

Abstract
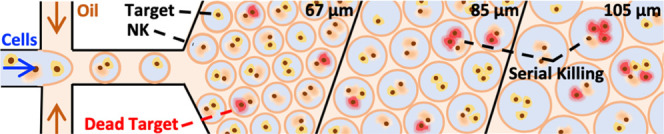
Natural killer (NK) cells are key players of the innate immune system. Due to their rapid cytotoxicity against infectious pathogens, hematologic malignancies, and solid tumors, NK cells represent solid candidates for cell-based immunotherapy. Despite the progress made in recent years, the heterogeneity in their cytotoxic behavior represents a drawback. With the goal of screening the intrinsic diversity of NK cells, droplet-based microfluidic technology is exploited to develop a single-cell time-efficient cytotoxicity assay. Toward this end, NK-92 cells are coencapsulated with hematological tumor cell lines in water-in-oil droplets of different sizes and their cytotoxic activity is evaluated. The effect of droplet-based confinement on NK cytotoxicity is investigated by controlling the droplet volume. The successful optimization of the droplet size allows for time efficiency compared to cytotoxicity assays based on flow cytometry. Additionally, the ability of individual NK-92 cells to kill multiple target cells in series is explored, expanding the knowledge about the serial killing process dynamics. The developed droplet-based microfluidic assay does not require the labeling of NK cells and represents a step toward developing of a forthcoming process for the selection of NK cells with the highest cytotoxicity against specific targets.
Introduction
Natural killer (NK) cells represent fundamental members of the innate immune system. These highly cytotoxic immune effectors mediate rapid and broad cytotoxicity against infectious pathogens, hematologic malignancies, and a number of different solid tumors without prior target recognition.1 Due to their characteristics, they have recently become of interest as potential substitutes for autologous T lymphocytes in cancer immunotherapy,2 exhibiting the potential to overcome some of the drawbacks related to T-cell-based immunotherapy. NK cells exert low cytotoxicity and have a shorter lifespan2 than T cells, along with a lower risk of causing an on-target/off-tumor effect upon injection. Moreover, NK cells could represent an “off-the-shelf” allogeneic product3−5 for transfer therapies, thus marking a pivotal breakthrough for cancer immunotherapy.
Despite its great potential, NK-cell-based immunotherapy still holds some challenges. One drawback consists in the lack of screening and sorting techniques for rapid and accurate ex vivo preselection of highly cytotoxic immune cells (i.e., cells with the ability to kill one or more target cells) among a heterogeneous population. Notably, concerning natural killer cells, approximately half of the population does not kill any target cells at all. In contrast, approximately 7–10% of NK cells exhibit the ability to kill repeatedly, up to seven times in a row having been observed.6−8 Current cytotoxicity assays are carried out in bulk and are based on the evaluation of radioactive9 or fluorescent10 labels released or changing within targeted cancer cells lysed by cytotoxic immune cells, respectively. These bulk assays deliver averaged results for an entire batch of cells, effectively camouflaging molecular-level differences between individual NK cells. Moreover, time-lapse monitoring of individual NK cells interacting with their respective targets in bulk is challenging and time-consuming due to the consistent motility of immune cells. Fluorescence-activated cell sorting (FACS) is commonly employed to differentiate between individual NK cytotoxic subsets, but it has the limitation of requiring labeling procedures.11−13 Staining immune cells helps in recognition but, nevertheless, can affect cell behavior and thereby compromise the potential clinical use. Towards this end, the development of single-cell, high-throughput ex vivo screening methodologies is fundamental to both investigate and fine-tune the immunological process.
