 100 patients per treatment group was estimated to provide 89% power to detect any difference vs placebo in the vIGA-AD (score of 0 or 1) response rates at week 16 (power calculations detailed in the eMethods in Supplement 2). The primary and secondary end points were analyzed according to the prespecified statistical analysis plan (eMethods in Supplement 2), namely, with a graphical testing procedure using a multiplicity adjustment to control for the overall familywise type l error rate at a 2-sided α = .05. These procedures were separately prespecified for the US and Japan as well as the European Union regulatory submissions based on different regional doses and end point objectives (eFigures 2 and 3 in Supplement 2). There were no adjustments for multiple comparisons for other analyses.
Efficacy and health outcomes data were analyzed with the intention-to-treat population, defined as all randomized patients. For categorical end points, a logistic regression model was used for comparisons with region, baseline disease severity (vIGA-AD), baseline value, and treatment group in the model except for the analysis on vIGA-AD, which included region, baseline severity (vIGA-AD), and treatment group in the model. For continuous end points, a restricted maximum likelihood–based mixed-effects model of repeated measures was used for comparisons with treatment, region, baseline disease severity (vIGA-AD), visit, and treatment × visit interaction as fixed categorical effects and baseline and baseline × visit interaction as fixed continuous effects. For daily diary assessments, the model for analyses up to week 16 included all weekly assessments.
Safety analyses included all randomized patients who received 1 dose or more of study drug and who did not discontinue participation in the study for the reason of lost to follow-up at the first postbaseline visit. Adverse events were inclusive of the treatment period. Comparisons for AEs, discontinuation, and other categorical safety data were assessed with the Fisher exact test. Continuous safety variables were analyzed using analysis of covariance with baseline value and treatment group in the model.
Unless noted, efficacy and health outcomes data were analyzed using the primary censoring rule, which censored data after permanent discontinuation of use of the study drug or after rescue therapy (equivalent to using all data up to rescue). For categorical end points, a nonresponder imputation was applied after censoring; for continuous end points, data after rescue were censored and set as missing, and mixed-effects models of repeated measures analyses were performed. All statistical tests of treatment effects were 2-sided with a significance level of P = .05 unless otherwise stated (ie, graphical multiple testing strategy). Statistical analyses were performed using SAS software, version 9.4 (SAS Institute Inc).
Results
Patients
Of 378 patients screened, 329 patients (mean [SD] age, 33.8 [12.4] years; 216 [66%] male) were randomized, and 309 patients (94%) completed the study (Figure 1). No patients in the intention-to-treat population were excluded from the efficacy and health outcomes analyses. One patient failed screening and was randomized to the placebo group in error. The patient did not receive study treatment and was excluded from safety analyses. Protocol deviations are given in the eResults in Supplement 2. Baseline demographics and disease characteristics were balanced among treatment groups (Table 1 and eTable 1 in Supplement 2).
Figure 1. CONSORT Diagram.

TCS indicates topical corticosteroid.
aOne patient failed screening and was randomized in error, and 1 patient was noncompliant with study visits.
Table 1. Baseline Demographics and Disease Characteristicsa.
| Characteristic | Placebo and TCSs (n = 109) | 2 mg of Baricitinib and TCSs (n = 109) | 4 mg of Baricitinib and TCSs (n = 111) |
|---|---|---|---|
| Age, mean (SD), y | 33.7 (13.2) | 33.8 (12.8) | 33.9 (11.4) |
| Female | 38 (35) | 39 (36) | 36 (32) |
| Race | |||
| Asian | 57 (52) | 57 (52) | 54 (49) |
| White | 46 (42) | 50 (46) | 54 (49) |
| Otherb | 6 (6) | 2 (2) | 3 (3) |
| Time since AD diagnosis, mean (SD), y | 22.0 (12.2) | 24.6 (14.8) | 25.5 (13.2) |
| Weight, mean (SD), kg | 73.0 (15.8) | 72.4 (15.5) | 73.3 (17.8) |
| BMI, mean (SD) | 25.5 (4.6) | 25.2 (4.7) | 25.1 (5.1) |
| Geographic region | |||
| Europe | 38 (35) | 38 (35) | 39 (35) |
| Japan | 21 (19) | 20 (18) | 22 (20) |
| Otherc | 50 (46) | 51 (47) | 50 (45) |
| vIGA-AD score of 4d | 48/108 (44) | 50 (46) | 50 (45) |
| EASI score, mean (SD)e | 28.5 (12.3) | 29.3 (11.9) | 30.9 (12.6) |
| SCORAD score, mean (SD)f | 66.6 (13.8) | 66.8 (14.0) | 68.3 (13.2) |
| Body surface area affected, mean (SD) | 48.1 (24.4) | 50.6 (21.6) | 52.1 (23.3) |
| Itch NRS score, mean (SD)g | 7.4 (1.7) | 7.0 (2.1) | 7.0 (2.0) |
| Skin Pain NRS score, mean (SD)h | 6.8 (2.3) | 6.3 (2.5) | 6.0 (2.5) |
| ADSS Item 2 score, mean (SD)i | 1.8 (2.0) | 1.9 (2.3) | 1.8 (2.3) |
| POEM score, mean (SD)j | 20.9 (6.7) | 21.0 (6.3) | 21.4 (6.0) |
| DLQI score, mean (SD)k | 15.0 (7.9) | 15.0 (7.7) | 14.7 (7.9) |
| PGI-S-AD score, mean (SD)l | 4.2 (0.8) | 3.9 (0.8) | 4.0 (0.8) |
| HADS score, mean (SD)m | |||
| Anxiety | 6.8 (4.3) | 6.4 (4.0) | 6.7 (4.4) |
| Depression | 5.8 (4.3) | 5.3 (3.7) | 5.5 (4.1) |
| WPAI-AD score, mean (SD)n | |||
| Absenteeism | 10.9 (25.5) | 9.1 (20.3) | 8.8 (21.6) |
| Presenteeism | 43.0 (26.5) | 49.4 (24.6) | 45.1 (26.7) |
| Overall impairment (work productivity loss) | 45.8 (28.5) | 51.7 (25.5) | 47.0 (27.6) |
| Activity impairment | 52.9 (28.0) | 57.1 (25.3) | 52.2 (26.0) |
| EQ-5D-5L score, mean (SD)o | |||
| VAS | 57.2 (23.1) | 58.0 (22.3) | 57.4 (22.6) |
| Health State Index | |||
| US | 0.7 (0.2) | 0.7 (0.2) | 0.7 (0.2) |
| UK | 0.6 (0.2) | 0.6 (0.3) | 0.6 (0.3) |
Abbreviations: AD, atopic dematitis; ADSS, Atopic Dermatitis Sleep Scale; BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); DLQI, Dermatology Life Quality Index; EASI, Eczema Area and Severity Index; EQ-5D-5L, European Quality of Life-5 Dimensions–5 Levels; HADS, Hospital Anxiety Depression Scale; NRS, Numeric Rating Scale; PGI-S-AD, Patient Global Impression of Severity for Atopic Dermatitis; POEM, Patient Oriented Eczema Measures; SCORAD, SCORing Atopic Dermatitis; TCS, topical corticosteroid; VAS, visual analog scale; vIGA-AD, validated Investigator Global Assessment for Atopic Dermatitis; WPAI-AD, Work Productivity and Activity Impairment for Atopic Dermatitis.
