

Abstract
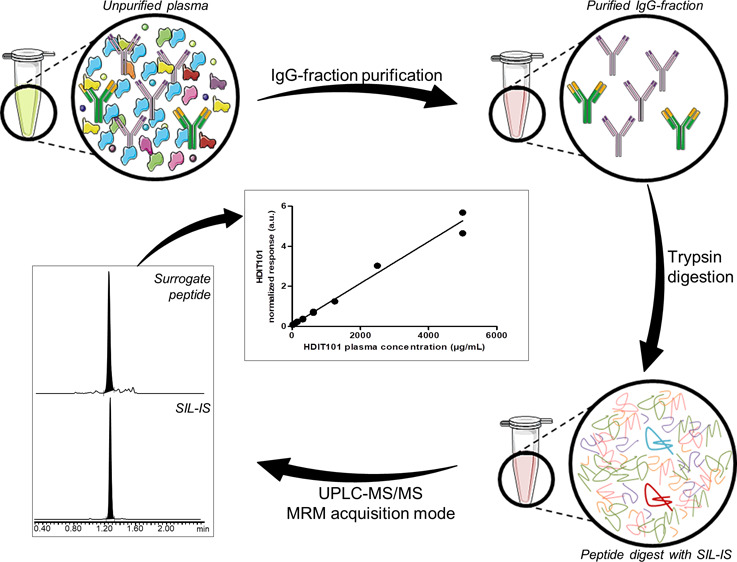
Multiple therapeutic monoclonal antibodies (mAbs) are currently under development or in (pre)clinical study phases to reach regulatory approval. Among these, a new mAb against herpes simplex virus, HDIT101, was recently tested in healthy volunteers during a phase I clinical trial (first-in-human, dose escalation). In the frame of the pharmacokinetic evaluation of this new therapy, a mass spectrometric (MS)-based method was developed for the quantification of HDIT101 in human plasma using liquid chromatography coupled to tandem mass spectrometry. In this work, we describe the development of this bioanalytical assay using the quantification of a HDIT101 surrogate peptide, the assay validation procedure according to the FDA guidelines within the calibration range from 20 to 5000 μg/mL, and its application to plasma samples from the first-in-human clinical trial. This work presents a generic workflow for the development of MS-based quantification assays of new therapeutic antibodies that allows reaching high immunopurification recovery (>98% for HDIT101 over the full calibration range with a precision of 6.9% CV). Surrogate peptide and stable isotopically labeled internal standard were stable, and batch-to-batch accuracies and precisions at the four quality standard levels ranged between −2 and 5% bias and 8 and 11% CV, respectively.
Introduction
Among immunotherapies, monoclonal antibodies (mAbs) have gained rapidly increasing importance since the approval of the first therapeutic mAb by the US Food and Drug Administration (FDA) 30 years ago to reach more than 60 FDA-approved therapeutic mAbs in 2018.1 As for any new systemic therapy to be regulatory approved, pharmacokinetic (PK) evaluation is necessary in humans and requires selective and accurate bioanalytical assays to quantify the total fraction of the therapeutic compound in the central compartment (usually plasma). Traditionally, mAb quantification was mostly performed using ligand binding assays, which enables high sensitivity with fast sample preparation. However, these immunoassays often lack selectivity because of cross-reactivity with endogenous antibodies or other endogenous compounds, are time-consuming for the development of the assay, are compound-specific, and thus cannot be used for multiplexing methods.2,3 Over the last decade, liquid chromatography (LC)–mass spectrometry (MS)-based methods have thus arisen as an alternative for the quantification of therapeutic mAbs in plasma for PK evaluation in preclinical and clinical studies,4−7 as for trastuzumab,8 nivolumab,9,10 infliximab,11,12 or adalimumab12 among others.13−15 LC–MS-based assays enable to reach high specificity, broad dynamic ranges, and versatility, are usually faster to develop, can be easily standardized, and also offer the possibility to monitor several compounds simultaneously.12,15 As a first development step in such assays, a specific investigation of the targeted antibody sequence and complementarity-determining regions (CDR) is necessary to guarantee the specificity of the analysis. Such investigations mostly lead to the selection of at least one surrogate peptide to monitor, which should be unique (proteotypic peptide)5,6 to the targeted mAb in the studied species. Then, sample preparation comprises the proteolytic digestion of plasma proteins without or with the purification of the immunoglobulin G (IgG)-like compounds, or—more specifically—of the targeted mAb itself. The mAb of interest is subsequently quantified by targeting its surrogate peptide in the presence of an internal standard (IS) using LC coupled to tandem MS (MS/MS) analysis in the multiple reaction mode (MRM). Such bioanalytical assays for the quantification of complex biomolecules, such as antibodies require a thorough sample preparation and analytical method optimization, together with the careful selection of an adapted IS, to be able to successfully reach the recommendations of the FDA and of the European Medicines Agency (EMA) for bioanalytical method validation.16,17 In most of these works, when protein-level sample clean-up is used, this is either performed using protein precipitation or using immune-affinity purification with only one elution fraction, which could lead to some lack of reproducibility.5
In the present work, we focused on maximizing the target extraction recovery from plasma samples and on balancing between extraction recovery and surrogate peptide stability. We developed, optimized, and validated our LC–MS-based bioanalytical assay for the absolute quantification of a humanized IgG1 therapeutic mAb, HDIT101, recently developed by the company Heidelberg ImmunoTherapeutics for therapy and prophylaxis of herpes simplex virus (HSV). With approximately 4 billion infected people worldwide,18 the various forms of HSV infections represent an important viral threat. The development of efficient targeted therapies against HSV would therefore have a major universal sanitary impact. Our bioanalytical assay allowed for HDIT101 quantification in plasma from 20 μg/mL (lower limit of quantification, LLOQ) to 5000 μg/mL using ultraperformance LC (UPLC) coupled to MS/MS in MRM mode and was finally applied to assess the PK parameters in healthy volunteers participating in a first-in-human clinical trial.
Materials and Methods
Chemicals and Reagents
UPLC/MS-grade acetonitrile (ACN), water (H2O), and formic acid (FA) were purchased from Biosolve Chimie SARL (Dieuze, France), tris(hydroxymethyl)-aminomethane (tris, purity > 99.9%) from Carl Roth GmbH (Karlsruhe, Germany), and hydrochloric acid (HCl) 37% from Honeywell Fluka (Charlotte, NC, USA). Pierce concentrated phosphate buffer saline and Pierce IgG elution buffer (pH 2) used for the immunopurification (IP) steps were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Digestion reagents were provided with the ProteinWorks eXpress Digest kit purchased from Waters (Milford, MA, USA). Intact HDIT101 sterile-filtered solution at 11.03 mg/mL in buffer (11 mM His-HCl, 5.5% mannitol, pH 6) was provided by Heidelberg ImmunoTherapeutics GmbH (Heidelberg, Germany), 50.4 mg/mL solutions of intact HDIT101 antibody were provided by Celonic Deutschland GmbH (Heidelberg, Germany), and synthetic peptides used for the method development and as stable isotopically labeled IS (SIL-IS) were obtained as pure solid compounds from PSL Peptide Specialty Laboratories GmbH (Heidelberg, Germany).