Droplet-based microfluidic technology has emerged as a powerful tool for handling and investigating single-cell genetics and behavior in a high-throughput manner.14 Surfactant-stabilized, microsized droplets of water-in-oil emulsion generated in microfluidic devices have been tested and recently used as picoliter biocompatible three-dimensional (3D) confinements for short- (i.e., few hours) and long-time (i.e., few days) spatiotemporal screening of mammalian cells.14−22 Besides its high-throughput screening capabilities, this technology permits the integration of multiple functional modules into one single microfabricated chip, including picoinjection,23 droplet content manipulation,24 and sorting.25 Moreover, droplet-based microfluidics requires minimal manual user intervention and minimal sample consumption. The flexible design of microfluidic devices allows for the formation and manipulation of monodisperse droplets with easily varied diameters ranging upward from 10 μm, hence allowing for the production of well-defined confinements necessary for long-term monitoring of specific cell–cell interactions. Due to its versatility, this technology has already been applied for the investigation of basic immunological behaviors of T, NK, and antigen-presenting cells,26 such as activation mechanisms,27 cytokine release,15,17,19 T-cell receptor (TCR) screening,28 and cytotoxicity.21,29 In the case of droplet-based cytotoxicity assessment, the studies have focused mainly on the dynamics of effector and target cell interactions leading to cytotoxic events and provided general statistics behind the cytolytic activity of immune cells. However, no information regarding the effect of different confinement sizes on the cytolytic activity of NK cells and the “serial killing” behavior of NK cells has been provided. In this study, we exploit droplet-based microfluidic technology to develop a time-efficient single and serial NK cell cytotoxicity assay. By tuning droplet dimensions, we aimed to characterize the effect of different confinements on the cytolytic activity of NK cells, leading to the identification of highly cytotoxic natural killer cells among a heterogeneous population. Toward these goals, NK-92 cells were coencapsulated with different hematological cancer cell types (i.e., K562, Jurkat, and KG1a) inside droplet-based confinements of different sizes (ddroplets = ∼105, 85, and 67 μm). Following coencapsulation, both single and serial cytotoxic events caused by NK-92 cells were recorded and dynamically analyzed over a period of 12 h. We demonstrated how the impact of droplet confinement is capable of boosting the dynamics of NK-92 cytotoxicity against several target cell types in the early stage of the assay time in comparison to flow cytometry methods. Moreover, in contrast to current flow cytometry approaches, we determine the cytolytic dynamics of individual label-free NK-92 cells against multiple targets with higher spatiotemporal resolution. We envision that the developed technology represents a fundamental step toward future selection processes in which highly cytotoxic NK cells and/or T lymphocytes for cancer immunotherapy have to be identified.
Results and Discussion
NK-92 Cytotoxicity Assay inside Droplet-Based Confinements
By developing a single-cell cytotoxicity assay inside droplet-based confinements we aimed to overcome the spatial- and temporal-related issues that affect standard bulk live imaging (see the Supporting Information, Video S1) and flow cytometry assays, respectively. Moreover, by investigating the effect of different confinement sizes on NK-92 single and serial cytotoxicity, against three different hematological malignant cell lines (K562, Jurkat, and KG1a), we intended to assess whereas confinement will enhance the dynamics of NK-92 cytolytic activity, thereby accelerating the integrated assay. When choosing droplet diameters, we considered several conditions such as close proximity between effector and target cells, preserved immune cell motility, and overall cell viability. Figure 1 represents a theoretical evaluation of the expected fitting of different ratios of effector and target cells coencapsulated within droplets of different diameters. We expected that upon encapsulation, effector (red) and target cells (gray) would sediment at the bottom of the droplets. Therefore, the available volume for the effector–target cell complexes (1:1, 1:2, and 1:3, effector-to-target cell ratio) is reduced in comparison with the full volume of the droplets. Based on the theoretical estimation of available space, we assumed that the smallest confinements (∼67 and 85 μm) would provide a higher probability for effector/target cell encounters and interactions in comparison to bigger droplets (∼105 μm) and other cytotoxicity assays.30 In addition to the close proximity, some effector cells require enough space to engage in “on–off conjugation” or “transient scanning” interactions with their targets.21,29,31,32 Notably, in previously published studies,21,29,32 it was reported that, instead of forming a stable immunological synapse, some of the effector cells transiently perform a “scansion” on target cells by engaging in multiple (i.e., 2–10) contacts and dissociations before releasing the cytolytic hit.21 Additionally, we considered that activated effector cells undergo morphological changes and alternate high and low phases of motility, as shown in the studies of Sarkar and Khorshidi.21,29,31 Therefore, if either proximity, motility, or “screening” (i.e., on–off conjugation) is restricted, the cytotoxic behavior of the effector cells could be potentially compromised. Finally, sufficient supply with nutrients to maintain cell viability and functionality during the entire assay time is another important factor.33,34 Altogether, we selected three confinement sizes (∼67, 85, and 105 μm) to find an optimal balance between cell viability, effector–target proximity, and immune cell motility through physical confinement. We expected that, for droplet diameters smaller than 50 μm, aspects such as immune cellular motility and transient “scanning” would have started to compromise the gain in boosting the dynamics of NK cell cytotoxicity.