Data are presented as number (percentage) of patients unless otherwise indicated.
American Indian or Alaska native, Black or African American, native Hawaiian or other Pacific Islander, or multiple races.
Argentina, Australia, South Korea, and Taiwan.
Measures the investigator global assessment of disease severity based on a static 5-point scale ranging from 0 (clear skin) to 4 (severe disease).
Scores range from 0 to 72, with higher scores indicating greater severity.
Combined score of investigator-reported disease severity and affected body surface area and patient-reported symptoms of itch and sleep dysfunction; scores range from 0 to 103, with higher scores indicating greater disease severity.
Scores range from 0 (no itch) to 10 (worst itch imaginable).
Scores range from 0 (no pain) to 10 (worst pain imaginable).
Assesses the frequency of nighttime awakenings attributable to itch the previous night on a scale of 0 to 29, with higher scores indicating a greater number of awakenings owing to itch each night.
Composite measure of patient-reported symptoms, including the effect of symptoms on sleep, and evaluates the frequency of symptoms (including itch) and the effect of atopic dermatitis on sleep on a scale of 0 to 28, with higher scores indicating greater disease severity.
Evaluates health-related quality of life on a scale of 0 to 30, with higher scores indicating a greater effect on a patient's life.
Evaluates patient-reported severity, ranging from no symptoms to severe.
Evaluates symptoms of anxiety and depression; scores range from 0 to 21, with higher scores indicating greater anxiety or depression.
Assesses overall work productivity and impairment; scores are calculated as percentages of impairment, with higher scores indicating greater impairment and less productivity.
Evaluates health-related quality of life consisting of 2 components: a descriptive system of the respondent’s health and a rating of his/her current health state using a 0-mm (worst imaginable health state) to 100-mm (best imaginable health state) VAS.
Efficacy
The primary end point and key secondary end points were assessed in separate graphical testing procedures for the US and Japan as well as the European Union (Table 2 and eFigures 2 and 3 in Supplement 2). The proportion of patients who achieved the primary end point of vIGA-AD score of 0 or 1 and had a 2-point or greater improvement from baseline at week 16 was significantly higher for patients treated with 4 mg of baricitinib vs placebo (34 of 111 [31%]; P = .004) (Figure 2A and Table 2). The primary end point for 2 mg of baricitinib was not met (26 of 109 [24%]; P = .08); further statistical testing was stopped for the 2-mg group; therefore, no key secondary end point achieved statistical significance within the graphical testing procedures for 2 mg (Table 2). The 4 mg of baricitinib group had significant improvement compared with the placebo group (P < .001) for the following key secondary end points with the US and Japan graphical testing procedure: proportion of patients who achieved an EASI75 response at week 16 (53 of 111 [48%] in the 4-mg group, 47 of 109 [43%] in the 2-mg group, and 25 of 109 [23%] in the placebo group) (Figure 2B and Table 2), percent CFB in the total EASI score at week 16 (−67% in the 4-mg group, −58% in the 2-mg group, and −45% in the placebo group) (Table 2 and eFigure 4A in Supplement 2), proportion of patients who achieved a 4-point or greater improvement on the Itch NRS at week 4 (52 of 100 [52%] for the 4-mg group, 33 of 97 [34%] for the 2-mg group, and 11 of 104 [11%] for the placebo group) and week 16 (44 of 100 [44%] for the 4-mg group, 37 of 97 [38%] for the 2-mg group, and 21 of 104 [20%] for the placebo group) (Figure 2C and Table 2), and mean (SE) CFB on the Skin Pain NRS at week 16 (−3.7 [0.2] for the 4-mg group, −3.2 [0.2] for the 2-mg group, and −2.1 [0.2] for the placebo group) (Table 2 and eFigure 7 in Supplement 2).
Table 2. Primary and Key Secondary Outcomes in the Intention-to-Treat Populationa.