Plasma Sample Collection
Blank plasma samples from healthy individuals were obtained from the local blood bank (IKTZ Heidelberg GmbH, Heidelberg, Germany) and from the Clinical Trial Center (KliPS) of the Department of Clinical Pharmacology and Pharmacoepidemiology (Heidelberg University) for research purposes.
Clinical Study Samples
Study samples were obtained within the first-in-human trial HDIT101-01. The trial was performed following the principles of good clinical practice and in accordance with the ethical principles described in the then applicable version of the Declaration of Helsinki (6th revision, 2008). The trial was registered with EudraCT (2017-004452-37; date of first approval Jan 31 2018) and the protocol and subsequent amendments were approved by the responsible Ethics Committee of the Medical Faculty of Heidelberg (ethical vote AFmo-538/2016) and the relevant regulatory authority (Paul Ehrlich Institute, Langen, Germany). All volunteers were fully informed about the trial and gave their written consent prior to any study procedures. This phase I dose-escalation trial was performed using six different doses (50, 150, 450, 1350, 4050, and 12,150 mg), intravenously administered to healthy volunteers. For each of the volunteers, 20 1 mL venous plasma samples were collected corresponding to 20 different time points from 15 min before the start of infusion (SOI) to 29 days after SOI (SOI – 15 min = 0 h, SOI + 30 min, SOI + 60 min = end of infusion (EOI), EOI + 15 min, EOI + 30 min, EOI + 60 min, EOI + 90 min, EOI + 2 h, EOI + 3 h, EOI + 4 h, EOI + 6 h, EOI + 8 h, EOI + 10 h, EOI + 12 h, EOI + 24 h, EOI + 48 h, EOI + 72 h, 8, 15, and 29 days).
Bioinformatics Tools for the Preselection of Surrogate Peptide Candidates
The HDIT101 amino acid sequence was digested in silico using the ExPASy tool PeptideMass (Swiss Institute of Bioinformatics, SIB, Lausanne, Switzerland) with the following parameters: cysteines treated with iodoacetamide, digestion with trypsin, and no miscleavage. Each theoretical peptide with 10 to 30 amino acids was compared to amino acid sequences of proteins referenced in the UniProt and Swiss-Prot databases using the Protein Basic Local Alignment Search Tool (BLAST) from the National Center for Biotechnology Information (NCBI, Bethesda, MD, USA). The following research parameters were used: human taxonomy and protein–protein BLAST (Blastp) algorithm.
Standard Solutions
For the optimization of the LC and MRM methods, the synthetic peptides were dissolved in H2O. For each peptide, a 1 mg/mL stock solution was prepared and then diluted to 1 μg/mL. Calibration samples were freshly prepared before each validation and analytical batch by spiking blank plasma with intact HDIT101 antibody solution. First, a 10 mg/mL HDIT101 working solution was prepared in plasma by diluting the 50 mg/mL HDIT101 stock solution 5-fold in blank plasma. Calibration standards (CAL) were subsequently prepared by serial dilution in plasma from 5000 μg/mL (CAL I) to 20 μg/mL (CAL A). QC samples were prepared from an independent HDIT101 solution similarly to the calibration standards. A working HDIT101 solution at 10 mg/mL in plasma was also serially diluted in plasma to prepare four QC levels at 20, 60, 2000, and 4000 μg/mL.
A 1 mg/mL stock solution of the SIL-IS peptide was prepared in water and further diluted to 7.5 μg/mL in water. The IS stock solution was stored at −25 °C, and the stability was proven for up to 2 months by comparing a 2 month IS solution to a newly prepared IS stock solution.
Immuno-Affinity Enrichment of IgG Fraction from Human Plasma
In order to reduce the matrix complexity and select only the IgG fraction of plasma samples (blank plasma, HDIT101-spiked plasma, or plasma study samples), an IP step was performed before the reduction–alkylation–digestion step (Figure 1). Purification was carried out on spin plates (Pierce, Thermo Fisher Scientific) with protein A-coated agarose beads, which bind noncovalently and with high affinity to the Fc fraction of human IgG (except human IgG3). Sample purification was carried out according to the manufacturer’s instructions with some modifications. For all samples, 25 μL of plasma was processed. First, the loading of the sample on the protein A resin was performed twice to avoid any loss of material. Then, at the end of the three elution steps, the three IP fractions from one sample were processed separately or pooled, frozen with liquid nitrogen (15 min), and freeze-dried overnight at 0 °C.
Figure 1.
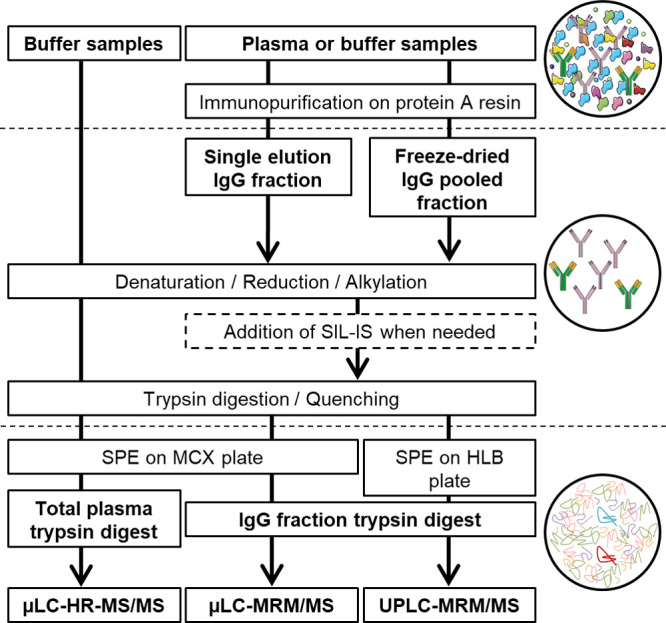
Graphical summary of the different workflows applied to HDIT101 plasma and buffer samples for method development and validation of the HDIT101 quantification assay. Circles show schemed sample compositions during the different steps of the workflow. In the last circle, the HDIT101 surrogate peptide is pictured in dark red and SIL-IS peptide in light blue.
Digestion of Affinity-Purified Plasma Samples for Method Development and Validation
The reduction–alkylation–digestion workflow for the method development and validation steps was performed according to the ProteinWorks eXpress Digest kit’s (Waters) instructions, with the following modifications. As illustrated in Figure 1, unpurified antibody buffer solutions and single IgG elution fractions (200 μL) were processed as they were, or freeze-dried IgG fractions were reconstituted in 120 μL of digestion buffer and subsequently transferred to the cluster tubes containing the RapiGest SF surfactant for denaturation. For quantification (last method development phase, validation, and analytical batches), SIL-IS at 7.5 μg/mL in H2O was added (5 μL) after the alkylation step, prior to the digestion step. Peptide digests were finally transferred to Oasis μElution MCX plates (Waters, mixed-mode polymeric cation exchange sorbent, particle size 30 μm, pore size 80 Å, sorbent weight 2 mg), eluted with 100 μL of 25% ACN in 2% NH4OH, pH 10, and analyzed by μLC–MS/MS. Alternatively, peptide digests were transferred to Oasis μElution HLB plates (Waters, polymeric reversed-phase sorbent, particle size 30 μm, pore size 80 Å, sorbent weight 2 mg), and eluted with 100 μL of 50% ACN in 0.1% FA. Samples were then diluted with 100 μL H2O 0.1% FA and analyzed by μLC–MS/MS or UPLC–MS/MS.