Figure 1.
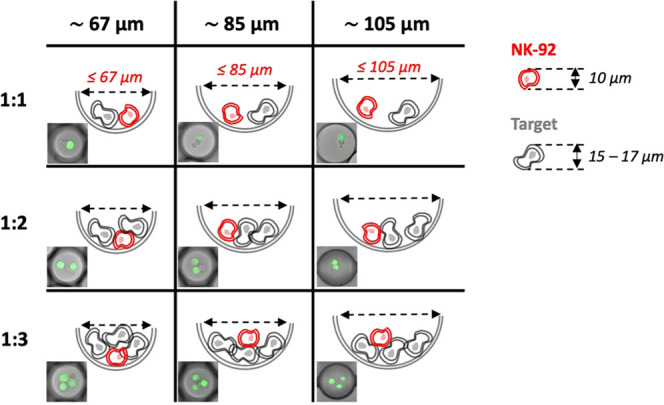
Defining the droplet diameters for single and serial NK-92 cytotoxicity assays. The table depicts the schematic drawings of the lateral view of the three droplet sizes (∼105, 85, and 67 μm), each containing one effector cell (NK-92 in red) and one, two, or three target tumor cells (gray). Each drawing is accompanied by a corresponding representative merged fluorescence and bright-field images of a droplet matching in size and content. Target cells are labeled with Cell Tracker Green. Upon encapsulation, cells precipitate at the bottom of the droplets. The dashed double-sided arrow delimits the potential space available for interactions between effector and target cells in different-sized droplet confinements.
The droplet size was controlled by tuning the pressure of the aqueous and oil phase inlets (Table 1) of the microfluidic device (Figure 2A,C), reaching, on average, droplet diameters of ∼105, 85, and 67 μm (for further information concerning the measured diameters for all of the different coencapsulated target cells, see the Supporting Information, Figure S1). To investigate the dynamics of NK-92 cell cytotoxicity, we began by examining droplets containing a single NK-92 effector cell confined with a single or several same-type target cells (K562, Jurkat, or KG1a) (Figure 2D). Therefore, we investigated the cell-to-cell interaction of two, three, and four cells (i.e., one NK-92 and one, two, or three target cells) within ∼105, 85, and 67 μm droplets. To be able to distinguish between the effector and target cells inside the droplets, we labeled only the target cells using Cell Tracker Green. Droplets containing only NK-92 or only target cells were used as controls to evaluate cellular viability (see the Supporting Information, Figure S2). Cell counts within droplets followed the Poisson distribution (see the Supporting Information, Figure S3). By adjusting the cell density in the aqueous phase prior to coencapsulation, we were able to control, on average, cell counts in the different droplet populations (Table 1).
Table 1. Microfluidic Settings for the Formation of Surfactant-Stabilized Water-in-Oil Droplets of Different Sizesa.
| droplet diameter (μm) | pressure oil phase (bar) | pressure aqueous phase (bar) | number of NK/tumor cells (106 cells/mL) |
|---|---|---|---|
| 105 ± 2 | ∼0.55 | ∼0.5 | ∼1.5–2/1.5–2 |
| 85 ± 2 | ∼1.5 | ∼1.4 | ∼2–2.5/2–2.5 |
| 67 ± 2 | ∼2 | ∼1.75 | ∼2.5–3/2.5–3 |
The table lists the combinations of settings (aqueous and oil phase pressures, NK-92 cell/tumor cell concentration) employed for droplet generation and cell encapsulation.
Figure 2.