| Outcome | Placebo and TCSs (n = 109) | 2 mg of Baricitinib and TCSs (n = 109) | 4 mg of Baricitinib and TCSs (n = 111) | ||||
|---|---|---|---|---|---|---|---|
| All | Comparison with placebo (95% CI) | P valueb | All | Comparison with placebo (95% CI) | P valueb | ||
| Primary outcome | |||||||
| vIGA-AD score of 0 or 1c | 16 (15) | 26 (24) | 1.9 (0.9 to 3.9) | .08 | 34 (31) | 2.8 (1.4 to 5.6) | .004 |
| Key secondary outcomes | |||||||
| EASI75c | 25 (23) | 47 (43) | 2.6 (1.4 to 4.8) | NA | 53 (48) | 3.3 (1.8 to 6.0) | <.001 |
| Percent change from baseline in total EASI score, LSM (SE)d | −45.1 (3.8) | −58.2 (3.7) | −13.1 (−23.4 to −2.7) | NA | −67.2 (3.7) | −22.1 (−32.5 to −11.8) | <.001 |
| ≥4-Point improvement in Itch NRS scorec | |||||||
| Day 2 | 2/104 (2) | 5/97 (5) | 2.4 (0.6 to 10.5) | NA | 8/100 (8) | 3.9 (1.0 to 15.8) | NA |
| Week 1 | 4/104 (4) | 8/97 (8) | 2.1 (0.7 to 6.8) | NA | 9/100 (9) | 2.5 (0.8 to 8.0) | NA |
| Week 2 | 16/104 (15) | 23/97 (24) | 1.8 (0.9 to 3.8) | NA | 33/100 (33) | 3.1 (1.5 to 6.2) | NA |
| Week 4 | 11/104 (11) | 33/97 (34) | 4.7 (2.2 to 10.0) | NA | 52/100 (52) | 10.4 (4.9 to 22.2) | <.001 |
| Week 16 | 21/104 (20) | 37/97 (38) | 2.9 (1.5 to 5.6) | NA | 44/100 (44) | 3.8 (2.0 to 7.5) | <.001 |
| Change from baseline, LSM (SE)d | |||||||
| Skin Pain NRS | −2.1 (0.2) | −3.2 (0.2) | −1.2 (−1.8 to −0.5) | NA | −3.7 (0.2) | −1.7 (−2.3 to −1.0) | <.001 |
| Item 2 of the ADSS at week 1 | −0.5 (0.1) | −0.7 (0.1) | −0.2 (−0.5 to 0.0) | NA | −0.9 (0.1) | −0.4 (−0.7 to −0.2) | NA |
| Item 2 of the ADSS at week 16 | −0.5 (0.2) | −1.3 (0.1) | −0.8 (−1.2 to −0.4) | NA | −1.4 (0.1) | −0.9 (−1.3 to −0.5) | NA |
| EASI90c | 15 (14) | 18 (17) | 1.2 (0.6 to 2.6) | NA | 27 (24) | 2.1 (1.0 to 4.2) | NA |
| SCORAD75c | 8 (7) | 12 (11) | 1.5 (0.6 to 3.8) | NA | 20 (18) | 2.7 (1.2 to 6.3) | NA |
Abbreviations: ADSS, Atopic Dermatitis Sleep Scale; EASI, Eczema Area and Severity Index; LSM, least-squares mean; NA, not applicable; NRS, Numeric Rating Scale; SCORAD, SCORing Atopic Dermatitis; TCS, topical corticosteroid; vIGA-AD, validated Investigator Global Assessment for Atopic Dermatitis.
Data are presented as number (percentage) of patients unless otherwise indicated. Data were assessed at week 16 unless otherwise specified. Data were analyzed with a logistic regression model with nonresponder imputation for response rates and mixed-models repeated-measure analysis for LSM change and percent change from baseline. P values are for comparisons of 2 or 4 mg of baricitinib with placebo.
To control the overall familywise type l error rate for multiple comparisons, the primary and key secondary end points were analyzed according to the prespecified statistical analysis plan with a graphical testing procedure. Results are shown for the US and Japan graphical testing procedure.
Comparisons are odds ratios.
Comparisons are LSM difference.
Figure 2. Efficacy and Health Outcomes.
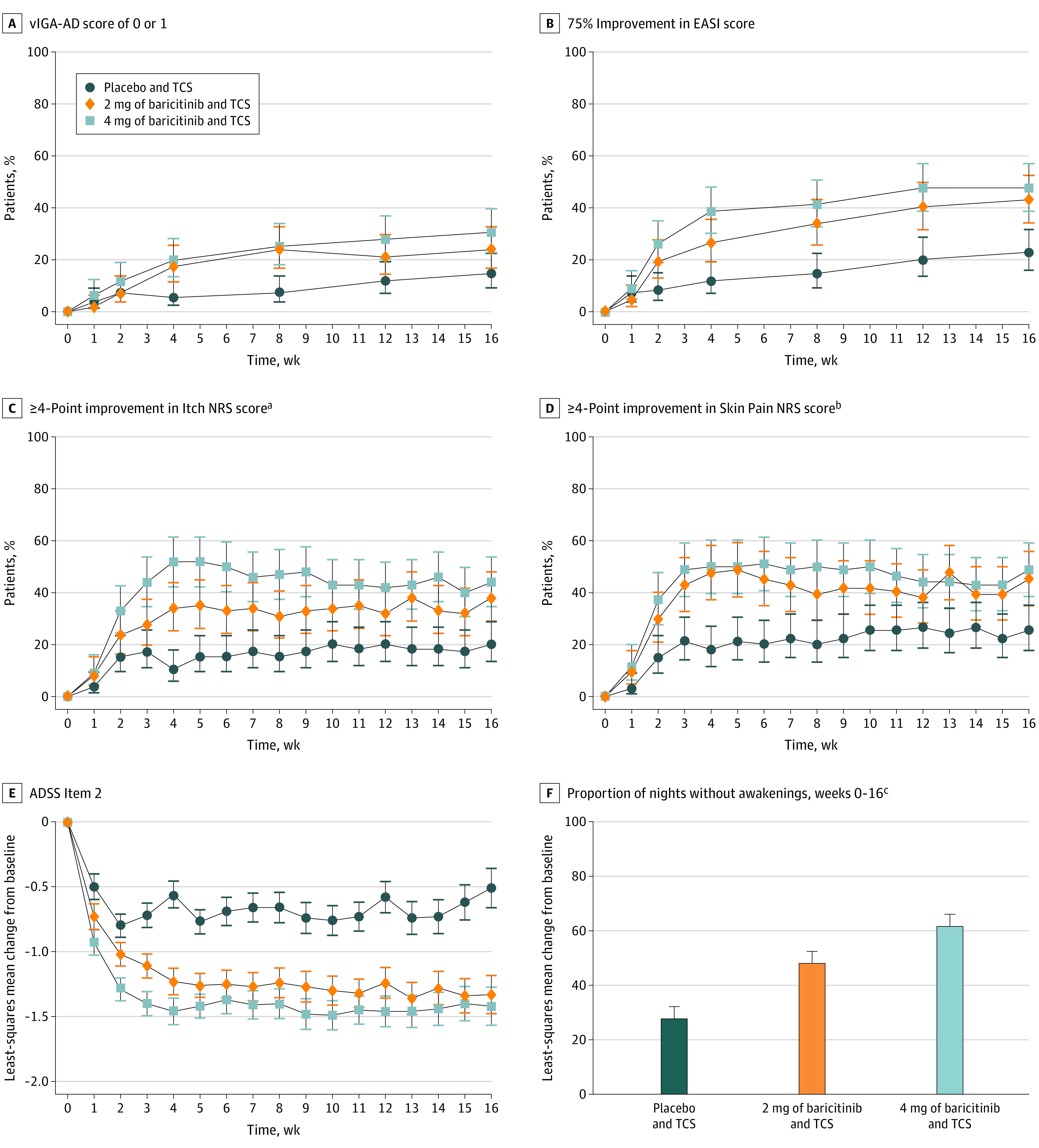
Data were assessed for patients in the intention-to-treat population and analyzed with a logistic regression model with nonresponder imputation for response rates and mixed-models repeated-measure analysis or analysis of covariance for least-squares mean change from baseline. ADSS indicates Atopic Dermatitis Sleep Scale; EASI, Eczema Area and Severity Index; NRS, Numeric Rating Scale; TCS, topical corticosteroid; and vIGA-AD, validated Investigator Global Assessment for Atopic Dermatitis.