High-Resolution-μLC–MS/MS for Signature Peptide Discovery
The surrogate peptide discovery analyses were performed on a Waters Acquity M-class μLC system coupled to a Waters Xevo G2-XS Q-TOF mass spectrometer featured with an IonKey/MS separation device (Waters). The solid-phase-extracted (SPE) desalted peptide digests were injected (two analyses for each sample with injection volumes of 2 and 5 μL) onto a trapping C18 column (Waters Acquity UPLC M-Class Trap Symmetry, 300 μm × 50 mm, 100 Å, 5 μm). Flow was then inverted toward the analytical C18 column (Waters Peptide CSH C18 iKey, 150 μm × 50 mm, 130 Å, 1.7 μm). μLC and electrospray ionization (ESI)–MS/MS parameters used for the peptide discovery experiments are detailed in Supporting Information part I (Table S1). Data were acquired in data-independent acquisition mode MSe, for which all incoming precursor ions are fragmented in the collision cell and all fragment ions are subsequently detected. Acquired MSe data were interpreted using Biopharmalynx software (Waters) to search for tryptic peptides from the HDIT101 sequence. From these results, sequence coverage of light and heavy chains and global sequence coverage were calculated using the data-processing parameters given in Table S1.
μLC–MRM/MS for Final Method Optimization
Final choice of HDIT101 surrogate peptides and further workflow optimizations were performed on a Waters Xevo TQ-S mass spectrometer equipped with a Waters Acquity M-class μLC system and an IonKey/MS separation device. The final chromatographic conditions such as mobile-phase composition, analytical column, and initial conditions were set as described in the previous part and in Table S1. ESI parameters are detailed in Table S1, as well as MRM acquisition parameters, which were optimized on synthetic peptides using MassLynx V4.1 IntelliStart optimization procedures (Waters) with argon as collision gas for collision-induced dissociation. Optimized collision energies were set for each transition as described in Table S1, and dwell time was set to 52 ms for the MS method with six MRM transitions to record and to 80 ms for the MS method with four MRM transitions to record. MRM data were subsequently transferred to TargetLynx V 4.1 software (Waters) to compute peak intensity, peak area, response, calibration parameters, calculated concentrations, and residual standard deviations.
UPLC–MRM/MS for Quantification Method Validation
The validation of the optimized quantification method for HDIT101 in human plasma and study sample analyses were performed on a Waters Xevo TQ-XS mass spectrometer with Z-spray ionization and step-wave source optimization equipped with a Waters Acquity classic UPLC system. A sample volume of 10 μL was injected and transferred onto the analytical C18 column (Waters BEH300 C18 Acquity, 300 Å, 2.1 × 50 mm, 1.7 μm) with integrated filter disc, and UPLC–MRM/MS analyses were performed using the parameters, as summarized in Table 1. For MRM measurements, the four transitions given in Table 1 for the surrogate peptide LC1 and its IS SIL-LC1 were optimized using IntelliStart software and argon as collision gas for collision-induced dissociation. Optimized collision energy was set to 24 V and dwell time to 79 ms. Acquired MRM data were subsequently transferred to Waters TargetLynx v. 4.2 software.
Table 1. Final LC–MS Parameters Used for the Last Method Development and Validation Stepsa.
| UPLC-MRM/MS | |||
|---|---|---|---|
| LC eluent | A: H2 0 5% ACN 0.1% FA | ||
| B: ACN 0.1% FA | |||
| analytical separation | BEH C18, 2.1 × 50 mm, 1.7 μm, 0.5 mL/min, 5% B, 40 °C on column, 5% B for 0.1 min, 5 to 60% B in 1.9 min, 60 to 98% B in 0.5 min, 98 to 5% B in 0.5 min and 5% B for 0.5 min | ||
| ESI parameters | ESI positive mode | ||
| capillary voltage: +3 kV, cone voltage: 48 V | |||
| source at 150 °C, desolvation at 250 °C | |||
| cone gas flow at 150 L/h, desolvation gas at 1000 L/h, collision gas flow at 0.15 mL/min and nebulizer gas at 7 bar | |||
| acquisition parameters | MRM mode transitions [m/z] | collision energy | |
| LC1 | 896.06 > 807.55 | 24 V | |
| 896.06 > 982.26 | 24 V | ||
| SIL-LC1 | 899.60 > 814.44 | 24 V | |
| 899.60 > 989.27 | 24 V | ||
| data analyses | Targetlynx v. 4.1 | ||
| regression model: linear with 1/x weighting and origin exclusion. Savitzky–Golay smoothing (iterations: 2, width: 2) | |||
| integration using apex track algorithm | |||
ACN: acetonitrile; ESI: Electrospray ionization; n.a. not applicable.
Validation of the Analytical Method
The bioanalytical assay was validated according to FDA and EMA guidelines on bioanalytical method validation.16,17 Validation was carried out with four validation batches and two stability batches. Each batch comprised duplicate samples of blank value (BV, nonspiked control plasma), zero-level calibrators (CAL 0, blank plasma spiked only with SIL-IS), and the nine nonzero calibration levels (CAL A to CAL I). Validation batches also included six replicates of all four QC levels (LLOQ, QC A, QC B, and QC C) and stability batches included duplicates of low, middle, and high QC levels (QC A, QC B, and QC C). QC data from individual validation batches were used to compute within-run and between-run accuracy and precision of the method. Accuracy (% bias) was calculated as the ratio of the difference between the mean measured concentration and the nominal concentration to the nominal concentration. Precision (% CV) was calculated as a percentage of the determined standard deviation of the mean measured concentration.
To prove the specificity of the assay, blank plasma samples from six healthy individuals were evaluated for interfering signals at the surrogate peptide and IS retention times. Likewise, eluent samples measured after the highest calibration standards were investigated to assess potential carry-over effects during analyses.
Analyte recovery of the IP method was assessed by comparing the peak area of HDIT101 in QC samples (QC A, QC B, and QC C, three-fold each) to the peak area in immunopurified blank plasma spiked post-processing with HDIT101 (100%-QC A, 100%-QC B, and 100%-QC C, three-fold each). Likewise, a potential matrix effect of human plasma was evaluated by comparing the peak areas of HDIT101 and its IS from the 100%-QC samples to the peak areas from immunopurified buffer spiked post-processing with HDIT101 (eluent-QC A, eluent-QC B, and eluent-QC C, three-fold each).
Freeze/thaw stability over three cycles and five-month stability were both tested at −80 °C using triplicates of low and high QC-levels, and autosampler stability for 24 h at 10 °C was tested on all calibration standards and QC samples of one stability batch.
