

Abstract
Cerasus serrulata is a flowering cherry germplasm resource for ornamental purposes. In this work, we present a de novo chromosome-scale genome assembly of C. serrulata by the use of Nanopore and Hi-C sequencing technologies. The assembled C. serrulata genome is 265.40 Mb across 304 contigs and 67 scaffolds, with a contig N50 of 1.56 Mb and a scaffold N50 of 31.12 Mb. It contains 29,094 coding genes, 27,611 (94.90%) of which are annotated in at least one functional database. Synteny analysis indicated that C. serrulata and C. avium have 333 syntenic blocks composed of 14,072 genes. Blocks on chromosome 01 of C. serrulata are distributed on all chromosomes of C. avium, implying that chromosome 01 is the most ancient or active of the chromosomes. The comparative genomic analysis confirmed that C. serrulata has 740 expanded gene families, 1031 contracted gene families, and 228 rapidly evolving gene families. By the use of 656 single-copy orthologs, a phylogenetic tree composed of 10 species was constructed. The present C. serrulata species diverged from Prunus yedoensis ~17.34 million years ago (Mya), while the divergence of C. serrulata and C. avium was estimated to have occurred ∼21.44 Mya. In addition, a total of 148 MADS-box family gene members were identified in C. serrulata, accompanying the loss of the AGL32 subfamily and the expansion of the SVP subfamily. The MYB and WRKY gene families comprising 372 and 66 genes could be divided into seven and eight subfamilies in C. serrulata, respectively, based on clustering analysis. Nine hundred forty-one plant disease-resistance genes (R-genes) were detected by searching C. serrulata within the PRGdb. This research provides high-quality genomic information about C. serrulata as well as insights into the evolutionary history of Cerasus species.
Subject terms: Genome assembly algorithms, Genetic variation, Comparative genomics
Introduction
Cerasus serrulata (Lindley) Loudon belongs to Cerasus Mill. in the Rosaceae family1,2. The corymbose-racemose or subumbellate inflorescences of this species usually have 2–3 (5) flowers, and most flowers are white or pale pink, with single petals3. Trees of C. serrulata (Cerasus serrulata) have dark green leaves with acuminate serrate teeth on the margins, dense flowers with unfolding petals, and black fruits4,5 (Fig. 1). Cerasus serrulata is the parent of many ornamental cherry varieties and is an important flowering cherry germplasm resource used for ornamental purposes6.
Fig. 1. Images of a Chinese flowering cherry tree and its flowers and berries from March to April 2019.

This Chinese flowering cherry tree grows in Baohua mountain, Jiangsu Province. The lower right corner shows a close-up of cherry flowers and berries
Cerasus serrulata is widely distributed in the midwestern and eastern regions of China and possibly also is present on the Korean Peninsula and in Japan3,5. China has the most abundant germplasm resources of C. serrulata, where various natural and artificial hybrids have been developed and selected7,8. More than 45 species (31 endemics) and nine varieties occur in Southwest China, along the middle and lower reaches of the Yangtze River, in Northeast China, and in Chinese Taipei (there are ~16 species and 11 varieties in Japan)4,9. Due to a long history of cultivation along with the naturalization of wild flowering cherry species and interspecific hybridization in China, there has been much taxonomic debate over the name, origin, and delimitation among different populations in C. serrulata (especially different populations in alpine areas). Numerous investigations into C. serrulata, including the collection of specimens, development of morphological markers and molecular markers, and assessment of evolutionary and phylogeographic relationships, have been carried out to determine its origin10,11. The interpretation of Li Chao-Luang and Bruce Bartholomew in the Flora of China was adopted here in dealing with taxonomic disputes4. Considering that genomics-based research could be fundamentally helpful in overcoming this drawback to some extent, a de novo genome sequencing project was launched in this study, aiming to provide a scientific and theoretical basis for understanding C. serrulata.
The acquisition of a high-quality genome may provide more detailed insights into the evolutionary history and contribute toward the settlement of disputes over the classification. In the past decade, whole-genome sequencing has been widely performed on a number of Rosaceae species, including the fruit crop species sweet cherry12, peach13, apple14, Chinese plum15, strawberry16, apricot17, and black raspberry18, as well as the ornamental species Prunus x yedoensis19, plum blossom20 Chines rose21, and multiflora rose22. In this study, we successfully assembled the genome of C. serrulata (2n = 2x = 16) and compared it with the genomes of closely related species in terms of the gene family, positive gene selection, and phylogeny. This work provides a foundation for clarifying the genetic variation, genetic diversity, and genealogical structure of C. serrulata.
Results
Genome estimation and assembly
The genome size of C. serrulata was estimated by k-mer analysis to be 256.65 Mb with repeat and heterozygosity percentages of 46.55% and 1.67%, respectively (Supplementary Fig. S1). In this work, 1.88 million clean reads (~47.99 Gb) were obtained, with a read N50 of 32.08 bb and a read mean length of 25.49 bb (Table 1). The results of BLAST with randomly selected reads aligned to the nucleotide sequence database (Nt) were used to assess contamination. A total of 43.60% and 25.59% of mapped reads on the alignment were represented by P. mume and P. persica, respectively (Supplementary Fig. S2). A Nanopore long-read distribution was computed using different gradient lengths (Supplementary Table S1). Primary assembly was employed by Canu v1.5, WTDBG v1.1, and SMARTdenovo, with adjusted parameters, resulting in an optimized primary assembled genome of 265.38 Mb distributed across 182 contigs.
Table 1.
Statistics and characteristics of the genome of C. serrulata
| Characteristics | Number | Size | Sequence coverage (X) | Percentage |
|---|---|---|---|---|
| Illumina reads | 38.14 Gb | 148.62 | ||
| Nanopore reads | 47.99 Gb | 180.84 | ||
| Hi-C reads | 36.77 Gb | 143.28 | ||
| The estimate of genome size | 256.65 Mb | |||
| Final-assembly genome size | 265.40 Mb | |||
| Contig number and N50 | 304 | 1.56 Mb | ||
| Maximum contig | 7.34 Mb | |||
| Scaffold number and N50 | 67 | 31.12 Mb | ||
| Maximum scaffold | 49.87 Mb | |||
| GC content | 38.51% | |||
| Heterozygosity percentage | 1.67% | |||
| Total repetitive sequences | 130.11 Mb | 49.02% | ||
| Total protein-coding genes | 29,094 | 104.75 Mb | ||
| Annotated protein-coding genes | 27,611 | 94.90% | ||
| MicroRNA | 94 | |||
| Ribosomal RNA (rRNA) | 315 | |||
| Transfer RNA (tRNA) | 472 |
In total, 122.84 million paired reads were generated from Hi-C, of which 95.72 million paired reads (77.92%) were mapped to the primary assembly, and 53.35 million paired reads (43.43%) were uniquely mapped. Furthermore, 41.20 million valid interaction pairs were strongly mapped to the unique mapped paired reads, accounting for 77.23% (Supplementary Table S2). A total of 182 primary contigs were fragmented and reassembled using the Lachesis combined Hi-C data. A total of 263.16 Mb genomic sequences, accounting for 99.16% of total contig sequences, were located. Two hundred and eighty-nine corrected contigs were clustered into eight groups using the agglomerative hierarchical clustering method. Among them, 245 ordered contigs (252.25 Mb) were anchored with a defined order and orientation (Supplementary Table S3).
