

Abstract
We studied three patients with mutations in the senataxin gene (SETX). One had juvenile onset of ALS. The second case resembled hereditary motor neuropathy. The third patient had an overlap syndrome of ataxia-tremor and motor neuron disease, phenotypes previously associated with SETX mutations. Our patients were all apparently sporadic, with no other affected relative. Two relatives of patient no. 2 carried the SETX c.4660T > G transversion but did not manifest motor neuron disease, abnormal eye movements, ataxia, or tremor suggesting that genetic or environmental modifiers may influence expression of this SETX polymorphism. Relatives of patients 1 and 3 were not available for examination or SETX mutation screening. Mutations causing ALS4 may be more frequent and heterogeneous than expected. Screening for SETX mutations should be considered in patients with apparently sporadic juvenile-onset ALS, hereditary motor neuropathy, and overlap syndromes with ataxia and motor neuron disease.
Keywords: Amyotrophic lateral sclerosis, senataxin, mutation, ALS4, genetics
Introduction
Amyotrophic lateral sclerosis (ALS) research advances have accelerated; for instance, a review of research advances over 18 months in 2009 – 2010 was based on 125 reference citations (1). ALS is a syndrome, not a single disease. For example, mutations in 12 different genes cause symptoms and signs of a composite disorder that may differ in minor specifics but include the essential clinical elements of ALS – namely, evidence of dysfunction of both upper and lower motor neurons. The basic clinical findings are similar, no matter which gene is altered, which means there are at least 12 different molecular pathways to the pathogenesis of ALS (2–4). Moreover, these familial forms, which account for 5% of all cases, do not differ clinically from sporadic cases, which account for 95% of all cases (5). The most common mutations affect the superoxide dismutase gene (SOD1), which is found in about 20% of all familial cases. The other known mutations are each responsible for 1 – 2% (2–4).
One difference that divides familial ALS from sporadic is the age at onset of symptoms. Sporadic ALS starts before age 20 years in only 5 – 10% of all cases. SOD1-ALS may also start after age 40 years, but juvenile onset is seen more often in familial ALS, as exemplified by the following three case histories, which were all simplex cases; they were all the only symptomatic members of their families, and all carried mutations in the senataxin gene (SETX).
Description of patients
Patient 1
At age 37 years, in 2004, this right-handed hospital biochemistry laboratory technician came for evaluation of weakness in the right leg. He reported normal birth and early development. No-one else in the family was similarly affected. He had no siblings and his maternal great grandparents were first cousins. In adolescence, scoliosis became prominent but he functioned well until September 2002 when his right leg seemed weak and he limped. In September 2003, he noted right foot-drop. Subsequently, his right hand became clumsy in writing or buttoning a shirt, but he used the hand for typing. He also had difficulty raising the right arm. Left arm and leg seemed normal. He had no dysarthria, dysphagia, cramps, bladder symptoms, or paresthesias. He felt and saw occasional ‘fasciculations’ (his word). He lived alone and was independent in activities of daily living.
Examination.
His height was 5 feet 11 inches and he weighed 140 pounds. He looked healthy but had micrognathia and a high palatal arch. The marked scoliosis was concave to the left and the right shoulder was higher than the left. His head was tilted, with the right ear lower than the left. All cranial nerve functions were normal. There was marked winging of the right scapula but there was no weakness of the trapezii, rhomboids, supraspinati or infraspinati. The right deltoid and forearm were smaller than the left. He had 4/5 weakness of the right hand intrinsic muscles and finger flexors, with no muscle atrophy in the hands or visible fasciculation. The right quadriceps was mildly wasted. There was bilateral 4/5 weakness of the anterior and posterior tibials, peroneals, and extensors hallicus longus muscles. Alternating movements were impaired in the right arm and leg but not the left. Gait was normal and he could walk on his toes but could not stand or walk on his heels. He could hop clumsily on the left foot but not at all on the right. Tendon reflexes were overactive on both sides, more so on the right. Marked ankle clonus was evoked on the right and a few beats of clonus on the left, with right Hoffmann and right Babinski signs. Sensation was intact and pedal arches were not overtly abnormal.