PK Parameter Calculations
PK parameter evaluation was performed using Kinetica software V5.0 (Thermo Fisher Scientific) with the following computing parameters: noncompartmental analysis assistant for intravenous infusion, AUC set to log linear, AUC0 set to c0 = 0, AUCinf set to computed Clast/Lz, AUCcum set to “interpolated”, below LLOQ (BLQ) data treatment set to default, dose set as infusion variable, and Cmax set to “use first”. Computed PK parameters were subsequently transferred to Prism V5.01 (GraphPad Software Inc., San Diego, CA, USA) for graphical analyses.
Results and Discussion
Method Development
As the first step, candidate surrogate peptides suitable for subsequent quantification by UPLC–MS/MS were identified. Good candidates for surrogate peptides should be present only in the compound of interest and in no other protein of the targeted organism (humans). This implies that candidates should also include sequences of CDR of the targeted antibody. Ideally and whenever possible, surrogate peptides should be peptides of 10 to 30 amino-acid length19 and comprise no post-translational modification site5 such as cysteine residues, which are readily modified by alkylation thus escaping detection. BLAST search of in silico tryptic peptides against human proteins from UniProt and Swiss-Prot databases revealed three peptides unique to HDIT101 in the heavy chain and two candidates in the light chain (Table 2). However, two of them comprised a cysteine residue, and only one out of five had an ideal length of 14 amino acids. All were nevertheless further experimentally tested to avoid ruling out any valid surrogate peptide only for theoretical reasons.
Table 2. HDIT101 surrogate peptide Candidates and Their Characteristics.
| HC/LC | amino acid sequence | peptide reference | length (amino acids) | cysteine | CDR sequence |
|---|---|---|---|---|---|
| HC | ALEWLAHIWWNNDK | HC1 | 14 | no | yes |
| HC | ESGPALVKPTQTLTLTCTFSGFSLSTSGMSVGWIR | HC2 | 35 | yes | yes |
| HC | IYYGYRPYAMDYWGQGTLVTVSSASTK | HC3 | 27 | no | yes |
| LC | SSQSIVHSBGBTYLEWYLQKPGQSPQLLIYKa | LC1 | 31 | no | yes |
| LC | VEAEDVGVYYCFQGSHVPWSFGQGTK | LC2 | 26 | yes | yes |
The underlined leucine residues indicate the positions for the stable isotope labeling of the LC1 peptide. CDR: complementarity-determining region; HC: heavy chain; LC: light chain.
We first tested whether these five candidate peptides could be detected in digested HDIT101 buffer samples using high-resolution μLC–MS/MS with MSe acquisition. To this aim, HDIT101 in buffer was prepared in four technical replicates as described in the Materials and Methods section (Figure 1), and each replicate was analyzed twice. As detailed in Table 3, no validated MS/MS spectrum could be matched to the HC2 peptide, and the HC1 peptide was matched to validated MS/MS spectra in <40% of the analyses. Given these poor MS responses, these two peptides were excluded from further analyses. For the last peptide remaining from the heavy chain, HC3, matched MS/MS spectra were validated in >60% of the analyses with 6 to 17 b/y ions detected. Because it was also the best remaining candidate when looking at the theoretical characteristics, HC3 was further investigated together with LC1 and LC2, for which matching MS/MS spectra were systematically validated with 12 to 27 b/y ions detected and validated in seven out of eight analyses with 5 to 12 b/y ions detected, respectively. To make sure that none of the remaining candidate peptides could be detected in blank human plasma, digests of purified IgG fractions of blank plasma were also analyzed using high-resolution μLC–MS/MS with MSe acquisition to search for HDIT101 proteotypic peptides. None of the three remaining candidates positively matched the MS/MS spectra from the blank plasma, suggesting that all three candidates were either absent or not detectable in the blank plasma IgG fraction (Table 3).
Table 3. Peptide Discovery Results from MSe Data for the Validation of MS-Compatible HDIT101 Surrogate Peptide Candidates.
| number
of b/y ions in MS/MS spectra matched with HDIT101 theoretical peptidesa |
|||||||
|---|---|---|---|---|---|---|---|
| replicate | HC coverage (%) | LC coverage (%) | HC1 | HC2 | HC3 | LC1 | LC2 |
| 1 | 63.9 | 88.6 | 7 | n.d. | 12 | 23 | 6 |
| 75.7 | 84.9 | 3 (n.v.) | n.d. | 7 | 26 | 7 | |
| 2 | 70.6 | 90.0 | 6 | n.d. | 17 | 27 | 12 |
| 66.6 | 89.0 | 4 (n.v.) | 1 (n.v.) | 1 (n.v.) | 16 | 6 | |
| 3 | 67.3 | 83.6 | 1 (n.v.) | n.d. | 7 | 22 | 6 |
| 40.9 | 79.5 | 2 (n.v.) | n.d. | 4 (n.v.) | 12 | 4 (n.v.) | |
| 4 | 64.4 | 90.0 | 5 | n.d. | 2 (n.v.) | 23 | 5 |
| 67.5 | 88.1 | 4 (n.v.) | n.d. | 6 | 12 | 6 | |
| plasma | 54.2 | 60.3 | n.d. | n.d. | 1 (n.v.) | n.d. | 1 (n.v.) |
| 39.2 | 49.3 | n.d. | n.d. | n.d. | n.d. | 1 (n.v.) | |
| suitable | no | no | yes | yes | yes | ||
A MS/MS spectrum matching with one HDIT101 theoretical peptide must have ≥ 5 b/y ions to be validated. HC: heavy chain; LC: light chain; n.d.: not detected; n.v.: not validated.
The three selected peptides, HC3, LC1, and LC2, were further tested for detection in buffer samples spiked with 200 μg/mL of intact HDIT101, processed in triplicates through the described digestion/clean-up workflow, and analyzed by μLC–MRM/MS. Because it was not possible to clearly identify LC2 peaks in any of the buffer samples spiked with intact HDIT101 (Figure 2), LC2 was removed from the list of candidates. Moreover, MRM data highlighted that only the LC1 peptide could be unambiguously detected from the purified IgG fraction of plasma samples spiked with 200 μg/mL HDIT101 (Figure 3). LC1 was therefore selected as the final surrogate peptide for HDIT101 quantification in human plasma, and its SIL version (SIL-LC1, LC1 peptide with two labeled [13C6, 15N]-leucine residues as described in Table 2) was synthetized for further method optimization.
Figure 2.

MRM traces of transition m/z 1027 > m/z 780 for the HC3 peptide in buffer spiked with 200 μg/mL HDIT101 and in blank buffer (A). MRM traces of transition m/z 896 > m/z 982 for the LC1 peptide in buffer spiked with 200 μg/mL HDIT101 and in blank buffer (B). MRM traces of transition m/z 964 > m/z 1125 for LC2 peptide in buffer spiked with 200 μg/mL HDIT101 and in blank buffer (C). Absolute intensities of the most intense peak of each MRM trace are indicated on the baseline of each trace. Retention time of the targeted compounds is given in bold character on the top trace.