The final unambiguous chromosomal-level genome of C. serrulata with no obvious assembly errors was composed of eight clusters, as indicated in the Hi-C interaction heatmap (Fig. 2). The chromosomal genome of C. serrulata is 265.40 Mb in length and is characterized by 304 contigs and 67 scaffolds, with a contig N50 of 1.56 Mb and a scaffold N50 of 31.12 Mb (Table 1).
Fig. 2. Hi-C interaction heatmap for Cerasus serrulata.

Chr01–08 are the abbreviations of chromosome 1–8
Genome assessment and annotation
BUSCO analysis showed that, of the 1614 expected embryophytic genes (embryophyta_odb10), 1528 (94.67%) complete BUSCO genes were identified, and 1352 genes were complete single-copy genes (Supplementary Table S4). CEGMA analysis indicated that of the 458 conserved core genes in eukaryotes, 447 (97.60%) core genes were identified based on sequence similarity (Supplementary Table S4). Approximately 254.77 million reads (98.08%) for Illumina were mapped to the assembly, and the number of mapped paired reads was ~230.62 million reads (88.79%) (Supplementary Table S4).
A high degree of consistency among the BUSCO and CEGMA data and the mapping ratio for Illumina confirms the high quality of the assembled genome. The repetitive sequences were 130.11 Mb in total length, representing 49.02% of the genome (Table 1). Approximately 63.20 Mb of long terminal repeats (LTRs), representing 48.57% of all repeat types, were dominant. Copia (25.27 Mb) and Gypsy (34.23 Mb) were the most abundant subtypes, representing 19.42% and 26.31%, respectively. The characteristics of the repeats, including the number, length, and percentage of each repeat type, were captured in detail (Supplementary Table S5).
Ab initio-, homology-based-, and transcriptome-based predictions were achieved using various workflows (Supplementary Table S6). The results of the predictions were integrated into the final nonredundant protein-coding genes using EVM (Table 1). These 29,094 (104.75 Mb) coding genes were composed of 157,063 exons, with an average gene length of 3.61 kb. For genome annotation, 27,611 genes, accounting for 94.90% of predicted coding genes, were annotated in at least one functional database. Gene annotation numbers and percentages were calculated by aligning to all the databases (Supplementary Table S7), and each gene annotation is provided in great detail in Supplementary Table S8. In addition, the C. serrulata genome was characterized by 2170 pseudogenes, 94 microRNAs, 315 rRNAs, and 472 tRNAs (Supplementary Table S9).
Synteny analysis between C. serrulata and both P. persica and C. avium
Two highly consistent colinear maps were constructed by comparing the C. serrulata genome with the P. persica and C. avium genomes. The colinear map indicated that the sequences of the C. serrulata genome were practically identical to those of the P. persica genome, as indicated by the red lines on the diagonal (Fig. 3a). Only a few spots of blue color represent identical sequences of opposite orientation between the two genomes. Interestingly, the colinear map of C. serrulata and C. avium showed that they possessed numerous identical sequences on different chromosomes (Fig. 3b). Among their differences, identical sequences in opposite orientations constituted a relatively high proportion. The map indicated that, compared with the P. persica genome, the C. serrulata genome is of extraordinarily high quality. The spots on the colinear map may exist and might have caused some functional changes between the two species or otherwise could have resulted from assembly errors. The syntenic blocks generated by comparing the C. avium genome with the C. serrulata genome were distributed across eight similar chromosomes (Fig. 3c). We obtained 333 syntenic blocks, which were composed of 14,072 genes, across the two genomes (Supplementary Tables S10 and S11). Among the 333 syntenic blocks, there are 193 common blocks with an accurate match on the same chromosome, representing 57.96% of the total blocks. Thirty-six blocks on chromosome 04 of C. serrulata were discovered in C. avium, of which 33 occurred on chromosome 04 of C. avium. The results also indicated that 58 blocks on chromosome 01 of C. serrulata were distributed on all of the chromosomes of C. avium and were not found on other chromosomes in C. serrulata. These findings imply that chromosome 04 of C. serrulata is relatively conserved, while chromosome 01 is the most ancient or has been the most active chromosome throughout evolution.
Fig. 3. Synteny analysis via comparisons of Cerasus serrulata with Prunus persica and Cerasus avium.

a Colinear map of the C. serrulata genome and P. persica genome. The red color represents identical sequences in the same orientation, while the blue color represents identical sequences in the opposite orientation. b Colinear map of the C. serrulata genome and C. avium genome. The red color represents identical sequences in the same orientation, while the blue color represents identical sequences in the opposite orientation. c Syntenic blocks of C. serrulata and C. avium
Evolutionary analysis of C. serrulata
Based on the combined results from BLASTP and Pfam searches, gene families were identified and subjected to comparative genomic analysis to reveal their evolutionary relationships. The results revealed that there were 14,792 gene families in the C. serrulata genome and that clustered with the annotated 29,094 protein-coding genes. Among the 14,792 gene families, 4451 gene families were unique or specific to the C. serrulata genomes compared with the genomes of C. avium and P. yedoensis (Supplementary Fig. S3a). The 4451 gene families clustered with 5671 genes annotated by Pfam and other databases (Supplementary Table S12). The enrichment results for GO functional analysis involved mainly transmembrane transport, endonuclease activities, and triphosphate biosynthetic processes (Supplementary Table S13). KEGG functional analysis revealed enrichment in terms related to oxidative phosphorylation and meiosis (Supplementary Table S13). These results suggest that C. serrulata has a tendency for cell division and reproduction and undergoes phosphorylation processes. Furthermore, we obtained all types of features of the gene families, including single-copy orthologs, multiple-copy orthologs, other orthologs, and unique genes, present in the related species included in the phylogenetic tree construction (Supplementary Fig. S3b).
Seven hundred forty expanded gene families and 1031 contracted gene families were identified by analyzing the differences between the ancestral species and C. serrulata using CAFE v4.0 (Fig. 4a and Supplementary Table S14). The expanded and contracted family genes were significantly enriched (q < 0.05) in 232 and 266 GO terms of the three categories, respectively (Supplementary Tables S15 and S16). A total of 228 rapidly evolving gene families consisting of 2023 genes were significantly enriched (q < 0.01) in 269 GO terms (Supplementary Table S17). The top functional terms involve oxidation–reduction, glycerolipid biosynthetic processes, glycerolipid and lipid metabolism, and so forth. The KEGG enrichment analysis indicated that rapidly evolving genes were relevant to the metabolism of glycine, serine, threonine, phenylalanine, beta-alanine, and tyrosine (Supplementary Fig. S4).
Fig. 4. Analysis of the evolution of the Cerasus serrulata genome.