Laboratory data.
Brain and spine MRI showed no abnormality other than the scoliosis and a protruding disc at C6 – 7 with no cord compression. Motor and sensory nerve conductions studies were normal. EMG showed no evidence of denervation in the arms, but the right gastrocnemius muscle showed occasional sharp waves with normal motor units and mildly reduced recruitment. Somatosensory evoked potentials were normal. DNA analysis excluded Friedreich ataxia, as well as hereditary spastic paraplegia genes SPG3A, SPG4, and NIPAI. In 2008, DNA analysis (Athena Diagnostics) revealed a novel heterozygous c.6085A > G variant in SETX, resulting in a lysine to glutamic acid substitution at amino acid 2029 (p.2029K > E) and was absent in 100 controls (Table II). The mutation altered a highly conserved threonine in the helicase domain of senataxin (Figure 1).
Figure 1.

Sites of senataxin protein mutations (highlighted in bold text) in patients 1 – 3 with phylogenetic homologies.
Course.
All symptoms became slowly worse. By 2006, he had switched all hand activities to his left except for writing, and used his left foot instead of the right to drive. Despite a right ankle-foot orthosis, he fell about once a week. Findings on examination remained fundamentally unchanged except for more overt weakness and wasting of muscles of the right hand, but no fasciculation. In 2007, he had to stop working in the laboratory. The scoliosis, lower motor neuron signs and upper motor neuron signs have persisted until 2009. He could still walk alone, wore orthoses on both legs and drove an automobile. He lived with his mother, still independent in activities of daily living.
Patient 2
In May 2009, this man was examined at age 17 years. His birth and early development had been normal. In early childhood, he was a toe-walker and had high pedal arches. He was a slow runner and could never do pull-ups. At age six years, while playing basketball, he had exercise-induced shortness of breath with audible inspiratory stridor and leg weakness. He continued full activities at school. In the fall of 2006, he joined a school wrestling team but, in training, jogging for 5 – 15 min brought on leg weakness, shortness of breath, and numbness in the feet as well as stridor, all relieved by rest for a few minutes. Evaluation by pulmonologists revealed normal arterial blood gases, pulmonary function tests and vocal cord functions by endoscopy. He never had pigmenturia. His 41-year-old mother, 38-year-old father and two sisters, aged 12 and 14 years, were asymptomatic.
Examination.
Cranial nerve functions were normal. Limb muscle bulk was normal, with no visible fasciculation. Achilles tendon contractures and hammer toes were evident (Figure 2). Muscle bulk and tone were normal. Strength was normal for the neck flexors, extensors, and all limb muscles except the infraspinati, which were 4/5, ankle flexors 4+/5 and toe extensors 4−/5. He could stand on his toes, but not on his heels. Sensation was normal. Tendon reflexes were normal at the biceps, triceps, knees, and ankles. A Babinski sign was evident on the right, but not the left.
Figure 2.