Figure 3.

MRM traces of transition m/z 1027 > m/z 780 for the HC3 peptide after complete sample processing of blank plasma and plasma spiked with 200 μg/mL HDIT101 (A). MRM traces of transition m/z 896 > m/z 982 for the LC1 peptide after complete sample processing of blank plasma and plasma spiked with 200 μg/mL HDIT101 (B). Absolute intensities of the most intense peak of each MRM trace are indicated on the baseline of each trace. Retention time of the targeted compounds is given in bold character on the top trace.
SIL-proteins are considered as the gold standard for absolute protein quantification, because they enable comprehensive assessment of the recovery and matrix effect occurring during the full sample preparation workflow.5,6,20−22 Such SIL-standards are, however, often expensive, and SIL-versions of the surrogate peptide of the targeted protein are usually the most frequently used alternative. As a standard for absolute protein quantification, SIL-peptides can be added either after the digestion process and help evaluating the impact of the desalting step, if present, or it can already be added before digestion to also give insights into possible degradations of the surrogate peptide during tryptic digestion. To make sure that our SIL-peptide could be used as a reliable IS when added before digestion, the synthetic SIL-version of LC1 was added to immunopurified fractions of calibration standards before and after the digestion step, and processed samples were subsequently desalted on a MCX μElution plate and analyzed by μLC–MRM/MS. Our SIL-peptide was added to a zero-level calibration standard and two non-zero level calibration standards at 5 and 5000 μg/mL (each in duplicate) to monitor its behavior in the blank matrix and in the presence of low and high concentrations of the targeted antibody. SIL-LC1 yielded good reliability when added before the digestion step with an acceptable peak area difference (<25% signal loss) between IS added after digestion and the same IS added before. Likewise, the variation coefficients between replicates when SIL-LC1 was added before digestion were similar for the two monitored transitions to those obtained between replicates when SIL-LC1 was added after digestion (between 13 and 21% CV). Therefore, in the final sample processing workflow, SIL-LC1 was added before the digestion step.
For the first development steps, each IP elution fraction was processed as one separate sample to digest. During method development, it turned out that HDIT101 was detected in the three fractions from IP with the major part of HDIT101 mAb being present in the two first elution fractions. To improve the reproducibility of the workflow and enable the process of each sample in one tube without losing any targeted antibody, a freeze-drying process was designed to pool all three IP fractions from one plasma sample and facilitate the next steps of the sample processing. In comparison to response linearity obtained from 10 to 1000 μg/mL HDIT101 in plasma for separated IP fractions (r2 < 0.80 for both transitions, Figure 4A), pooling and freeze-drying of IP elution fractions enabled to reach better response linearity (r2 > 0.95 for both HDIT101 transitions, Figure 4B) over the same calibration range. Therefore, pooling and freeze-drying of IP elution fraction was kept for the optimized workflow. Furthermore, these experiments also enabled to choose the first LC1 MRM transition, m/z 896 > m/z 807, as final quantification trace, associated to its equivalent trace for SIL-LC1, m/z 899 > m/z 814. Within the freeze-dried samples, this transition combination for response calculations gave an improved response linearity with an r2 coefficient of 0.993 against only 0.952 for the combination m/z 896 > m/z 982/m/z 899 > m/z 989. For the first transition combination, all back-calculated concentrations using mean responses from duplicate samples were within the ±15% bias accuracy limits or ±20% bias at LLOQ (10 μg/mL), whereas only two out of five levels reached the criteria for the second transition combination.
Figure 4.

HDIT101 response linearity of the two monitored transitions m/z 896 > m/z 807 (■ and ●) and m/z 896 > m/z 982 (□ and ○) from the processed first single IP fraction (A) and from processed pooled freeze-dried fractions (B). Responses are given in arbitrary units (a.u.).
During the last steps of method development, MRM traces of both surrogate peptides, LC1 and IS SIL-LC1, exhibited LC peak instability in processed samples after MCX μElution (Supporting Information part II, Figure S1A). This instability originated from deamidations of the surrogate peptide probably because of basic pH conditions during MCX μElution after trypsin digestion and storage in the autosampler (10 °C) for 24 h (Table S2), as already described in similar protein digestion conditions.23 The observed peak instability made the peak integration for HDIT101 quantification more challenging because integration needed to be manually changed for each transition to include all LC1-related LC peaks in order to prevent underestimating the LC1 amount in the samples. This also greatly increased the risk of integrating peaks from other peptides and then affected the specificity of our assay. The SPE μElution protocol was thus changed to HLB μElution with 50% ACN in 0.1% FA as elution solvent. This greatly improved the signal stability of both LC1 and SIL-LC1 peptides and thus improved the peak integration reproducibility and specificity (Supporting Information part II, Figure S1B).
Further analyses of calibration standards processed in plasma at ten nonzero calibration levels (10, 20, 40, 75, 150, 315, 625, 1250, 2500, and 5000 μg/mL) revealed response linearity from 20 to 5000 μg/mL (Supporting Information part III, Figure S2). LLOQ was set to 20 μg/mL, which was the lowest concentration level fulfilling the <20% CV precision limit between duplicates, and the upper limit of quantification (ULOQ) was set to 5000 μg/mL to avoid saturation of the IP resin. Response linearity in this concentration range was proven with an r2 coefficient of 0.998 and 100% of back-calculated concentrations within the ±15% bias accuracy limits or ±20% bias at the LLOQ (Figure S2 and Table S3).
With this optimized sample preparation workflow and the MRM/MS method, the HDIT101 quantification assay was then transferred from μLC–MRM/MS to UPLC–MRM/MS for method validation and study sample analysis. Using μLC–MRM/MS, it was necessary to use a long gradient over ≥20 min, which makes analyses of large batches of study samples extremely long (≥3 day run for duplicate analysis of 40 study samples with associated calibration standards, QC samples, and eluent samples to avoid carry-over when needed). Micro-LC dimensions made also the analyses more challenging because of the significant risk of carry-over arising from the nature of the HDIT101 surrogate peptide. For all these reasons, we decided to perform our developed bioanalytical assay with higher flow LC using UPLC separation to shorten the analysis time to 3.5 min and minimize carry-over effects. This also resulted in cleaner MRM traces further improving reproducibility of peak integration (Figure 5). Using the UPLC–MRM/MS system enabled confirming linearity from 20 to 5000 μg/mL with r2 determination coefficients >0.98 and fulfilling the FDA’s and EMA’s relevant criteria (Figure 6).
Figure 5.

Comparison of MRM traces obtained for quantification from both LC1 and SIL-LC1 in a 20 μg/mL HDIT101 plasma sample after μLC–MRM/MS analysis (A) and after UPLC–MRM/MS analysis (B).
Figure 6.

Calibration curve of HDIT101 spiked in human plasma highlighting the response linearity over the concentration range from 20 to 5000 μg/mL (A). Representative MRM traces of HDIT101 surrogate peptide LC1 (B) and its IS SIL-LC1 (C) in a blank plasma sample (BV) and at LLOQ (20 μg/mL) and ULOQ (5000 μg/mL) levels. Absolute intensity of the most intense peak of each MRM trace is indicated on the baseline of each trace. The retention time of the targeted compounds is given in bold character on the top trace.