a Phylogenetic tree; divergence time; and profiles of gene families that underwent expansion, contraction, and rapid evolution in ten species. b Ka/Ks distribution of paralogs and orthologs of C. serrulata compared with that of C. avium and P. yedoensis. c Distribution of 4DTv distances in C. serrulata, C. avium, and P. yedoensis
Gene family analysis revealed that 3754 genes were similarly present in all the species, and 656 of these genes were single-copy genes. The phylogenetic tree composed of ten species was generated using 656 single-copy orthologs. The tree indicated that C. serrulata was most closely related to P. yedoensis, which speciated ~17.34 million years ago (Mya), while the divergence time estimated between C. serrulata and C. avium was ~21.44 Mya (Fig. 4a). These results demonstrated the close evolutionary relationship between C. serrulata and P. yedoensis. The phylogenetic tree convincingly showed that the time scale of Cerasus and Prunus was close, diverging ~28.14 Mya. This verified the taxonomic claim that Cerasus is independent of Prunus. In addition, the relationship of Rosa L., represented by R. chinensis and R. multiflora, to Rubus L., represented by R. occidentalis, suggested speciation from ~90.80 Mya. All of the genera shared common ancestors and diverged at ~175.51 Mya. These results are consistent with the observed morphological similarities and differences in the genera.
The homologous gene pairs obtained by MUSCLE were subjected to the KaKs Calculator to calculate each gene pair value. As reflected by the Ka/Ks-density curves of C. serrulata vs. P. yedoensis and C. serrulata vs. C. avium, the differences in numbers for the two groups were not large (Fig. 4b). The positively selected genes (Ka/Ks >1) for C. serrulata vs. C. avium and for C. serrulata vs. P. yedoensis were composed of 136 and 133 gene pairs, respectively (Supplementary Tables S18 and S19). Transcription factors with definitively matched Pfam domains were identified from these gene pairs and screened to detect their functions. Ten types of transcription factors were found for C. serrulata compared to C. avium, and eight types were found for C. serrulata compared to P. yedoensis (Tables 2 and 3). The common transcription factors, including ERF, B3, TCP, bHLH, and NAC types, suggest that these genes are more likely to be positively selected in the process of growth and development, physiological metabolism, and stress responses in C. serrulata. The 4DTv values were estimated by comparison with themselves or with each other and indicated that they had experienced only one WGD event (Fig. 4c). Self-comparison peaks at 0.014 (C. serrulata), 0.011 (P. yedoensis), and 0.028 (C. avium) indicated a common WGD event. The peaks of the mutual comparisons appeared slightly earlier than those of the self-comparisons, implying that they had begun to differentiate after the end of the whole-genome replication event. The divergence times of the three species were close: the peak of C. serrulata vs. C. avium was calculated to be 0.0088, and the peak of C. serrulata vs. P. yedoensis was calculated to be 0.0069. This suggests that the divergence of C. avium occurred earlier than did that of P. yedoensis, which is in accordance with the phylogenetic tree results.
Table 2.
Positively selected genes by C. serrulata compared to C. avium
| Transcription factor | Gene ID | Pfam | Annotation |
|---|---|---|---|
| HD-ZIP | EVM0006705 | HD-ZIP_N | Homeobox-leucine zipper protein HAT14 |
| LBD | EVM0013307 | Epimerase | Tetraketide alpha-pyrone reductase 2 |
| NAC | EVM0010591 | NAM | NAC domain-containing protein 72 |
| M-type_MADS | EVM0017730 | RuBisCO_small | Ribulose bisphosphate carboxylase small chain |
| TCP | EVM0020820 | TCP | Transcription factor TCP12 |
| ERF | EVM0025239 | AP2 | Ethylene-responsive transcription factor protein At4g13040 |
| B3 | EVM0026745 | B3 | B3 domain-containing protein |
| G2-like | EVM0027330 | Myb_DNA binding | Probable transcription factor KAN4 |
| bHLH | EVM0004714 | HLH | Transcription factor bHLH62 |
| EVM0021371 | HLH | Transcription factor BIM1 | |
| WRKY | EVM0011053 | WRKY | WRKY DNA-binding transcription factor 70 |
| EVM0017221 | NB-ARC/LRR_8 | Disease-resistance protein At4g27190 |
Table 3.
Positively selected genes by C. serrulata compared to P. yedoensis
| Transcription factor | Gene ID | Pfam | Annotation |
|---|---|---|---|
| NAC | EVM0004149 | NAM | NAC domain-containing protein 96 |
| TCP | EVM0005613 | Cullin | cullin-1 |
| NF-YB | EVM0006103 | Lectin_legB | putative L-type lectin domain-containing receptor kinase S.7 |
| TALE | EVM0008637 | KNOX2/ELK/Homeobox_KN/KNOX1 | homeobox protein knotted-1 |
| bHLH | EVM0008718 | HLH | Transcription factor ILR3 |
| MYB | EVM0010759 | 4HBT | acyl-coenzyme A thioesterase 13 |
| EVM0016863 | Myb_DNA binding | Transcription factor MYB | |
| B3 | EVM0011074 | B3 | B3 domain-containing transcription factor VAL3 |
| EVM0027593 | B3 | B3 domain-containing protein REM5 | |
| ERF | EVM0017051 | GST_C_2/GST_N_3 | Probable glutathione S-transferase DHAR2 |
| EVM0018545 | AP2 | Dehydration-responsive element-binding protein | |
| EVM0023070 | AP2 | AP2/EREBP family transcription factor |
MADS-box gene family in C. serrulata
MADS-box transcription factors control multiple traits in plants and are best known for their regulation of plant floral organ development by type II MIKCc MADS-box genes23. The MADS-box gene family has been thoroughly described in Rosaceae genomes, such as those of P. mume20, P. persica24, and M. domestica25. The MADS-box family was identified by aligning to A. thaliana using MAFFT 7.026, and the tree was constructed by FastTree v227, with the default parameters. Here, we identified a total of 148 members of MADS-box genes in the genome of C. serrulata, including 44 type I MADS-box genes and 104 type II MADS-box genes (Supplementary Table S20). Compared with the number of previously reported MADS-box genes in other Rosaceae species, such as P. persica (79) and M. domestica (146), the number of MADS-box genes in C. serrulata was evidently greater than that in P. persica. This suggests that the C. serrulata floral organs are relatively well developed, while P. persica floral organ development may have been limited by the deletion of some MADS-box genes. The type II MADS-box genes were classified into SVP, AGL15, ANR1, AGL12, AG, STK, AP1, AGL6, SEP1/3, SEP2/4, FLC, TM3, TM8, AP3, and PI subfamilies, in accordance with the classification of A. thaliana (Fig. 5). However, we did not detect any members of the AGL32 subfamily in C. serrulata, suggesting the loss of several functions that may affect seed development occurred, such as the maternal role in fertilization. In addition, the most expanded subfamily was the SVP subfamily, which expanded to 32 members in C. serrulata (there are only four members in A. thaliana). Considering that this family plays important roles in early flowering in early spring, we postulate that the expansion of the SVP subfamily is correlated with flowering time control in C. serrulata.