aAssessed for patients with a baseline Itch NRS score of 4 or greater.
bAssessed for patients with a baseline Skin Pain NRS score of 4 or greater.
cAssessed for patients with a baseline ADSS Item 2 score greater than 1.
Summary statistics are provided for the remaining key secondary end points. End points assessed at week 16 included the proportion of patients who achieved an EASI90 response (27 of 111 [24%] in the 4-mg group, 18 of 109 [17%] in the 2-mg group, and 15 of 109 [14%] in the placebo group) (Table 2 and eFigure 4B in Supplement 2) and a SCORAD75 response (20 of 111 [18%] in the 4-mg group, 12 of 109 [11%] in the 2-mg group, and 8 of 109 [7%] in the placebo group) (Table 2). The proportion of patients who achieved a 4-point or greater improvement on the Itch NRS was also assessed at day 2 (8 of 100 [8%] in the 4-mg group, 5 of 97 [5%] in the 2-mg group, and 2 of 104 [2%] in the placebo group) (Table 2 and eFigure 6 in Supplement 2), week 1 (9 of 100 [9%] in the 4-mg group, 8 of 97 [8%] in the 2-mg group, and 4 of 104 [4%] in the placebo group), and week 2 (33 of 100 [33%] in the 4-mg group, 23 of 97 [24%] in the 2-mg group, and 16 of 104 [15%] in the placebo group) (Figure 2C and Table 2). The mean (SE) CFB in the ADSS Item 2 was assessed at week 1 (−0.9 [0.1] in the 4-mg group, −0.7 [0.1] in the 2-mg group, and −0.5 [0.1] in the placebo group) and week 16 (−1.4 [0.1] in the 4-mg group, −1.3 [0.1] in the 2-mg group, and −0.5 [0.2] in the placebo group) (Figure 2E and Table 2).
Outside the multiplicity-adjusted analysis, baricitinib treatment improved multiple other prespecified secondary efficacy outcomes as described in the eResults and eTable 2 in Supplement 2 and showed improvements in itch (as early as 2 days after initiating treatment for CFB and percent CFB on the Itch NRS [eTable 2 in Supplement 2] and as early as 4 days for the proportion of patients achieving a 4-point or greater improvement on the Itch NRS [4 mg only] compared with placebo [Figure 2C and eFigures 5 and 6 in Supplement 2]), skin pain (Figure 2D and eFigure 7 in Supplement 2), and sleep disturbance due to itch (Figure 2E and F and eFigure 8 in Supplement 2) over time. Baricitinib-treated patients had an increase in the proportion of TCS-free days during 16 weeks compared with those given placebo and a reduction in the amount of background TCSs used (eFigure 11 and eTables 3 and 4 in Supplement 2). Overall, few baricitinib-treated patients (11 of 220 [5%]) required rescue treatment during the treatment period (eTable 5 in Supplement 2).
Safety
A higher proportion of patients reported 1 or more treatment-emergent AEs with 4 mg of baricitinib (64 of 111 [58%]) and 2 mg of baricitinib (61 of 109 [56%]) than with placebo (41 of 108 [38%]) (Table 3). The most frequently reported (≥2% in any treatment group) treatment-emergent AEs for both baricitinib doses compared with placebo were nasopharyngitis, folliculitis, oral herpes, upper respiratory tract infection, acne, diarrhea, and back pain (Table 3). Frequencies of serious AEs were similar across treatments (Table 3), with no clinically meaningful differences among treatments when grouped by system organ class and preferred term (eTable 6 in Supplement 2). A higher proportion of patients treated with 4 mg of baricitinib had AEs that required permanent discontinuation (5 of 111 [5%]) vs 2 mg of baricitinib (0 patients) or placebo (1 of 108 [1%]) (Table 3). One pulmonary embolism (1%) was reported 10 weeks after starting baricitinib therapy in a 51-year-old patient treated with 4 mg of baricitinib who was receiving oral contraceptives and was a previous smoker (half a pack per day for 14 years) (Table 3). The patient discontinued treatment per protocol requirements and recovered from the event. No malignant tumors, major adverse cardiovascular events, or deaths were reported (Table 3).
Table 3. Safety Overview in the Safety Populationa.