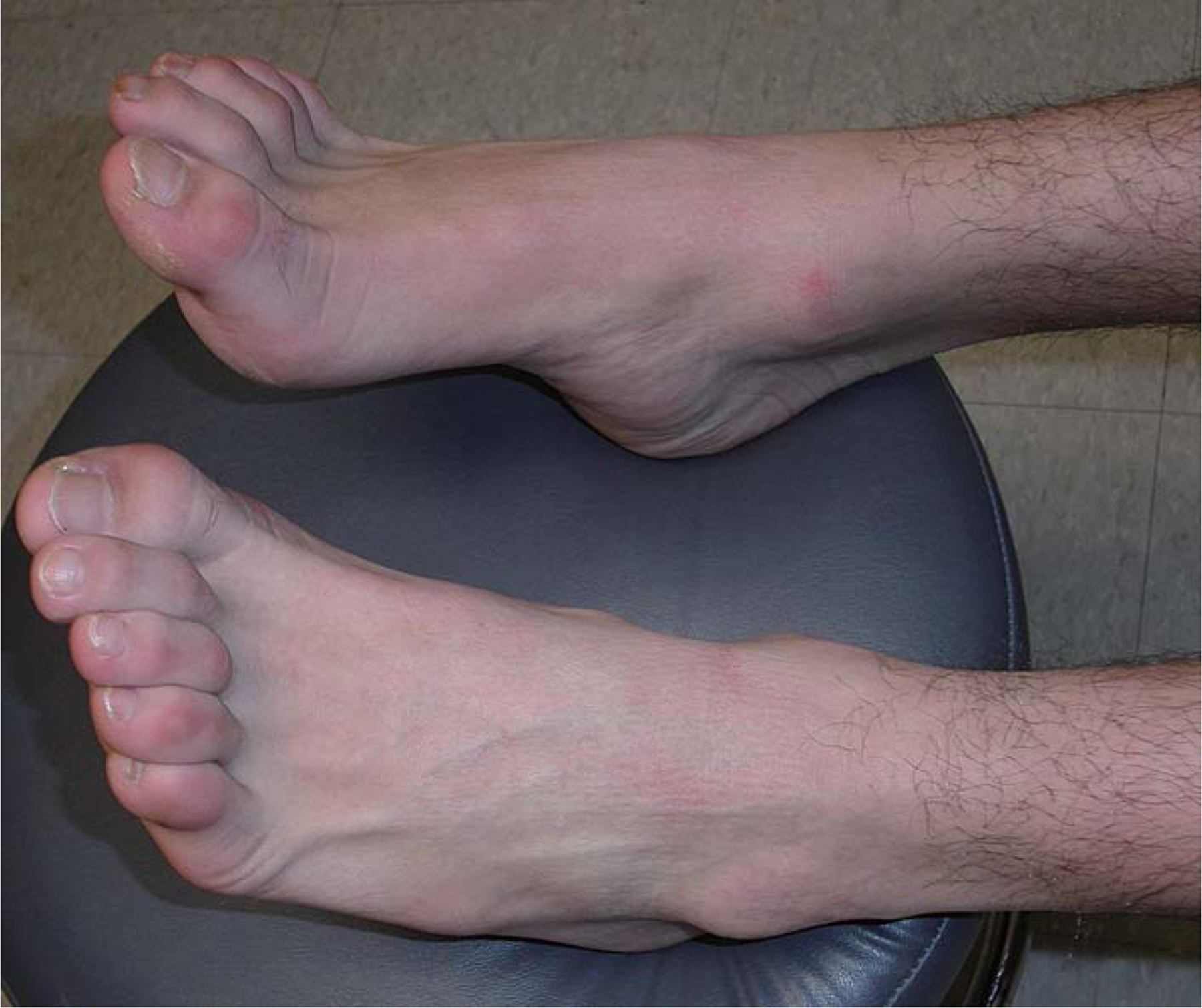
Photograph demonstrating hammer toes and contractures of Achilles tendons in patient 2.
Laboratory data.
Genetic tests gave negative results for Charcot-Marie-Tooth (CMT) disease, including PMP22, duplication deletion and sequencing, connexin 32, MPZ, EGR2, NFL, PRX, GDAP1, LITAF, and MFN2. Serum creatine kinase (CK) was normal. Motor nerve conduction studies revealed low amplitude evoked responses in the tibial nerves and borderline evoked response amplitudes in the peroneal nerves with normal velocities and distal latencies. Sensory nerve conduction studies were normal. Needle EMG showed long-duration motor units and spontaneous activity in the left tibialis anterior indicating denervation, but was normal in the proximal leg and arm muscles. Brain and spine MRI in 2007 showed no pathogenic changes. Somatosensory evoked potentials of the median and posterior tibial nerves were normal.
SETX gene sequencing (Athena Diagnostics) revealed a heterozygous c.4660T > G transversion that changed amino acid 1554 from cysteine to glycine (p.1554C > G). The patient’s asymptomatic mother and paternal grandfather carried the same SETX polymorphism, which was absent in > 100 controls. The site of this mutation is highly conserved in mammals (Figure 1).