Fig. 5. The MADS-box gene family in Cerasus serrulata and Arabidopsis thaliana.

The red color represents genes from Cerasus serrulata, and the green color represents genes from Arabidopsis thaliana
MYBandWRKYgene families in C. serrulata, C. avium, and P. yedoensis
The members of other gene families, such as those of MYB and WRKY transcription factors, play important roles in many biological functions and are widely distributed in plant roots, stems, leaves, and flowers. Members of the MYB and WRKY gene families containing distinct protein domains were identified by alignment to the Pfam and Swiss-Prot database using Pfam and phmmer searches. Phylogenetic trees for the MYB and WRKY gene families were constructed using C. serrulata, C. avium, and P. yedoensis data. The members of the MYB gene family grouped into seven subfamilies (subfamily a–g) containing 1132 genes (Fig. 6). Among 1132 genes, 372 originated from C. serrulata, 429 originated from C. avium, and 331 originated from P. yedoensis. Specifically, subfamily a has 71 members with the same or similar protein domain, and subfamilies b–g have 89, 60, 145, 171, 96, and 500 members, respectively (Supplementary Table S21). For the WRKY gene family, the same method was adopted to ensure the domain and classify the subfamilies. Eight subfamilies (subfamilies a–h) composed of 221 genes were generated and clustered with regularity (Supplementary Fig. S5). Sixty-six genes in C. serrulata were present in eight subfamilies, while 77 and 78 genes were found in C. avium and P. yedoensis, respectively (Supplementary Table S22). The acquisition of a gene family implies species-specific tandem duplication and gene loss events during evolution that may have led to species-specific functional deterioration.
Fig. 6. The MYB gene family could be divided into seven subfamilies, namely, subfamilies a–g.
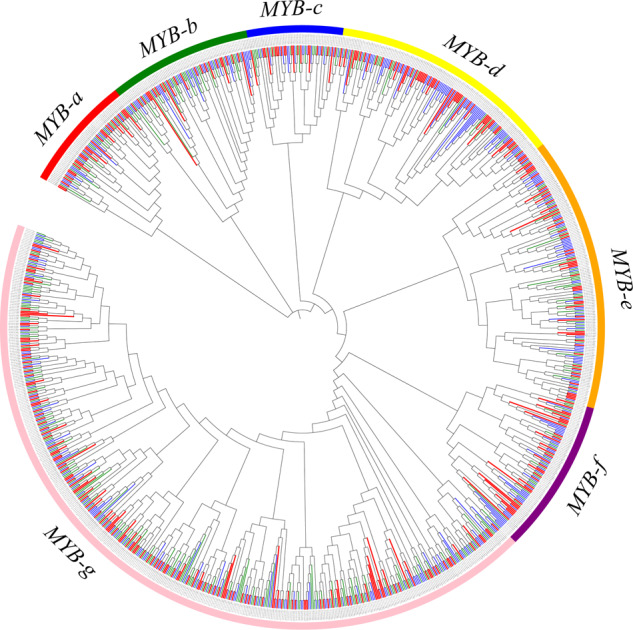
The red members represent genes from C. serrulata, the blue members represent genes from C. avium, and the green members represent genes from P. yedoensis
Plant disease-resistance genes (R-genes) in the Rosaceae
To identify and classify plant disease-resistance genes (R-genes) in Rosaceae species, nine species involved in the gene family analysis were analyzed by aligning to the PRGdb (http://prgdb.crg.eu/wiki/Main_Page) using the BLAST program. In total, 941 R-genes were identified in C. serrulata, which was more than that identified in P. yedoensis (555) but less than that identified in C. avium (1149) (Supplementary Table S23). The common and functionally confirmed R-gene types mainly included five major types: CNLs, TNLs, RLKs, RLPs, and Mlos (Fig. 7). Among all of the types, 161 and 180 TNL R-genes are present in C. serrulata and C. avium, while only four genes were found in P. yedoensis. Of all the representative species, P. persica has 1123 R-genes, and R. occidentalis has 187 R-genes, which are two species with the most and the least R-genes, respectively. For many plants, resistance is considered one of the most important traits that is controlled by R-genes and is affected by the environment. Our results provide fundamental information on the R-genes in these species, which are a prerequisite for resistance.
Fig. 7. Plant disease-resistance genes (R-genes) identified in nine representative species of Rosaceae.
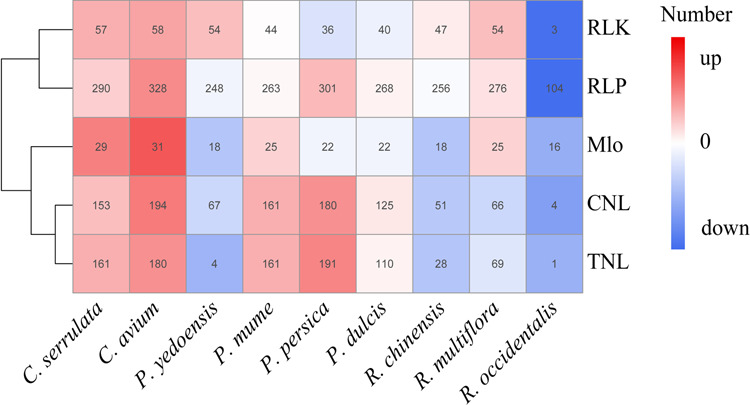
The major types, CNLs, TNLs, RLKs, RLPs, and Mlos were identified in nine species
Discussion
The complete genomes of C. avium and P. yedoensis were previously sequenced and published12,19. As an important germplasm resource of Cerasus, C. serrulata is cultivated worldwide for its beautiful blossoms and superior ornamental characteristics. Resolving the genome sequence of C. serrulata is thus of great significance. Genome assembly can be relatively challenging due to high heterozygosity percentages and high proportions of repeat sequences. However, with the development of long-read sequencing techniques, the assemblies of heterozygous genomes have been largely successful28–30. In this work, we de novo assembled the C. serrulata genome with a chromosomal-level assembly using a combination of Nanopore and Hi-C sequencing techniques. The assembled genome is 265.4 Mb in length, which is smaller than both the C. avium genome (272.4 Mb) and P. yedoensis genome (323.8 Mb). The contig and scaffold N50 values of 1.56 and 31.12 Mb, respectively, are higher than those of C. avium (scaffold N50 = 291.57 kb) and P. yedoensis (contig N50 = 132.59 kb, scaffold N50 = 198.95 kb). These results demonstrate that a high-quality C. serrulata genome was generated and could serve as a credible whole-genome reference sequence for research into the flowering cherry.