Method Validation
The HDIT101 quantification assay was validated following the FDA and EMA guidelines for bioanalytical method validation.16,17 Validation included four batches, and its results are summarized in Table 4.
Table 4. Result Summary for the Validation of HDIT101 Quantification Assay.
| validation components | summary | |||||||
|---|---|---|---|---|---|---|---|---|
| analyte | HDIT101 mAb using its surrogate peptide LC1. MRM transition: 896.06 > 807.55 | |||||||
| IS | stable-isotopically-labelled version of LC1 peptide (SIL-LC1) | |||||||
| matrix | human plasma | |||||||
| sample volume | 25 μL | |||||||
| analytical method | UPLC–MRM/MS, 10 μL injection, 3.5 min LC gradient | |||||||
| sensitivity | LLOQ at 20 μg/mL, six replicates in every batch. | |||||||
| precision (% CV) | accuracy (% bias) | |||||||
| within-batch | 9.0 | 7.2 | 12.3 | 13.9 | –3.0 | 4.8 | –8.9 | –2.1 |
| batch-to-batch | 11.0 | –2.0 | ||||||
| specificity | no significant interfering peak for LC1 (<6.9% of lowest LLOQ signal) and SIL-LC1 (<2.3% of mean SIL-LC1 signal) observed in blank human plasma samples | |||||||
| carry-over | no significant interfering peak for LC1 (<12.9% of lowest LLOQ signal) and SIL-LC1 (<0.2% of mean SIL-LC1 signal) observed in eluent samples following CAL I standards | |||||||
| calibration range | 20–5000 μg/mL | |||||||
| QC within-batch results | precision (% CV) | accuracy (% bias) | ||||||
| QC A—60 μg/mL | 7.2 | 7.9 | 9.2 | 12.0 | 3.4 | 6.1 | 8.6 | –0.1 |
| QC B—2000 μg/mL | 5.1 | 7.8 | 12.6 | 12.6 | 8.2 | 6.1 | 3.8 | –0.3 |
| QC C—4000 μg/mL | 5.9 | 10.1 | 11.3 | 8.4 | 2.7 | 4.8 | 0.3 | 0.1 |
| QC batch-to-batch results | precision (% CV) | accuracy (% bias) | ||||||
| QC A—60 μg/mL | 9.1 | 4.0 | ||||||
| QC B—2000 μg/mL | 9.0 | 4.8 | ||||||
| QC C—4000 μg/mL | 8.4 | 1.7 | ||||||
| freeze-and-thaw stability | demonstrated over three freeze-and-thaw cycles from –80 °C to room temperature on the three QC levels in triplicates. | |||||||
| processed sample stability | demonstrated for 24 h at 10 °C on full calibration range and QC samples with <11% CV between reinjected and original results. | |||||||
| long-term stability | demonstrated over four months at –80 °C for the three QC levels in duplicates. | |||||||
| recovery | average of 98.2% of recovery for HDIT101 for the three QC levels in triplicates using protein A IP (6.9% CV) | |||||||
| matrix effect | normalizeda matrix-effect consistency for each QC level in duplicate. | |||||||
| QC A—60 μg/mL | 36% (11.4% CV) | |||||||
| QC B—2000 μg/mL | 40% (6.1% CV) | |||||||
| QC C—4000 μg/mL | 71% (6.3% CV) | |||||||
LC1 matrix effect was normalized by the SIL-LC1 matrix effect.
The method LLOQ was set to 20 μg/mL, and sensitivity was shown across the four validation batches using six replicates of LLOQ level plasma samples. As recommended in the guidelines for LC–MS/MS bioanalytical method validation for the LLOQ level, within-batch and batch-to-batch precisions were below 20% CV, with within-batch precisions between 9 and 14% CV, and a batch-to-batch precision of 11% CV. Similarly, within-batch and batch-to-batch accuracies fell into the recommended ±20% bias limits, with within-batch accuracies between −9% bias and 5% bias and batch-to-batch accuracy at −2% bias. The specificity of the method was also proven for the HDIT101 surrogate peptide LC1 and the IS, SIL-LC1, using blank plasma samples from six different individuals. In these samples, both targeted signals were <7% of the LC1 signal at the LLOQ level and <3% of the mean SIL-LC1 signal from calibration standards and QC samples. Furthermore, in all four batches, no carry-over occurred after the higher concentrated calibration standard CAL I, with targeted signals always lower than 13% of the LC1 signal at the LLOQ level and lower than 1% of the mean SIL-LC1 signal from calibration standards and QC samples.
Over the full validation procedure, four calibration curves were computed using Targetlynx data processing software. All four curves were drawn between 20 and 5000 μg/mL using a linear regression model with 1/x weighting. Linearity was proven for every batch on at least eight nonzero calibration levels with r2 determination coefficients >0.98 and >75% of calibration standards within the ±15% bias accuracy limits and below the 15% CV limit between duplicates. All validation runs passed acceptance criteria (15% CV for precision and ±15% bias for accuracy) with within-batch accuracies between −1 and 9% bias, within-batch precisions between 2 and 13% CV, batch-to-batch accuracies between 1 and 5% bias, and batch-to-batch precision between 8 and 10% CV on ≥ 5 replicates per level for the three QC levels.
Recovery was tested on the three QC levels and was proven to be reproducibly close to 100% on triplicate experiments with an average HDIT101 recovery from IP of 98% and a precision of 7% CV. Matrix effects were tested on all three levels, and <15% CV of the normalized matrix effect was proven on duplicate experiments.
Stability experiments were conducted on two additional batches. Freeze-and-thaw stability of QC samples was proven on three cycles from −80 °C to room temperature with >75% of tested QC samples within the ±15% bias accuracy limits. Autosampler stability of processed samples was proven at 10 °C for 24 h with 100% of calibration standards (nine nonzero concentration levels in duplicates) and QC samples (three concentration levels in duplicates) < 11% CV. This confirmed the possibility to reinject processed samples within 24 h. Finally, long-term stability of QC samples was proven at −80 °C for 4 months with >75% of tested QC samples within the ±15% bias accuracy limits.
Measurements of Plasma Samples from the Phase I Clinical Trial
The sets of 20 plasma samples from six healthy volunteers—one for each dose level—were analyzed using the validated assay within six different batches, each consisting of the study samples blank plasma samples without and with SIL-LC1, calibration standards at nine nonzero levels, QC samples at three concentration levels, and the study samples. Each blank, calibration standard, QC sample, and study sample was processed in duplicate, and all plasma samples from one healthy volunteer were processed within the same analytical batch.
Analytical runs were accepted according to FDA regulatory guidelines with >75% of calibration standards within the ±15% bias accuracy limits and a proven linearity in the calibration range (20–5000 μg/mL) using ≥eight nonzero calibration levels. The complete set of QC samples from analytical batches yielded within-batch accuracy and precision on the three QC levels between −2 and 4% bias and between 10 and 11% CV, respectively.