In August 2007, studies showed normal nerve conduction and EMG showed large amplitude long-duration motor units in the left tibialis anterior, vastus lateralis, and extensor digitorum communis with reduced recruitment in the tibialis anterior and deltoid indicating chronic neurogenic changes. Gracilis muscle biopsy showed significant fiber-type grouping (APH).
Patient 3
At age 30 years, this female patient noted stiffness of her fingers, with muscle twitching and tremor, as well as a feeling of imbalance and a sense of motion when she looked down or to either side. There was no family history of neurological disease. Examination was notable for full extraocular movements but with downbeat nystagmus on down-gaze as well as right and left gaze. Scattered fasciculations were visible in both arms and legs and the paraspinal muscles, and both hands showed muscle weakness and atrophy. Muscle tone was normal. Strength was 4/5 for the finger extensors bilaterally, but normal elsewhere. Alternating movements were slow in the right arm, with a mild bilateral postural tremor as well as a tremor of the head and right head tilt. Tandem gait was slightly impaired. Tendon reflexes were moderately increased at the left knee and ankle, with no Babinski sign. At age 12 years, she had Graves disease, which was treated medically at first and then by surgery at age 14 and again at 32 years.
Laboratory data.
Brain and cervical spine MRI, with and without contrast, were normal. CSF showed normal cell count, protein, and glucose without oligoclonal bands. EMG/NCV in 2006 showed fibrillations and positive sharp waves along with fasciculations and chronic neurogenic changes in the limb and paraspinal muscles. MR spectroscopy of the motor cortex gave normal ratios of N-acetylaspartate/creatine (> 2.50). Laboratory evaluation showed normal or negative CBC, CRP, lipid panel, ESR, ANA, SSA, SSB, RPR, FTA, Lyme, ACE, B12, anti-HU antibodies, anti-YO antibodies, anti-RI antibodies, T3, T4, TSH, PTH, copper, anti-GAD antibodies, vitamin E, and alpha-feto-protein. Abnormal tests included anti-thyroglobulin antibodies 640 (normal < 10), thyroperoxidase antibodies > 2.9 (0 – 2), ceruloplasmin 24 mg/dl (25 – 63), and lactate 2.6 mM/l (0.5 – 1.6).
DNA testing (Athena Diagnostics) showed a senataxin sequence variant in exon 26 with a heterozygous c.7640T > C transition at nucleotide 7640 predicting an amino acid change of isoleucine to threonine at position 2547 (p.2547I > T). This change has not been previously reported and was absent in 100 controls. The site of the mutation is poorly conserved in other organisms (Figure 1). Testing DNA showed no mutations of SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA10, SCA17, DRPLA, FRDA1, APTX, or SCA14.
Discussion
Juvenile ALS was first reported in 1990 (6). The next year, Siddique et al. mapped the first autosomal dominant form of familial ALS (FALS) in adults to chromosome 21 (7), leading to the identification of pathogenic mutations in the superoxide dismutase-1 gene (SOD1) (8). This became ALS1. Other familial forms have been identified and the list now goes up to ALS12 (3,9). ALS4 was first described by Chance et al. (10) as an autosomal dominant disease that mapped to chromosome 9q34. Other FALS categories include recessive and possibly X-linked forms (3,9).
The ALS4 gene proved to be SETX (11,12), which encodes senataxin and first came into prominence in 2004 when Moreira et al. (13) identified mutations of this gene in the syndrome of ataxia and oculomotor apraxia type 2 (AOA2), which was first considered a late-onset form of the syndrome but the average age at onset is 14 years. AOA1 was thought to be of juvenile onset but may vary from two to 18 years (14). The name, senataxin, was chosen because of homologies with the yeast (Saccharomyces cerevisiae) putative RNA/DNA helicase Sen1p. Although senataxin is widely expressed in mammals, SETX mutations can cause AOA2, ALS, sensorimotor axonal neuropathy without overt amyotrophy, distal hereditary motor neuropathy (HMN, also known as distal spinal muscular atrophy or the spinal form of CMT) or a tremor-ataxia syndrome (Table I) (15–20). Recessive SETX mutations cause AOA2 and dominant mutations cause FALS (21).