The magnum opus of Linnaeus, Species Plantarum, included the initial classification of Prunus, which included cherry, plum, peach, plum, apricot, and so on31. The earliest taxonomic history of Cerasus can be traced back to 1753, and Cerasus was suggested to be independent of Prunus in 175432. Since then, the parallel existence of Prunus and Cerasus has been accepted. However, the application of molecular techniques has indicated that the Cerasus subgenus should belong to a single genus4. The C. serrulata genome and the comparative analysis of related species suggest that P. yedoensis should be classified as C. yedoensis. Moreover, the phylogenetic tree demonstrated that C. serrulata, C. avium, and P. yedoensis clustered together with the shortest divergence time and were distinct from other species of Prunus. The relatively recent differentiation of C. serrulata and P. yedoensis suggests that they are evolutionarily closely related. 4DTv analysis revealed that C. serrulata, C. avium, and P. yedoensis experienced one common WGD event and that the divergence of C. avium occurred earlier than that of P. yedoensis. Thus, C. serrulata and P. yedoensis diverged relatively recently, while C. avium diverged from the ancestor of these two species much earlier.
The colinear map revealed a high chromosome-level assembly of the C. serrulata genome in comparison with the P. persica genome. The colinear map of the C. serrulata genome and C. avium genome showed that they had many identical sequences on different chromosomes, evidently more than that in the colinear map of C. serrulata and P. persica. A previous analysis based on a phylogenetic tree and 4DTv analysis convincingly demonstrated that the evolutionary relationship of C. serrulata and C. avium was closer than that of C. serrulata and P. persica. We postulate that, even though there are differences between the C. serrulata and C. avium genomes, they do not exceed the differences between the C. serrulata and P. persica genomes. The only reasonable explanation is that there could be assembly errors in the genomes of C. serrulata and C. avium. The assembly quality of C. serrulata was verified in several ways, including via BUSCO, the mapping ratio for Illumina, and the colinear map of C. serrulata and P. persica. However, further evidence or experiments are still needed to confirm this.
Cerasus serrulata is cultivated for its flowers, while C. avium is cultivated for its fruit; thus, an assessment of the characteristics of its genomes is warranted. Based on synteny analysis, we discovered 333 syntenic blocks sharing 14,072 syntenic genes. Among these genes, eight chromosomes containing 12,397 genes demonstrated a one-to-one correspondence. No large-scale chromosome translocation or chromosome rearrangement was observed between the two genomes, implying that C. serrulata is relatively evolutionarily conserved. In addition, we studied the commonness and intricacies of C. serrulata, C. avium, and P. yedoensis, including their gene families, positively selected genes, and plant disease-resistance genes (R-genes), and focused on the identification and clustering of the MYB and WRKY family genes in particular. We enriched specific family genes and positively selected genes to determine the key aspects of differentiating C. serrulata. The results showed that P. yedoensis had only 554 R-genes, while C. avium and P. yedoensis had 1149 and 941 R-genes, respectively. Among the five major types, the number of type TNL R-genes differed significantly, and C. serrulata, C. avium, and P. yedoensis were found to have 161, 180, and four TNL R-genes, respectively. Prunus yedoensis is a hybrid of P. speciosa and P. subhirtella, and is an important cultivar for ornamental purposes33,34. This remarkable contrast illustrates that many R-genes for P. yedoensis have been lost, such that its ability to survive in a harsh environment may decrease during the process of artificial selection and hybridization. R-genes play essential roles in breeding processes, and their acquisition enables us to determine on which resistant traits different types of R-genes govern.
Chinese flowering cherry is an important ornamental tree species during early spring and is widely planted in gardens. Understanding the regulation of floral organ development will help improve the varieties and explain the diversity in flowering cherry. The MADS-box gene family is known to be related to floral organ development. We identified 148 MADS-box members in the genome of C. serrulata; these members included 44 type I MADS-box genes and 104 type II MADS-box genes. Within the type II MADS-box genes, the loss of the AGL32 subfamily might affect seed development. The SVP subfamily expanded to 32 members in C. serrulata, whereas only four members disappeared in A. thaliana. The functions of the SVP subfamily members are related to early flowering, and the expansion event implies that these members may influence or control flowering time. Our results presented here may provide new insights into the current problems associated with genetic diversity and offer valuable information for evolutionary studies on C. serrulata and other Cerasus species.
Materials and methods
Plant materials and extraction of DNA
C. serrulata trees were grown in Baohua Provincial Nature Reserve, Jurong, Jiangsu (119.05 E, 32.06 N, 240 m above sea level). Fresh leaves and other tissues were collected for the deposition of specimens at the Herbarium of Nanjing Forestry University (specimen number: YXG18032501) and for sequencing at Biomarker Technologies Corporation, Beijing. High-quality genomic DNA was extracted from the leaves using a DNeasy Plant Mini Kit (Qiagen, Germany). The concentration and purity of the extracted DNA were assessed using a Nanodrop 2000 spectrophotometer (Thermo, MA, USA) and Qubit 3.0 (Thermo, CA, USA), and the integrity of the DNA was measured using pulsed-field electrophoresis with 0.8% agarose gel.
Library preparation and sequencing
A short-read library was prepared with a Nextera XT Library Prep Kit (Illumina, CA, USA). Approximately 1 μg of genomic DNA was extracted for short-read library construction and sheared to short fragment sizes of ~350 bp using a Covaris S2 sonicator (Covaris, MA, USA). The qualified library was sequenced on an Illumina X-ten platform (Illumina, CA, USA) with a paired-end 150 (PE150) read layout. In total, 38.14 Gb of clean data (~148.62 × the assembled genome) were generated and used for genome survey and correction.
Approximately 10 μg of genomic DNA was prepared for long-read library construction using an ONT Template Prep Kit (Nanopore, Oxford, UK). According to the manufacturer’s instructions, the total DNA was sheared to sizes of ~20 kb, and the sheared fragments were recovered by a BluePippin™ System using the processes of damage repair, end repair, and blunt-end adapter ligation via a NEBNext FFPE DNA Repair Mix Kit. The qualified library was subsequently sequenced on the Nanopore PromethION platform with an R9 cell sequencing reagent kit.
For Hi-C library construction, samples were digested with a restriction enzyme (HindIII), in situ labeled with a biotinylated residue, and end repaired. Purified DNA was sheared to a length of 300–700 bp using a NEBNext Ultra II DNA Library Prep Kit (Illumina). The remaining data consisted of 122.84 million clean reads (~36.77 Gb) following the removal of low-quality reads.
Four tissues (leaves, buds, flowers, and roots) collected from the same C. serrulata tree were mixed together for transcriptome sequencing. All of the Illumina and Nanopore sequencing data were deposited in the SRA database under NCBI BioProject ID PRJNA596558. The assembly and annotation data that supported the findings of this study have been deposited in the Figshare database, accessible via the following respective URLs: 10.6084/m9.figshare.12431846.v3 and 10.6084/m9.figshare.12431864.v3.
Estimation of genomic features
Short reads for Illumina were filtered by Fastp v0.19.335, after which they were randomly selected and prepared for contamination assessment using BLAST v2.2.3136 with an E-value = 1e−05. We estimated overall genomic features, namely, size, heterozygosity, and repeats, by plotting the 19-mer depth distribution (k = 19) using Jellyfish v237. In brief, the average k-mer depth was divided by the total k-mer numbers to calculate the genomic features according to the formula. Repetitive sequences were estimated where the depth of the k-mer was more than two times that of the main peak, and heterozygous sequences were estimated where the depth was half of the main peak.