| Variable | Patients, No. (%) | ||
|---|---|---|---|
| Placebo and TCSs (n = 108) | 2 mg of Baricitinib and TCSs (n = 109) | 4 mg of Baricitinib and TCSs (n = 111) | |
| Any treatment-emergent adverse event | 41 (38) | 61 (56) | 64 (58) |
| Treatment-emergent adverse event by severity | |||
| Mild | 20 (19) | 34 (31) | 33 (30) |
| Moderate | 18 (17) | 21 (19) | 25 (23) |
| Severe | 3 (3) | 6 (6) | 6 (5) |
| Serious adverse event | 4 (4) | 2 (2) | 4 (4) |
| Adverse event leading to permanent treatment discontinuationb | 1 (1) | 0 | 5 (5) |
| Death | 0 | 0 | 0 |
| Infectious adverse events with frequency of at least 2% in any treatment groupc | |||
| Nasopharyngitis | 13 (12) | 12 (11) | 17 (15) |
| Upper respiratory tract infection | 2 (2) | 8 (7) | 3 (3) |
| Folliculitis | 0 | 4 (4) | 6 (5) |
| Oral herpes virus infection | 0 | 4 (4) | 4 (4) |
| Herpes simplex virus infection | 3 (3) | 1 (1) | 3 (3) |
| Vaginal infectiond | 0 | 1/39 (3) | 0 |
| Non-infectious adverse events with frequency of at least 2% in any treatment groupc | |||
| Oropharyngeal pain | 3 (3) | 2 (2) | 2 (2) |
| Acne | 1 (1) | 1 (1) | 4 (4) |
| Back pain | 1 (1) | 0 | 4 (4) |
| Diarrhea | 1 (1) | 1 (1) | 3 (3) |
| Pyrexia | 3 (3) | 0 | 1 (1) |
| Allergic rhinitis | 3 (3) | 0 | 0 |
| Blood CPK level increase | 0 | 3 (3) | 0 |
| Oligomenorrhoead | 0 | 0 | 1/36 (3) |
| Adverse event of special interest | |||
| Any treatment-emergent infection | 26 (24) | 41 (38) | 37 (33) |
| Serious infection | 2 (2) | 0 | 0 |
| Herpes zoster virus infection | 1 (1) | 2 (2) | 0 |
| Opportunistic infection | 1 (1) | 0 | 0 |
| Tuberculosis | 0 | 0 | 0 |
| Deep vein thrombosise | 0 | 0 | 0 |
| Pulmonary embolisme | 0 | 0 | 1 (1) |
| MACEe,f | 0 | 0 | 0 |
| Gastrointestinal perforation | 0 | 0 | 0 |
| Malignant tumors other than NMSC | 0 | 0 | 0 |
| NMSC | 0 | 0 | 0 |
Abbreviations: CPK, creatine phosphokinase; MACE, major cardiovascular event; NMSC, nonmelanoma skin cancer; TCS, topical corticosteroid.
Data were assessed from weeks 0 to 16.
Includes the following preferred terms: placebo: postoperative abscess (serious; n = 1); and 4 mg of baricitinib: pulmonary embolism (serious, n = 1), asthma (serious, n = 1), abdominal pain (n = 1), and toxicoderma (n = 2).
Events are listed according to the preferred term in the Medical Dictionary for Regulatory Activities.
Denominator adjusted because this is a sex-specific event for women: placebo, n = 38; 2 mg of baricitinib, n = 39; and 4 mg of baricitinib, n = 36.
Adjudicated by an independent clinical event committee.
A MACE was defined as cardiovascular death, myocardial infarction, or stroke as adjudicated by an independent clinical event committee.
There was a greater proportion of patients with 1 or more treatment-emergent infections in the 4 mg of baricitinib group (37 of 111 [33%]) and 2 mg of baricitinib group (41 of 109 [38%]) than in the placebo group (26 of 108 [24%]) (Table 3). Increases in the proportion of infection with baricitinib were primarily attributable to folliculitis, oral herpes, and upper respiratory tract infection. There were 2 reports of serious infection, including 1 opportunistic infection (ocular toxoplasmosis), with placebo; none were reported with baricitinib (Table 3). Two cases of nonserious, localized herpes zoster virus infection were reported with 2 mg of baricitinib, and 1 case was reported with placebo (Table 3). There were 15 cases of herpes simplex virus infection reported across treatment groups; of those, 8 were reported as oral herpes, and 7 were reported as herpes simplex virus infection (Table 3). Area affected by herpes simplex virus infections was reported in 6 of the 7 cases; all cases reported 0.5% or less body surface area involvement. The proportion of patients developing skin infections that required antibiotic treatment at week 16 was similar across treatments (3 of 111 [3%] in those treated with 4 mg of baricitinib, 5 of 109 [5%] for those treated with 2 mg of baricitinib, and 3 of 108 [3%] for those treated with placebo). There were 3 cases (3%) of conjunctivitis reported with 2 mg of baricitinib and 2 cases (2%) reported with placebo; none were reported with 4 mg of baricitinib.
Creatinine phosphokinase levels were elevated with baricitinib compared with placebo, with most increases being Common Terminology Criteria for Adverse Events (CTCAE) grades 1 and 2 and few grade 3 or higher increases (eTable 7 in Supplement 2); elevations were not associated with AEs indicative of muscle injury. Hematologic changes that occurred during the study are described in eTable 8 in Supplement 2. One patient had CTCAE grade 3 or higher neutrophil count changes while taking 4 mg of baricitinib, and 2 patients in the 4 mg of baricitinib group and 1 patient in the placebo group had CTCAE grade 3 or higher lymphocyte count changes. Two patients in the 2 mg of baricitinib group had platelet count increases (≥600 billion/L) with no associated AEs; no patients in the 4 mg of baricitinib or placebo group had platelet count increases (≥600 billion/L). Changes were seen in lipid levels, including increases in high-density lipoprotein level (≥60 mg/dL [to convert to millimoles per liter, multiply by 0.0259]; 4-mg group, 28%; 2-mg group, 17%; and placebo group, 10%). Changes in low-density lipoprotein level were similar among treatment groups (≥160 mg/dL; 4-mg group, 3%; 2-mg group, 3%; and placebo group, 4%). There were no increases in alanine aminotransferase level greater than or equal to 3 times the upper limit of normal in the study.
Discussion
To our knowledge, this is the first double-blind, placebo-controlled, phase 3 randomized clinical trial of baricitinib in combination with TCSs for the treatment of adults with moderate to severe AD. Treatment with 4 mg of baricitinib plus TCSs achieved a statistically significant improvement compared with placebo plus TCSs in the proportion of patients achieving a vIGA-AD score of 0 or 1 with a 2-point or greater improvement from baseline at week 16 (primary end point) and successfully improved signs and symptoms of AD. Treatment with 2 mg of baricitinib plus TCSs did not meet the primary end point; thus, none of the key secondary end points achieved statistical significance within the graphical testing procedures for the 2-mg dose. Efficacy observed with baricitinib in this phase 3 trial confirms the results observed in a phase 2 TCS combination trial,22 although overall higher efficacy rates were observed in the current trial. Some differences are likely explained by changes in study design from phase 2 to phase 3, including higher baseline disease severity, implementation of a washout preceding randomization, and specific guidance for TCS use during the present study.