Table I.
SETX mutations causing motor neuron disease.
| Family | Geographic (ethnic) origin | SETX mutation | Amino acid substitution | Age at onset (years) | Number of affected/unaffected | Clinical features | Reference |
|---|---|---|---|---|---|---|---|
| K7000 | American (English) | c.H66T>C | p.389L>S | Ave. 17 (range 1–63) | 55/65 | Distal weakness and wasting with UMN signs (43/49 individuals), normal sensation (44/49). | 8 |
| CMT-61 | Belgian | C.6407OA | p.2136R>H | Average <6 | 5/3 | Distal weakness and wasting in legs before arms. ‘Brisk’ reflexes(6/7) and Babinski sign (4/7). Normal sensation. | 8 |
| CMT-106 | Austrian | C.8OT | p.3T>I | Average 8 (range < 5–15) | 7/10 | Distal weakness and wasting in legs before arms. ‘Brisk’ reflexes (4/5). No Babinski signs. Normal sensation. | 8 |
| Sporadic | Chinese (Han) | C.3353OT | p.H18T>I | 42 | 1/? | Distal limb and lingual weakness and wasting. No UMN signs. Normal sensation. | 9 |
| Patient 1 | American | C.6085OG | p.2029K>E | 35 | 1/? | Scoliosis. Asymmetric and distal>proximal weakness. Hyperactive reflexes and right Babinski and Hoffmann signs. Normal sensation. | |
| Patient 2 | American | c.4660T>G | P.1554OG | 6 | 1/2 | Achilles tendon contractures and hammer toes. Weakness of infraspinatus and distal leg muscles. Right Babinski. Normal sensation. | |
| Patient 3 | American | c.7640T>C | p.2547I>T | 30 | 1/? | End-gaze nystagmus. Fasciculations in limb and paraspinal muscles. Finger extensor weakness. Hyperactive reflexes in left leg. No Babinski sign. Mild ataxia and tremor. Normal sensation. |
Interestingly, the first family with ALS4 was originally described as having CMT syndrome due to childhood-onset distal weakness and atrophy (22), but the disorder was subsequently re-classified as autosomal dominant juvenile ALS (ALS4) due to the absence of sensory manifestations and corticospinal tract signs (11,23). ALS4 has been described as a ‘non-fatal’ form due to the indolent course (23). Our patient no. 2 has a similar juvenile-onset ALS with weakness in the distal legs, no sensory manifestations, a Babinski sign, and slow progression. By contrast, patient 1 had a relatively late onset (age 35 years) and a more rapid course that resembled sporadic ALS rather than typical ALS4. Patient 3 had a tremor-ataxia presentation with mild motor neuron signs. The various clinical presentations in our patients extend the phenotypic spectrum of SETX mutations.
Although it is difficult to prove pathogenicity of autosomal dominant mutations in apparently sporadic patients, we identified SETX polymorphisms that appear to be pathogenic based on the following criteria: 1) none of the polymorphisms has been previously reported; 2) all three DNA changes were absent in at least 200 control alleles; and 3) two of the three polymorphism sites are highly conserved in vertebrates. The normal functions of senataxin seem to involve RNA processing, participating in the unwinding of both RNA and DNA (24,25). Functional analysis of mutant SETX will be required to understand the pathogenic mechanism of ALS4 (26).