Genome assembly
The original offline data in FAST5 format obtained from the Nanopore PromethION platform were converted into fastq format using the Guppy procedure embedded in MinKNOW (Oxford Nanopore). Long reads that were low in quality and with short fragments (minimum length cutoff of 2000 bp) were filtered and removed using Fastp v0.19.335. Primary assemblies of the Nanopore long reads were mainly performed by Canu v1.538 and WTDBG v1.139, with adjusted parameters. Long reads were assembled into genomic contigs automatically using Canu v1.538, with the following parameters: genomeSize = 300 m and corOutCoverage = 100. SMARTdenovo v1.040 was the third tool used for the assembly. The results of the assemblies were efficiently merged using Quickmerge v0.2.241, and the redundant data were removed with Numer v4.0.042. The consensus of the merged assembly as input was polished and corrected three times using Racon30. Moreover, Illumina reads specifically for genome feature estimation were prepared for the correction of the final assembly using Pilon v1.2243, which also performed three corrections.
Hi-C scaffolding
An assembled genome can be perfectly identified by chromatin interactions using the Hi-C technique. A total of 122.84 million clean reads were mapped to the primary assembly by Nanopore long reads using BWA aligner v0.7.10-r78944 with the default parameters. In cases where both ends of the paired reads mapped to the assembly, only reads of the two pairs that were uniquely mapped were used for further analysis. There were two different types of mapped paired reads obtained by assessing self-circle ligation, dangling ends, and religation: valid interaction pairs and invalid interaction pairs. To obtain valid interactive Hi-C reads, reads of self-ligation, nonligation, dangling ends, and other invalid reads were filtered and removed using Hi-C-Pro v2.10.045.
The contigs of the primary assembly were broken into 50-kb fragments normalized by the restriction sites. Candidate chromosomes were then generated automatically, and the reassembled contigs were divided into ordered, oriented, and anchored groups using Lachesis v2e27abb46 with the following parameters: CLUSTER_MIN_RE_SITES = 37, CLUSTER_MAX_LINK_DENSITY = 2, CLUSTER_NONINFORMATIVE_RATIO = 2, ORDER_MIN_N_RES_IN_TRUN = 33, and ORDER_MIN_N_RES_IN_SHREDS = 31. To further improve the assembly of Hi-C, gaps in the Hi-C assembly were filled using LR GapCloser v1.147.
Genome-quality evaluation
BUSCO v4.0.648 and CEGMA v2.549 are two tools frequently used to evaluate the level of final genome completeness. The Illumina reads for the genome survey were mapped to the final assembled genome using SAMtools v0.1.1850. Considering that it had low conservativeness in interspecies repeat sequences, it was necessary to construct a specific repetitive sequence database of C. serrulata for repeat predictions. Repetitive sequences of the C. serrulata genome were first predicted using LTR FINDER v1.0551, RepeatScout v1.0.552, and PILER-DF v2.453, and DNA repeats were identified and classified using PASTEClassifier v1.054. When the predictions and Repbase results were combined, a final repeat database for only C. serrulata was generated using RepeatMasker v4.0.755.
Genome annotation
Protein-coding gene prediction was performed using three classic strategies: ab initio prediction, homology-based prediction, and transcriptome-based prediction. For ab initio prediction, five tools, GenScan v3.156, Augustus v3.157, GlimmerHMM v1.258, Gene ID v1.459, and SNAP v2006-07-2860, were used to predict the coding genes with model training by turn. For homology-based prediction, protein sequences from five representative species (Arabidopsis thaliana61, Prunus persica13, Malus domestica14, Prunus dulcis15, and Rubus occidentalis18) were downloaded from databases and aligned to the C. serrulata protein sequences using GeMoMa v1.3.162. For transcriptome-based prediction, transcriptome sequencing data obtained in a previous study were used for predicting genes using HISAT v2.0.463, StringTie v1.2.363, TransDecoder v2.064, and GeneMark v5.165 according to the respective workflows. Three classic prediction strategies were subsequently integrated into nonredundant protein-coding genes using EVM v1.1.166 and then modified by PASA v2.0.267.
Pseudogenes and noncoding RNAs, including microRNA, ribosomal RNA (rRNA), and transfer RNA (tRNA), were identified and assessed during this step. In the process of searching for putative pseudogenes, they were assessed based on the premature stop codons or frameshift mutations within the structure of genes using GenBlastA v1.0.468 and GeneWise v2.4.169. MicroRNA and rRNA were identified by Infernal 1.169 based on information from miRBase70 and Rfam71, respectively, and the tRNA was identified using tRNAscan-SE v1.3.172.
Based on the sequence similarity and domain conservation, protein-coding genes were annotated by aligning to the EuKaryotic Orthologous Groups (KOG)73, Kyoto Encyclopedia of Genes and Genomes (KEGG)74, TrEMBL75, Swiss-Prot75, and Nonredundant (Nr) databases76 using BLAST v2.2.3136, with a maximal E-value of 1e−05. Additionally, functional genes were identified and searched by aligning to the Pfam database77 sequences and Gene Ontology (GO) terms78 using HMMER V3.079 and the BLAST2GO v2.5 pipeline80, respectively.
Synteny analysis
Cerasus species can be divided into those that are cultivated for their flowers and those that are cultivated for their fruit, as represented by C. serrulata and C. avium (Cerasus avium), respectively. The genome of P. persica (Prunus persica) has been widely used for its high-quality assembly. In this analysis, the genome sequence of two species with a chromosomal-level assembly, those of P. persica (Prunus L.)13 and C. avium12, were downloaded from databases and compared with the genome sequence of C. serrulata. The final chromosomes of C. serrulata were considered to be arranged in the best order by referring to the recognized P. persica genome. For collinearity analysis, we compared the C. serrulata genome with the genomes of P. persica and C. avium using MUMmer (http://mummer.sourceforge.net), with the parameter l = 10,000. In addition, the genomes of C. serrulata and C. avium were subjected to a synteny analysis to reveal syntenic blocks in detail and the associated genes using BLASTP (E < 1e−05)36. Each syntenic block comprising at least five sequential genes with no obvious error is displayed in the synteny map.
Gene family identification
To identify gene families from the protein-coding genes, protein sequences from C. serrulata and other representative species, including C. avium12, P. persica13, P. dulcis (Prunus dulcis)15, P. mume (Prunus mume)20, P. yedoensis (Prunus yedoensis)19, R. chinensis (Rosa chinensis)21, R. multiflora (Rosa multiflora)22, and R. occidentalis (Rubus occidentalis)18, were compared with each other using BLASTP, with a maximal E-value of 10−05. Markov chain clustering was performed by all-to-all analysis. The proteins predicted from all species with sequence lengths >100 amino acids were queried against the Pfam database using Pfam scan77. The profiles of gene family expansion, contraction, and rapid evolution were analyzed by comparing the differences between the ancestor and each species using CAFE v4.081. Each gene module was extracted, and its contents were subjected to GO and KEGG functional enrichment analyses. There are usually multiple transcripts present for one gene, and only the longest transcript of each gene obtained from all of the species was considered a single-copy ortholog. The orthologous and paralogous genes of the protein datasets were condensed by removing redundancy via OrthoMCL v2.0.982.