Itch is a central, debilitating symptom of AD, with substantial effects on quality of life, sleep disturbance, and anxiety and depressive symptoms.4 Baricitinib provided benefits for itch, as assessed by CFB and percent CFB, as early as 2 days after initiating treatment. As early as 4 days after initiating treatment, the proportion of patients achieving a 4-point or greater improvement from baseline on the Itch NRS was higher in the 4-mg group compared with the placebo group. Prior studies5,17,23,24 indicate that burdensome AD symptoms extend beyond itch, including its effect on sleep disturbance and skin pain. Similar results were observed in the current study. Patients reported a high number of nighttime awakenings owing to itch and skin pain at baseline. Improvements in sleep and skin pain were observed early with baricitinib, with associated improvements in symptoms of anxiety, depression, quality of life, and work productivity.
The overall safety profile in the current study was consistent with that observed during the phase 3 baricitinib monotherapy AD trials.17 However, although the frequency of AEs with 2 mg and 4 mg of baricitinib were similar across studies (54%-58% in the monotherapy studies vs 56%-58% in the present study), the frequency of AEs with placebo was lower (38%) than that observed in the monotherapy studies (54%-56%).17 The frequency of serious AEs remained similar between the baricitinib and placebo groups in all studies. In this study, treatment-emergent infections were more frequently reported with both baricitinib doses compared with placebo. However, 2 serious infections were reported with placebo and none with baricitinib. Herpes simplex virus infection was commonly reported with baricitinib treatment and is a recognized infection associated with AD and with the use of baricitinib.25 Acne and folliculitis were reported with baricitinib treatment, but these are 2 unique conditions, with acne as a recognized adverse drug reaction of baricitinib.25 In addition, in the baricitinib monotherapy AD studies, folliculitis was observed in less than 2% of patients across baricitinib groups and did not increase in frequency compared with placebo.17 One pulmonary embolism was reported with 4 mg of baricitinib, which was the only venous thrombotic event reported during the placebo-controlled period of the phase 3 studies of more than 2400 patients. The patient was older than 50 years, was taking oral contraceptives, and had a history of smoking. The patient had no other known precipitating factors. There were no malignant tumors, major adverse cardiovascular events, or deaths reported in this study. An increased incidence of conjunctivitis was reported in patients with AD who received the anti–IL-4Rα antibody dupilumab compared with placebo in clinical trials26; in this study, conjunctivitis was reported infrequently, with reports only in the placebo (2 [2%]) and 2 mg of baricitinib groups (3 [3%]). Creatinine phosphokinase level elevations were observed with baricitinib treatment; however, these elevations were not associated with symptoms of myositis or rhabdomyolysis. In this study, consistent with previous baricitinib AD trials,17 there was a paucity of reported hematologic laboratory abnormalities (grade 3 or 4) compared with placebo, suggesting that at the doses studied in phase 3 trials, baricitinib treatment does not lead to clinically meaningful JAK2 inhibition that would result in potential increases in anemia, neutropenia, and thrombocytopenia. Changes in lipid levels were similar between the baricitinib and placebo groups except with increases in high-density lipoprotein levels. Safety data conclusions are based on 16-week, placebo-controlled data and do not indicate safety regarding events more likely to occur with long-term use, such as malignant tumors; long-term data are being evaluated in an ongoing phase 3 long-term extension trial.21
The current mainstay therapy for AD includes emollients and TCSs, which often provide inadequate efficacy in patients with moderate to severe disease.6,7,8 Furthermore, application of topical medication is often not sustainable in patients with a large body surface area impacted by AD.6 Patients for whom topical therapies alone are insufficient may still benefit from topical therapies when combined with systemic therapy. For example, in patients treated with dupilumab, addition of topical therapy increased response rates in clinical trials by approximately 10%.27 Thus, combination therapy can be regarded as the current standard of care in AD management. However, it is desirable that the need to add topical therapies decreases with ongoing systemic therapy. Data in this study suggest that patients with AD treated with baricitinib may be able to reduce the frequency and total quantity of concomitant TCSs used, thus mitigating concerns associated with continual or sustained application of topical treatments. Because AD is a chronic disease with recurrent flares, the addition of TCSs to baricitinib could allow patients and physicians to escalate and reduce TCS therapy based on symptoms.
The efficacy and safety data reported here expand on the results of the previously reported phase 3 AD monotherapy studies17 on baricitinib. In all 3 studies, 4 mg of baricitinib demonstrated a larger magnitude of treatment effect and a faster onset compared with 2 mg of baricitinib. The data presented here suggest an added clinical benefit of concomitant topical therapies despite all patients previously having inadequate disease control with these treatments. In the baricitinib AD monotherapy studies,17 4 mg and 2 mg of baricitinib achieved a significant improvement vs placebo in the proportion of patients achieving vIGA-AD scores of 0 or 1 (BREEZE-AD1: 4 mg of baricitinib, 17% [95% CI, 11.3%-24.3%]; 2 mg of baricitinib, 11% [95% CI, 6.9%-18.2%]; and placebo, 5% [95% CI, 2.8%-8.2%]; BREEZE-AD2: 4 mg of baricitinib, 14% [95% CI, 8.8%-21.0%]; 2 mg of baricitinib, 11% [95% CI, 6.3%-17.2%]; and placebo, 5% [95% CI, 2.5%-7.9%]). In this study, the overall response rates for vIGA-AD scores of 0 or 1 were higher (4 mg of baricitinib, 31% [95% CI, 22.8%-39.7%]; 2 mg of baricitinib, 24% [95% CI, 16.8%-32.7%]; and placebo, 15% [95% CI, 9.2%-22.5%]); however, only 4 mg of baricitinib achieved significant improvement in this outcome. The vIGA-AD end point is a stringent measure of response that takes into account the appearance of skin lesions; however, the assessment does not consider improvements in disease extent, itch, sleep disturbances, or quality of life.20 Thus, although the vIGA-AD is useful as a regulatory end point, the achievement of a vIGA-AD score of 0 or 1 most likely does not encompass the entire benefit that patients with AD may experience during treatment.