Acknowledgements
MH and CMQ are supported by grants from the NIH, Muscular Dystrophy Association, and by the Marriott Mitochondrial Disorder Clinical Research Fund.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Traub R, Mitsumoto H, Rowland LP. Research Advances in Amyotrophic Lateral Sclerosis, 2009 to 2010. Curr Neurol Neurosci Rep. 2011. (In press.) [DOI] [PubMed] [Google Scholar]
- 2.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–23. [DOI] [PubMed] [Google Scholar]
- 3.Dion PA, Daoud H, Rouleau GA. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet. 2009;10:769–82. [DOI] [PubMed] [Google Scholar]
- 4.Beleza-Meireles A, Al-Chalabi A. Genetic studies of amyotrophic lateral sclerosis: controversies and perspectives. Amyotroph Lateral Scler. 2009;10:1–14. [DOI] [PubMed] [Google Scholar]
- 5.Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2010. (In press.) [DOI] [PubMed] [Google Scholar]
- 6.Ben Hamida M, Hentati F, Ben Hamida C. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain. 1990;113:347–63. [DOI] [PubMed] [Google Scholar]
- 7.Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med. 1991;324: 1381–4. [DOI] [PubMed] [Google Scholar]
- 8.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. [DOI] [PubMed] [Google Scholar]
- 9.Donkervoort S, Siddique T. Amyotrophic Lateral Sclerosis. Overview. GeneReviews [Internet] 2009. July 28 [cited 2010 Nov 6]; available from: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=als-overview [Google Scholar]
- 10.Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, et al. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet. 1998;62:633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74:1128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao ZH, Chen WZ, Wu ZY, Wang N, Zhao GX, Chen WJ, et al. A novel mutation in the senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:118–22. [DOI] [PubMed] [Google Scholar]
- 13.Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–7. [DOI] [PubMed] [Google Scholar]
- 14.Le Ber I, Moreira MC, Rivaud-Pechoux S, Chamayou C, Ochsner F, Kuntzer T, et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003;126:2761–72. [DOI] [PubMed] [Google Scholar]
- 15.Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132:2688–98. [DOI] [PubMed] [Google Scholar]
- 16.Arning L, Schols L, Cin H, Souquet M, Epplen JT, Timmann D. Identification and characterization of a large senataxin (SETX) gene duplication in ataxia with ocular apraxia type 2 (AOA2). Neurogenetics. 2008;9:295–9. [DOI] [PubMed] [Google Scholar]
- 17.Bassuk AG, Chen YZ, Batish SD, Nagan N, Opal P, Chance PF, et al. In cis autosomal dominant mutation of senataxin associated with tremor/ataxia syndrome. Neurogenetics. 2007;8:45–9. [DOI] [PubMed] [Google Scholar]
- 18.Criscuolo C, Chessa L, di Giandomenico S, Mancini P, Sacca F, Grieco GS, et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology. 2006;66:1207–10. [DOI] [PubMed] [Google Scholar]
- 19.Dierick I, Baets J, Irobi J, Jacobs A, de Vriendt E, Deconinck T, et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study. Brain. 2008;131:1217–27. [DOI] [PubMed] [Google Scholar]
- 20.Haack T, Friday D, Bender A, Rolfs A, Klopstock T. Ataxia oculomotor apraxia type 2: course over 27 years and a novel stop mutation in the senataxin gene. J Neurol. 2009;256:1555–7. [DOI] [PubMed] [Google Scholar]
- 21.Chen YZ, Hashemi SH, Anderson SK, Huang Y, Moreira MC, Lynch DR, et al. Senataxin, the yeast Sen1p ortholog: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol Dis. 2006;23:97–108. [DOI] [PubMed] [Google Scholar]
- 22.Myrianthopoulos NC, Lane MH, Silberberg DH, Vincent BL. Nerve conduction and other studies in families with Charcot-Marie-Tooth disease. Brain. 1964;87:589–608. [DOI] [PubMed] [Google Scholar]
- 23.Rabin BA, Griffin JW, Crain BJ, Scavina M, Chance PF, Cornblath DR. Autosomal dominant juvenile amyotrophic lateral sclerosis. Brain. 1999;122:1539–50. [DOI] [PubMed] [Google Scholar]
- 24.Kolb SJ, Sutton S, Schoenberg DR. RNA processing defects associated with diseases of the motor neuron. Muscle Nerve. 2010;41:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strong MJ. The basic aspects of therapeutics in amyotrophic lateral sclerosis. Pharmacol Ther. 2003;98:379–414. [DOI] [PubMed] [Google Scholar]
- 26.Richard P, Manley JL. Effect of ALS4 associated mutations of SETX on human transcriptome. (In preparation.) [Google Scholar]