For the six healthy volunteers, the mean concentrations of each plasma sample were calculated using the duplicate analyses, and samples for which >20% mean deviation was observed between duplicates were excluded from PK evaluations. Overall, the complete set of samples from the healthy volunteer dosed with 50 mg of HDIT101 was excluded from the PK evaluation because all concentrations were below LLOQ.
Among the five remaining sample sets, >85% of the mean concentrations above LLOQ were accepted with <20% mean deviation. The PK curves from the evaluable five healthy volunteers were subsequently plotted using the mean concentrations of each sample set (Figure 7A,B), and PK parameters were computed using Kinetica software. Dose linearity of the exposure was proven (Figure 7C) with the slope of the linear regression being close to 1 (0.9111), and the clearance and half-life of HDIT101 were shown to be constant over the full dose range (13.9 ± 2.8 mL/h and 12.2 ± 1.4 days, respectively) with the slopes of the linear regression being not significantly different from 0 (0.08899 and 0.03294, respectively) (Figure 7D,E).
Figure 7.

PK profiles over 29 days after SOI of the five healthy volunteers dosed with 150, 450, 1350, 4050, and 12,150 mg, respectively, (A) and zoomed view on the first 48 h (B). Dose correlations with total area under the curve (AUC0–inf) (C), clearance (D), and half-life (Thalf), (E) showing dose linearity for the AUC0–inf, a stable clearance, and Thalf independent from dose.
Conclusions
The development of methods for absolute and specific quantification of therapeutic mAbs in biological matrices is challenging because of matrix complexity and the multiple sample preparation steps (i.e., IgG-fraction purification, digestion process, and SPE desalting) that are necessary for protein processing. Sub-optimization of each of these crucial steps can greatly impair the final results of the developed assay. In the present work, we detailed all steps that were necessary to establish an assay for absolute quantification of a new mAb against HSV (HDIT101) in plasma by monitoring its surrogate peptide, LC1. In particular, to maximize mAb extraction and substantially improve assay reproducibility, three IP elution fractions were collected from one plasma sample and were subsequently pooled and freeze-dried before digestion. The optimization of the solid-phase extraction step for the peptide digest clean-up was also critical. The initially used mixedmode cation-exchange μElution with basic pH elution conditions induced asparagine deamidations when stored for 24 h at 10 °C after processing. The final optimized μElution workflow using reversed phase μElution with acidic elution conditions enabled to prevent it. Finally, we showed that UPLC clearly improved data treatment reproducibility with the retrieval of a single peak for the HDIT101 surrogate peptide and a much shorter analysis time. Overall, each of these optimization steps illustrated the high importance of carefully adjusting biochemical and LC–MS/MS settings whenever possible to establish a reliable assay for mAb quantification in the biological matrix. Altogether, although specific attention for the properties of the antibodies of interest stays absolutely necessary to reach an optimal quantification, this work illustrates the development and optimization of a MS-based quantification assay that can be applied to other therapeutic antibodies with limited additional optimization steps, such as the specific choice of an IS and a MRM trace for quantification, as well as the choice of a μElution workflow to maximized the surrogate peptide recovery and stability.
The reliability of the present HDIT101 MS-based quantification assay was demonstrated by successful validation according to the guidelines of FDA and EMA for bioanalytical method validation. Therefore, the method is readily useable for further evaluation of HDIT101 PK properties from clinical study samples, as it has been started with the PK analysis of samples collected in the first-in-human trial.
Future developments can realistically be envisioned in order to overcome the limitations of the present method and for its universalization. SIL-peptides are unfortunately not enriched by IP and are not as affected by proteolytic digestion as the targeted mAb. Therefore, these processes cannot be taken into account for normalization. To overcome this limitation, generic antibodies designed for internal standardization could be spiked in samples before processing, subsequently extracted by immunoprecipitation and digested.24,25 Resulting proteotypic peptides could then be used for signal normalization. The use of generic antibodies instead of SIL-peptides as ISs would permit a more accurate normalization and would lead to the development of universal methods for the quantification of mAbs in plasma. Such adaptation would also permit multiplexing mAb quantification. Additional developments could also be considered to reduce time and complexity for sample preparation. Further processing of immunopurified antibodies in solution currently necessitates a step of freeze-drying that leads to multiple sample transfers and a sample preparation time of two days. Former development in proteomics demonstrated that proteolytic digestion is possible directly from filtration membranes.26 Following the same principle, proteolytic digestion may be performed directly from the immunoprecipitation cartridges. This would help avoiding multiple sample transfers and the need of a freeze-drying step, thus enabling to reduce sample preparation time to only one day.
The present developments, combined with further adaptation for sample preparation, may open the road for the universalization of methods for mAb quantification in plasma.
Acknowledgments
Kevin Steimel is warmly acknowledged for his excellent technical support. This research was funded in part by the German Cancer Consortium (DKTK), a joint initiative involving the German Federal Ministry of Education and Research (BMBF), participating German states and the German Cancer Research Center (DKFZ).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02547.
Technical parameters used on the LC–MS devices for method development, details of the observation, experiments, results obtained to highlight the process of deamidation in peptide samples after basic μElution, obtained calibration curve and accuracy and precision calculations that helped to set the final calibration range (PDF).