Limitations
This study has limitations. This 16-week trial had a positive outcome; however, this time frame limited long-term outcome assessment. The ongoing phase 3 long-term extension trial BREEZE-AD321 will provide a comprehensive evaluation of the long-term efficacy and safety of baricitinib in adults with moderate to severe AD. In addition, although 2 mg of baricitinib improved skin inflammation and multiple secondary end points in this study, it failed to show statistical significance for the primary end point of a vIGA-AD score of 0 or 1. In the context of a combination therapy study, placebo plus TCSs increase placebo response rates; thus, a larger study may have established significance.
Conclusions
In this randomized clinical trial, treatment with 4 mg of baricitinib in combination with background TCS therapy significantly improved the signs and symptoms of moderate to severe AD in adults compared with placebo, with a rapid onset of action and a safety profile consistent with prior findings from baricitinib clinical development in AD.17,22 Baricitinib may represent a potential novel oral treatment option in combination with background TCSs for patients with moderate to severe AD.
Trial Protocol
eAppendix. Study Investigators, Inclusion Criteria, and Exclusion Criteria
eMethods. Supplementary Methods
eFigure 1. Study Design
eFigure 2. Results for Graphical Multiple-Testing Procedure: US/Japan
eFigure 3. Results for Graphical Multiple-Testing Procedure: EU
eFigure 4. Percent Change From Baseline in the Total EASI Score and Proportion of Patients Achieving a 90% Improvement in the Total EASI Score Over Time
eFigure 5. Percent Change From Baseline in Itch NRS Over Time
eFigure 6. Proportion of Patients Achieving a ≥4-Point Improvement in the Itch NRS Response During the First Week of Treatment
eFigure 7. Change From Baseline in Skin Pain NRS Over Time
eFigure 8. Proportion of Patients Achieving a ≥1.5-Point Improvement in the ADSS Item 2 Score Over Time
eFigure 9. Change From Baseline in the Total POEM Score and the Proportion of Patients Achieving a ≥4-Point Improvement in the Total POEM Score Over Time
eFigure 10. Change From Baseline in the Total DLQI Score and the Proportion of Patients Achieving a DLQI Score of 0 or 1 Over Time
eFigure 11. Mean Proportion of Time Without Use of Background Topical Corticosteroids and Mean Gram Quantity of Sponsor-Provided Moderate Potency Topical Corticosteroids Used Over 16 Weeks in the Intent-To-Treat Population, Responders Population, and Nonresponders Population
eResults. Supplementary Results
eTable 1. Summary of Atopic Dermatitis Treatment History at Baseline
eTable 2. Other Efficacy and Health Outcomes Secondary Endpoints in the Intent-to-Treat Population
eTable 3. Mean Number of Days and Proportion of Time Without Use of Background Topical Corticosteroids Over 16 Weeks in the Intent-to-Treat Population, Responders Population, and Nonresponders Population
eTable 4. Mean Gram Quantity of Sponsor-Provided Topical Corticosteroids Used Over 16 Weeks in the Intent-to-Treat Population, Responders Population, and Non-Responders Population
eTable 5. Summary of Patients Rescued and Rescue Medications Used
eTable 6. Summary of Serious Adverse Events by System Organ Class and Preferred Term
eTable 7. Elevations in Creatinine Phosphokinase by CTCAE Grading
eTable 8. Important Changes in Laboratory Analytes
Data Sharing Statement
References
- 1.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73(6):1284-1293. doi: 10.1111/all.13401 [DOI] [PubMed] [Google Scholar]
- 2.Silverberg JI, Gelfand JM, Margolis DJ, et al. Atopic dermatitis in US adults: from population to health care utilization. J Allergy Clin Immunol Pract. 2019;7(5):1524-1532.e2. doi: 10.1016/j.jaip.2019.01.005 [DOI] [PubMed] [Google Scholar]
- 3.Boguniewicz M, Fonacier L, Guttman-Yassky E, Ong PY, Silverberg J, Farrar JR. Atopic dermatitis yardstick: practical recommendations for an evolving therapeutic landscape. Ann Allergy Asthma Immunol. 2018;120(1):10-22.e2. doi: 10.1016/j.anai.2017.10.039 [DOI] [PubMed] [Google Scholar]
- 4.Silverberg JI, Gelfand JM, Margolis DJ, et al. Patient burden and quality of life in atopic dermatitis in US adults: a population-based cross-sectional study. Ann Allergy Asthma Immunol. 2018;121(3):340-347. doi: 10.1016/j.anai.2018.07.006 [DOI] [PubMed] [Google Scholar]
- 5.Vakharia PP, Chopra R, Sacotte R, et al. Burden of skin pain in atopic dermatitis. Ann Allergy Asthma Immunol. 2017;119(6):548-552.e3. doi: 10.1016/j.anai.2017.09.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis, section 2: management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-132. doi: 10.1016/j.jaad.2014.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ring J, Alomar A, Bieber T, et al. ; European Dermatology Forum (EDF); European Academy of Dermatology and Venereology (EADV); European Federation of Allergy (EFA); European Task Force on Atopic Dermatitis (ETFAD); European Society of Pediatric Dermatology (ESPD); Global Allergy and Asthma European Network (GA2LEN) . Guidelines for treatment of atopic eczema (atopic dermatitis) part I. J Eur Acad Dermatol Venereol. 2012;26(8):1045-1060. doi: 10.1111/j.1468-3083.2012.04635.x [DOI] [PubMed] [Google Scholar]
- 8.Katayama I, Aihara M, Ohya Y, et al. ; Japanese Society of Allergology . Japanese guidelines for atopic dermatitis 2017. Allergol Int. 2017;66(2):230-247. doi: 10.1016/j.alit.2016.12.003 [DOI] [PubMed] [Google Scholar]
- 9.Sidbury R, Davis DM, Cohen DE, et al. ; American Academy of Dermatology . Guidelines of care for the management of atopic dermatitis, section 3: management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. doi: 10.1016/j.jaad.2014.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ring J, Alomar A, Bieber T, et al. ; European Dermatology Forum; European Academy of Dermatology and Venereology; European Task Force on Atopic Dermatitis; European Federation of Allergy; European Society of Pediatric Dermatology; Global Allergy and Asthma European Network . Guidelines for treatment of atopic eczema (atopic dermatitis) part II. J Eur Acad Dermatol Venereol. 2012;26(9):1176-1193. doi: 10.1111/j.1468-3083.2012.04636.x [DOI] [PubMed] [Google Scholar]