Phylogenetic analysis
Protein alignments with gaps removed for each single-copy gene family were acquired using the programs MAFFT83 and trimAL84. The best substitution model for the alignment was estimated using ModelFinder85, with the default settings. The single-copy orthologous genes generated were aligned to a superalignment matrix with the guidance of protein alignment using MUSCLE 3.8.3186. A phylogenetic tree comprising C. serrulata and eight related species, C. avium12, P. persica13, P. dulcis15, P. mume20, P. yedoensis19, R. chinensis21, R. multiflora22, and R. occidentalis18, was constructed using PhyML v3.087 with the default parameters and A. thaliana61 as an outgroup. The recommended divergence time from a fossil was obtained from the TimeTree database, and a molecular clock was employed to date the divergence event using PAML v4.888 with the approximate likelihood calculation method.
Whole-genome duplication (WGD) analysis
In general, the ratio of the nonsynonymous substitution rate (Ka) and the synonymous substitution rate (Ks) was used to assess gene selection. Protein sequences of homologous genes from C. serrulata vs. those of representative species were aligned using MUSCLE86. As input files, the sequences of the homologous genes were imported into the KaKs Calculator to calculate the gene pair values. Positively selected genes in which Ka/Ks > 1 were subsequently selected. In addition, CDS alignments obtained from previous protein sequence alignments were used to detect WGD based on the 4DTv values using the HKY model.
Supplementary information
Table S8 Integrated lists of gene annotation for Cerasus serrulata genome
Table S9 The family of miRNAs, rRNAs and tRNAs in Cerasus serrulata
Table S12 Specific family genes annotated in Cerasus serrulata
Table S14 Expanded and contracted gene families in Cerrasus serrulata
Table S15 Expanded genes in Cerasus serrulata
Table S16 Contracted genes in Cerasus serrulata
Table S17 GO enrichment of rapidly evolving genes in Cerasus serrulata
Table S18 Positively selected gene pairs annotated in Cerasus serrulata and Cerasus avium
Table S19 Positively selected gene pairs annotated in Cerasus serrulata and Prunus yedoensis
Table S20 MADS-box family genes in Cerasus serrulata
Table S21 MYB family genes in Cerasus serrulata, Cerasus avium and Prunus yedoensis
Table S22 WRKY family genes in Cerasus serrulata, Cerasus avium and Prunus yedoensis
Table S23 Detail information for plant disease resistance genes in representative species of Rosaceae
Acknowledgements
This research was supported by grants from the Science Foundation of Jiangsu Province, China (BE2017037), and the Forestry Science and Technology Innovation and Popularization Project in Jiangsu Province, China (LYKJ[2017]14, LYKJ[2018]29, LYKJ[2018]43). The work reported here was performed at the Cerasus Research Center, Nanjing Forestry University, Nanjing, and Biomarker Technologies Corporation, Beijing.
Author contributions
X.G.Y. and J.C. conceived the project; H.Z. and M.L. collected the samples; M.Z., Y.F.D., and L.C. performed the genome sequencing; X.G.Y., J.C., M.Z., and L.W. analyzed the data; Z.S.S., X.H.L., and T.K. participated in discussions and provided valuable advice; and X.G.Y., X.Q.Y., and L.W. wrote and revised the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Xian-Gui Yi, Xia-Qing Yu, Jie Chen
Contributor Information
Xian-Gui Yi, Email: yixiangui@njfu.edu.cn.
Xian-Rong Wang, Email: wangxianrong66@njfu.edu.cn.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-020-00382-1).
References
- 1.Du HK. Practical methods for rapid seed germination from seed coat-imposed dormancy of Prunus yedoensis. Scientia Hortic. 2019;243:451–456. doi: 10.1016/j.scienta.2018.08.039. [DOI] [Google Scholar]
- 2.Balsamo RA, et al. Leaf biomechanics, morphology, and anatomy of the deciduous mesophyte Prunus serrulata (Rosaceae) and the evergreen sclerophyllous shrub Heteromelesarbutifolia (Rosaceae) Am. J. Bot. 2003;90:72–77. doi: 10.3732/ajb.90.1.72. [DOI] [PubMed] [Google Scholar]
- 3.Liu ZX, et al. Development of stamens and carpels in single and double flowers of Cerasus serrulata. J. Beijing For. Univ. 2010;32:486–491. [Google Scholar]
- 4.Li CL, et al. Cerasus in Flora of China. Science Press. 2003;9:404–420. [Google Scholar]
- 5.Yi XG. The variation and phylogeography of Cerasus serrulata Mill. populations. J. Nanjing For. Univ. 2018;14:166–172. [Google Scholar]
- 6.Wang XR. An illustrated monograph of cherry cultivars in China. Science Press. 2014;12:24–28. [Google Scholar]
- 7.Ma H, Olsen R, Pooler M, Kramer M. Evaluation of flowering cherry species, hybrids, and cultivars using simple sequence repeat markers. SocHort Science. 2009;134:435–444. [Google Scholar]
- 8.Knight R. Abstract bibliography of fruit breeding and genetics to 1965, Prunus. Commonwealth Agricultural Bureau. 1969;3:752–824. [Google Scholar]
- 9.Iwatsuki K, Boufford DE, Ohba H. Flora of Japan. Science Press. 2001;2:435–148. [Google Scholar]
- 10.Meng LI, et al. Numeric and structural characteristics of Cerasus serrulata population around the high-elevation wetlands of Dayangshan. J. Nanjing For. Univ. 2013;37:40–44. [Google Scholar]
- 11.Hong Z, et al. Application of the molecular marker technology to Cerasus Mill. (Rosaceae) World For. Res. 2018;31:16–24. [Google Scholar]
- 12.Shirasawa K, et al. The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Research. 2017;24:499–508. doi: 10.1093/dnares/dsx020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verde I, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013;45:487–494. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- 14.Velasco R, et al. The genome of the domesticated apple (Malus x domestica) Nat. Genet. 2010;42:833–839. doi: 10.1038/ng.654. [DOI] [PubMed] [Google Scholar]
- 15.Velasco D, et al. Evolutionary genomics of peach and almond domestication. G3. 2016;32:116–121. doi: 10.1534/g3.116.032672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shulaev V, et al. The genome of woodland strawberry (Fragaria vesca) Nat. Genet. 2011;43:109–116. doi: 10.1038/ng.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang F, et al. The apricot (Prunus armeniaca L.) genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Hortic. Res. 2019;6:128–134. doi: 10.1038/s41438-019-0215-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanburen R, et al. The genome of black raspberry (Rubus occidentalis) Plant J. 2016;87:535–547. doi: 10.1111/tpj.13215. [DOI] [PubMed] [Google Scholar]
- 19.Seunghoon B, et al. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol. 2018;19:45–51. doi: 10.1186/s13059-018-1414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, et al. The genome of Prunus mume. Nat. Commun. 2012;3:1318–1325. doi: 10.1038/ncomms2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin W, et al. Characterization of the complete chloroplast genome of Chinese rose, Rosa chinensis (Rosaceae: Rosa) Mitochondrial DNA B. 2019;4:51–58. doi: 10.1080/23802359.2018.1536451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hibrand SL, et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants. 2018;4:36–43. doi: 10.1038/s41477-017-0082-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fei C, et al. Evolutionary analysis of MIKCc-type MADS-box genes in gymnosperms and angiosperms. Front. Plant Sci. 2017;8:895–899. doi: 10.3389/fpls.2017.00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wells CE, et al. A genome-wide analysis of MADS-box genes in peach. BMC Plant Biol. 2015;15:41–46. doi: 10.1186/s12870-015-0436-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian Y, et al. Genome-wide identification and analysis of the MADS-box gene family in apple. Gene. 2014;555:277–290. doi: 10.1016/j.gene.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019;4:20–26. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan NP, Paramvir SD, Adam PA. FastTree2-approximately maximum-likelihood trees for large alignments. PLOS ONE. 2010;3:5–12. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayliss SC, et al. The use of Oxford Nanopore native barcoding for complete genome\r, assembly. GigaScience. 2017;6:1–6. doi: 10.1093/gigascience/gix001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodwin S, et al. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015;25:1750–1758. doi: 10.1101/gr.191395.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaser R, et al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;5:27–32. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brummitt RK, et al. The species plantarum project, an international collaborative initiative for higher plant taxonomy. Taxon. 2001;50:1217–1230. doi: 10.2307/1224752. [DOI] [Google Scholar]