Author Contributions
M.F. and J.B. designed the bioanalytical assay development. T.S., M.A., J.K., A.B., and W.E.H. designed and performed the clinical study. M.F. and R.L. performed sample preparation. M.F., R.L., M.S., and J.B. performed the analyses and data treatment. M.F. and R.L. wrote the manuscript. All authors provided scientific input and read the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Tsumoto K.; Isozaki Y.; Yagami H.; Tomita M. Future perspectives of therapeutic monoclonal antibodies. Immunotherapy 2019, 11, 119–127. 10.2217/imt-2018-0130. [DOI] [PubMed] [Google Scholar]
- Hagman C.; Ricke D.; Ewert S.; Bek S.; Falchetto R.; Bitsch F. Absolute quantification of monoclonal antibodies in biofluids by liquid chromatography-tandem mass spectrometry. Anal. Chem. 2008, 80, 1290–1296. 10.1021/ac702115b. [DOI] [PubMed] [Google Scholar]
- Iwamoto N.; Takanashi M.; Shimada T.; Sasaki J.; Hamada A. Comparison of bevacizumab quantification results in plasma of non-small cell lung cancer patients using bioanalytical techniques between LC-MS/MS, ELISA, and microfluidic-based immunoassay. AAPS J. 2019, 21, 101. 10.1208/s12248-019-0369-z. [DOI] [PubMed] [Google Scholar]
- Heudi O.; Barteau S.; Zimmer D.; Schmidt J.; Bill K.; Lehmann N.; Bauer C.; Kretz O. Towards absolute quantification of therapeutic monoclonal antibody in serum by LC-MS/MS using isotope-labeled antibody standard and protein cleavage isotope dilution mass spectrometry. Anal. Chem. 2008, 80, 4200–4207. 10.1021/ac800205s. [DOI] [PubMed] [Google Scholar]
- van den Broek I.; Niessen W. M. A.; van Dongen W. D. Bioanalytical LC-MS/MS of protein-based biopharmaceuticals. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2013, 929, 161–179. 10.1016/j.jchromb.2013.04.030. [DOI] [PubMed] [Google Scholar]
- Jenkins R.; Duggan J. X.; Aubry A.-F.; Zeng J.; Lee J. W.; Cojocaru L.; Dufield D.; Garofolo F.; Kaur S.; Schultz G. A.; Xu K.; Yang Z.; Yu J.; Zhang Y. J.; Vazvaei F. Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. AAPS J. 2015, 17, 1–16. 10.1208/s12248-014-9685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladwig P. M.; Barnidge D. R.; Willrich M. A. V. Mass spectrometry approaches for identification and quantitation of therapeutic monoclonal antibodies in the clinical laboratory. Clin. Vaccine Immunol. 2017, 24, e00545-16 10.1128/cvi.00545-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhraja R. H.; Shah M. A.; Suthar M.; Yadav A.; Shah S. P.; Kale P.; Asvadi P.; Valan Arasu M.; Al-Dhabi N. A.; Park C. G.; Kim Y. O.; Kim H. J.; Agrawal Y. K.; Krovidi R. K. LC-MS/MS validation analysis of trastuzumab using dSIL approach for evaluating pharmacokinetics. Molecules 2016, 21, 1464. 10.3390/molecules21111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto N.; Shimada T.; Terakado H.; Hamada A. Validated LC-MS/MS analysis of immune checkpoint inhibitor nivolumab in human plasma using a Fab peptide-selective quantitation method: nano-surface and molecular-orientation limited (nSMOL) proteolysis. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1023–1024, 9–16. 10.1016/j.jchromb.2016.04.038. [DOI] [PubMed] [Google Scholar]
- Irie K.; Okada A.; Yamasaki Y.; Kokan C.; Hata A.; Kaji R.; Fukushima K.; Sugioka N.; Okada Y.; Katakami N.; Fukushima S. An LC-MS/MS method for absolute quantification of nivolumab in human plasma: Application to clinical therapeutic drug monitoring. Ther. Drug Monit. 2018, 40, 716–724. 10.1097/FTD.0000000000000558. [DOI] [PubMed] [Google Scholar]
- Kleinnijenhuis A. J.; Ingola M.; Toersche J. H.; van Holthoon F. L.; van Dongen W. D. Quantitative bottom up analysis of infliximab in serum using protein A purification and integrated μLC-electrospray chip IonKey MS/MS technology. Bioanalysis 2016, 8, 891–904. 10.4155/bio-2015-0015. [DOI] [PubMed] [Google Scholar]
- El Amrani M.; Bosman S. M.; Egas A. C.; Hack C. E.; Huitema A. D. R.; van Maarseveen E. M. Simultaneous quantification of free adalimumab and infliximab in human plasma using a target-based sample purification and liquid chromatography-tandem mass spectrometry. Ther. Drug Monit. 2019, 41, 640–647. 10.1097/ftd.0000000000000633. [DOI] [PubMed] [Google Scholar]
- Iwamoto N.; Umino Y.; Aoki C.; Yamane N.; Hamada A.; Shimada T. Fully validated LCMS bioanalysis of bevacizumab in human plasma using nano-surface and molecular-orientation limited (nSMOL) proteolysis. Drug Metab. Pharmacokinet. 2016, 31, 46–50. 10.1016/j.dmpk.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Millet A.; Lebert D.; Picard G.; You B.; Ceruse P.; Guitton J. Determination of cetuximab in plasma by liquid chromatography-high-resolution mass spectrometry orbitrap with a stable labeled 13C,15N-cetuximab internal standard. Ther. Drug Monit. 2019, 41, 467–475. 10.1097/ftd.0000000000000613. [DOI] [PubMed] [Google Scholar]
- Willeman T.; Jourdil J.-F.; Gautier-Veyret E.; Bonaz B.; Stanke-Labesque F. A multiplex liquid chromatography tandem mass spectrometry method for the quantification of seven therapeutic monoclonal antibodies: Application for adalimumab therapeutic drug monitoring in patients with Crohn’s disease. Anal. Chim. Acta 2019, 1067, 63–70. 10.1016/j.aca.2019.03.033. [DOI] [PubMed] [Google Scholar]
- EMA. European Medicines Agency . Guidelines on bioanalytical method validation. 2011, https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed July 22, 2019).
- FDA. US Food and Drug Administration . Bioanalytical method validation guidance for industry. 2018, https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf (accessed January 16, 2019).
- Looker K. J.; Magaret A. S.; May M. T.; Turner K. M. E.; Vickerman P.; Gottlieb S. L.; Newman L. M. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One 2015, 10, e0140765 10.1371/journal.pone.0114989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Zeng J.; Titsch C.; Voronin K.; Akinsanya B.; Luo L.; Shen H.; Desai D. D.; Allentoff A.; Aubry A.-F.; Desilva B. S.; Arnold M. E. Fully validated LC-MS/MS assay for the simultaneous quantitation of coadministered therapeutic antibodies in cynomolgus monkey serum. Anal. Chem. 2013, 85, 9859–9867. 10.1021/ac402420v. [DOI] [PubMed] [Google Scholar]
- Bronsema K. J.; Bischoff R.; van de Merbel N. C. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 893–894, 1–14. 10.1016/j.jchromb.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Nouri-Nigjeh E.; Zhang M.; Ji T.; Yu H.; An B.; Duan X.; Balthasar J.; Johnson R. W.; Qu J. Effects of calibration approaches on the accuracy for LC-MS targeted quantification of therapeutic protein. Anal. Chem. 2014, 86, 3575–3584. 10.1021/ac5001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderón-Celis F.; Encinar J. R.; Sanz-Medel A. Standardization approaches in absolute quantitative proteomics with mass spectrometry. Mass Spectrom. Rev. 2018, 37, 715–737. 10.1002/mas.21542. [DOI] [PubMed] [Google Scholar]
- Hao P.; Ren Y.; Datta A.; Tam J. P.; Sze S. K. Evaluation of the effect of trypsin digestion buffers on artificial deamidation. J. Proteome Res. 2015, 14, 1308–1314. 10.1021/pr500903b. [DOI] [PubMed] [Google Scholar]
- Kaur S.; Liu L.; Cortes D. F.; Shao J.; Jenkins R.; Mylott W. R. Jr.; Xu K. Validation of a biotherapeutic immunoaffinity-LC-MS/MS assay in monkey serum: ’plug-and-play’ across seven molecules. Bioanalysis 2016, 8, 1565–1577. 10.4155/bio-2016-0117. [DOI] [PubMed] [Google Scholar]
- Hashii N.; Tousaka Y.; Arai K.; Goda R.; Inoue N.; Murata K.; Okuzono T.; Sasahara S.; Shigeyama T.; Tachiki H.; Yamane S.; Saito Y.; Ishii-Watabe A. Generic MS-based method for the bioanalysis of therapeutic monoclonal antibodies in nonclinical studies. Bioanalysis 2020, 12, 231–243. 10.4155/bio-2019-0253. [DOI] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.