- 11.Dupixent. Prescribing information. Regeneron Pharmaceuticals; October 2017. Reference ID: 4337903. Accessed June 7, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761055s007lbl.pdf
- 12.Dupixent. Product information. Sanofi Deutschland GmbH. Accessed June 7, 2020. https://www.ema.europa.eu/en/documents/product-information/dupixent-epar-product-information_en.pdf
- 13.Fridman JS, Scherle PA, Collins R, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184(9):5298-5307. doi: 10.4049/jimmunol.0902819 [DOI] [PubMed] [Google Scholar]
- 14.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16(12):843-862. doi: 10.1038/nrd.2017.201 [DOI] [PubMed] [Google Scholar]
- 15.Howell MD, Fitzsimons C, Smith PA. JAK/STAT inhibitors and other small molecule cytokine antagonists for the treatment of allergic disease. Ann Allergy Asthma Immunol. 2018;120(4):367-375. doi: 10.1016/j.anai.2018.02.012 [DOI] [PubMed] [Google Scholar]
- 16.Renert-Yuval Y, Guttman-Yassky E. New treatments for atopic dermatitis targeting beyond IL-4/IL-13 cytokines. Ann Allergy Asthma Immunol. 2020;124(1):28-35. doi: 10.1016/j.anai.2019.10.005 [DOI] [PubMed] [Google Scholar]
- 17.Simpson EL, Lacour JP, Spelman L, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol. 2020;183(2):242-255. doi: 10.1111/bjd.18898 [DOI] [PubMed] [Google Scholar]
- 18.Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis, section 1: diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338-351. doi: 10.1016/j.jaad.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 20.Simpson E, Bissonnette R, Eichenfield L, et al. The Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD™): A Clinical Outcome Measure for the Severity of Atopic Dermatitis [abstract]. European Academy of Dermatology and Venereology; 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.A study of long-term baricitinib (LY3009104) therapy in atopic dermatitis (BREEZE-AD3). ClinicalTrials.gov identifier: NCT03334435. Updated March 17, 2020. Accessed August 15, 2020. https://clinicaltrials.gov/ct2/show/NCT03334435
- 22.Guttman-Yassky E, Silverberg JI, Nemoto O, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: a phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol. 2019;80(4):913-921.e9. doi: 10.1016/j.jaad.2018.01.018 [DOI] [PubMed] [Google Scholar]
- 23.Silverberg JI, Gelfand JM, Margolis DJ, et al. Pain is a common and burdensome symptom of atopic dermatitis in United States adults. J Allergy Clin Immunol Pract. 2019;7(8):2699-2706.e7. doi: 10.1016/j.jaip.2019.05.055 [DOI] [PubMed] [Google Scholar]
- 24.Thyssen JP, Halling-Sonderby AS, Wu JJ, Egeberg A. Pain severity and use of analgesic medication in adults with atopic dermatitis: a cross-sectional study. Br J Dermatol. 2020;182(6):1430-1436. doi: 10.1111/bjd.18557 [DOI] [PubMed] [Google Scholar]
- 25.Olumiant. Prescribing information. Eli Lilly and Company; 2018. Reference ID: 4271150. Accessed March 28, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207924s000lbl.pdf
- 26.Akinlade B, Guttman-Yassky E, de Bruin-Weller M, et al. Conjunctivitis in dupilumab clinical trials. Br J Dermatol. 2019;181(3):459-473. doi: 10.1111/bjd.17869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287-2303. doi: 10.1016/S0140-6736(17)31191-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
eAppendix. Study Investigators, Inclusion Criteria, and Exclusion Criteria
eMethods. Supplementary Methods
eFigure 1. Study Design
eFigure 2. Results for Graphical Multiple-Testing Procedure: US/Japan
eFigure 3. Results for Graphical Multiple-Testing Procedure: EU
eFigure 4. Percent Change From Baseline in the Total EASI Score and Proportion of Patients Achieving a 90% Improvement in the Total EASI Score Over Time
eFigure 5. Percent Change From Baseline in Itch NRS Over Time
eFigure 6. Proportion of Patients Achieving a ≥4-Point Improvement in the Itch NRS Response During the First Week of Treatment
eFigure 7. Change From Baseline in Skin Pain NRS Over Time
eFigure 8. Proportion of Patients Achieving a ≥1.5-Point Improvement in the ADSS Item 2 Score Over Time
eFigure 9. Change From Baseline in the Total POEM Score and the Proportion of Patients Achieving a ≥4-Point Improvement in the Total POEM Score Over Time
eFigure 10. Change From Baseline in the Total DLQI Score and the Proportion of Patients Achieving a DLQI Score of 0 or 1 Over Time
eFigure 11. Mean Proportion of Time Without Use of Background Topical Corticosteroids and Mean Gram Quantity of Sponsor-Provided Moderate Potency Topical Corticosteroids Used Over 16 Weeks in the Intent-To-Treat Population, Responders Population, and Nonresponders Population
eResults. Supplementary Results
eTable 1. Summary of Atopic Dermatitis Treatment History at Baseline
eTable 2. Other Efficacy and Health Outcomes Secondary Endpoints in the Intent-to-Treat Population
eTable 3. Mean Number of Days and Proportion of Time Without Use of Background Topical Corticosteroids Over 16 Weeks in the Intent-to-Treat Population, Responders Population, and Nonresponders Population
eTable 4. Mean Gram Quantity of Sponsor-Provided Topical Corticosteroids Used Over 16 Weeks in the Intent-to-Treat Population, Responders Population, and Non-Responders Population
eTable 5. Summary of Patients Rescued and Rescue Medications Used
eTable 6. Summary of Serious Adverse Events by System Organ Class and Preferred Term
eTable 7. Elevations in Creatinine Phosphokinase by CTCAE Grading
eTable 8. Important Changes in Laboratory Analytes
Data Sharing Statement