- 32.Miller P. containing the methods of cultivating and improving the kitchen, fruit and flower garden, as also the physick garden, wilderness, conservatory, and vineyard. Gardeners Dictionary. 1753;3:576–614. [Google Scholar]
- 33.Innan H, Terauchi R, Miyashita NT, Tsunewaki K. DNA fingerprinting study on the intraspecific variation and the origin of Prunus yedoensis (Someiyoshino) Jpn. J. Genet. 1995;70:185–196. doi: 10.1266/jjg.70.185. [DOI] [PubMed] [Google Scholar]
- 34.Kato S, et al. Origins of Japanese flowering cherry (Prunus subgenus Cerasus) cultivars revealed using nuclear SSR markers. Tree Genet. Genomes. 2014;10:477–487. doi: 10.1007/s11295-014-0697-1. [DOI] [Google Scholar]
- 35.Chen S, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:884–890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul SF, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 37.Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koren S, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Y, et al. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl Acad. Sci. USA. 2016;113:52–56. doi: 10.1073/pnas.1520394112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jue, R. smartdenovo: ultra-fast de novo assembler using long noisy reads. https://github.com/ruanjue/smartdenovo (2015). [DOI] [PMC free article] [PubMed]
- 41.Chakraborty M, et al. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016;44:147–151. doi: 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:12–15. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:29–32. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics. 2013;3:76–78. [Google Scholar]
- 45.Servant N, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:1–11. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu GC, et al. LR Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. Gigascience. 2018;8:157–160. doi: 10.1093/gigascience/giy157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simão FA, et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:19–23. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 49.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 50.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Z, Wang H. LTR FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:265–268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:351–358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 53.Edgar RC, Myers EW. PILER: identification and classification of genomic repeats. Bioinformatics. 2005;21:152–158. doi: 10.1093/bioinformatics/bti1003. [DOI] [PubMed] [Google Scholar]
- 54.Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 55.Smit, A., Hubley. R. & Green, P. RepeatMasker Open-4.0 (2013-2015). http://repeatmasker.org (2017).
- 56.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 57.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19:215–225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 58.Majoros WH, et al. Two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 59.Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr. Protoc Bioinform. 2007;18:1–28. doi: 10.1002/0471250953.bi0403s18. [DOI] [PubMed] [Google Scholar]
- 60.Korf I. Gene finding in novel genomes. BMC Bioinform. 2004;5:59–62. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyerowitz EM. Arabidopsis thaliana. Ann. Rev. Genet. 2003;21:93–111. doi: 10.1146/annurev.ge.21.120187.000521. [DOI] [PubMed] [Google Scholar]
- 62.Keilwagen J, et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44:89–95. doi: 10.1093/nar/gkw092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pertea M, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016;11:1650–1658. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haas, B. J. & Papanicolaou, A. TransDecoder (Find Coding Regions Within Transcripts). http://transdecoder.github.io (2015).
- 65.Tang S, Lomsadze A, Borodovsky M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015;43:78–85. doi: 10.1093/nar/gkv227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:7–15. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campbell MA, et al. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics. 2006;7:324–327. doi: 10.1186/1471-2164-7-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.She R, et al. genBlastG: using BLAST searches to build homologous gene models. Bioinformatics. 2011;27:2141–2143. doi: 10.1093/bioinformatics/btr342. [DOI] [PubMed] [Google Scholar]
- 69.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Griffiths JS, et al. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Griffiths JS, et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:121–124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNAgenes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tatusov RL, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boeckmann B, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marchler BA, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:225–229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.El GS, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:427–432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dimmer EC, et al. The UniProt-GO annotation database in 2011. Nucleic Acids Res. 2012;40:565–570. doi: 10.1093/nar/gkr1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eddy SR, Mitchison G, Durbin R. Maximum discrimination hidden Markov models of sequence consensus. J. Comput. Biol. 1995;2:9–23. doi: 10.1089/cmb.1995.2.9. [DOI] [PubMed] [Google Scholar]
- 80.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 81.De BT, et al. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- 82.Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Capella GS, Silla M, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kalyaanamoorthy S, Minh BQ, Wong TK, Von HA, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–592. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 88.Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S8 Integrated lists of gene annotation for Cerasus serrulata genome
Table S9 The family of miRNAs, rRNAs and tRNAs in Cerasus serrulata
Table S12 Specific family genes annotated in Cerasus serrulata
Table S14 Expanded and contracted gene families in Cerrasus serrulata
Table S15 Expanded genes in Cerasus serrulata
Table S16 Contracted genes in Cerasus serrulata
Table S17 GO enrichment of rapidly evolving genes in Cerasus serrulata
Table S18 Positively selected gene pairs annotated in Cerasus serrulata and Cerasus avium
Table S19 Positively selected gene pairs annotated in Cerasus serrulata and Prunus yedoensis
Table S20 MADS-box family genes in Cerasus serrulata
Table S21 MYB family genes in Cerasus serrulata, Cerasus avium and Prunus yedoensis
Table S22 WRKY family genes in Cerasus serrulata, Cerasus avium and Prunus yedoensis
Table S23 Detail information for plant disease resistance genes in representative species of Rosaceae