Construction of a cytotoxicity assay utilizing droplet-based microfluidics. (A) Representative QCAD design of the microfluidic device including a unit for the generation of cell-laden water-in-oil droplets and a unit for time-lapse analysis of droplets. For visualization purposes, the inlets of the water and oil channels are shown in blue and orange, respectively. (B) Representative bright-field image of the trapped droplets inside the drop-spot chamber. The scale bar is 120 μm. (C) Representative bright-field images of the flow-focusing junction used for the production of cell-laden droplets of different sizes. The scale bar is 80 μm. The ability to tune the pressures applied to the oil and aqueous inlet channels makes it possible to form droplets of different diameters (∼105, 85, and 67 μm). (D) Representative confocal fluorescence images of coencapsulated NK-92 effector and K562 target cells—in different effector/target cell ratios—inside droplets of different diameters. Target cells are labeled with Cell Tracker Green. The scale bars are 50 μm.
Following droplet generation and coencapsulation of effector and target cells, droplets were trapped in a microfluidic observation chamber (Figure 2B) for the cytotoxicity assessment. Pulled by gravity, cells settled at the bottom of the droplets only minutes after encapsulation, irrespective of the droplet size (Figure 2D).
Gravity-driven sedimentation of cells toward the bottom of the droplets enhanced the probability of cell–cell interactions, underscoring the importance of the confinement size. Upon sedimentation, NK-92 cells showed normal motility behavior around target cells as described previously.21,29,31 Briefly, even though no quantitative assessment has been made, NK-92 cells showed behavior such as migration toward the target cells and alternation of high, low, and no motility while changing their morphology and transiently scanning the target cells (see the Supporting Information, Figure S4).
To fully understand the cytotoxic behavior of single NK-92 cells against target cells, we attempted to individually assess the impact of time, confinement space, and target cell type. First, we examined the cytotoxic behavior of individual NK-92 against single target cells in droplets containing only these two cells (Figure 3A–C). For each condition—target cell type (K562, Jurkat, and KG1a) and different diameters—on average, 100 droplets have been analyzed. Cytotoxic events were detected as a change of fluorescence caused by the internalization of fluorescent intercalating agent propidium iodide (PI) into the target cells. Figure 3D–F shows NK-92 cytotoxicity following 12 h of coencapsulation. Regardless of the droplet diameter, after a certain time of culture, the percentage of target cell deaths within a specific population reached a plateau at the values of 57, 37, and 14% for K562, Jurkat, and KG1a cells, respectively. Remarkably, in the case of the smallest droplets (∼67 μm in diameter), the plateau values were, on average, reached within 6 h of coencapsulation of NK-92 with the K562 and Jurkat target cells. This outcome highlighted the impact of confinement size on the cytotoxic behavior of NK-92 cells. A closer look revealed that, regardless of the target cell line, roughly half of the killing events in 67 μm sized droplets happened within the first hours of incubation. The robustness of the developed assay lays in the fact that independently from the droplet size, after 12 h, we are able to quantify the number of NK-92 cells that killed their targets. Moreover, since the dynamics of the cytolytic activity of NK-92 cells is enhanced within the smaller confinement, by running the assay in droplets of ∼67–85 μm in diameter, we are able to determine the overall amount of cytotoxic NK cells in half of the assay time—6 h. Altogether, by investigating the effect of different droplet confinements on NK-92 cell cytotoxicity, we were able to optimize the assay, hence performing a time-efficient screening of cytolytic NK-92 cells. Notably, NK-92 cells and target cells coencapsulated alone showed low cell mortality throughout the entire assay (see the Supporting Information, Figure S2), thus excluding the risk of cell toxicity due to insufficient nutrition and exposure to cellular metabolic waste.
Figure 3.

Effect of confinement size on the dynamics of individual NK-92 cell cytotoxicity against one target cell. (A–C) Representative confocal images of the cytotoxic dynamics of NK-92 cells (contoured) against the coencapsulated (A) K562 (green), (B) Jurkat (blue), or (C) KG1a target cells (turquoise) in droplets of different sizes. White arrows mark target cell death, as determined by changes in PI fluorescence. For ∼105 and 85 μm droplets, the scale bars are 70 μm; for ∼67 μm droplets, the scale bar is 60 μm. The time point 0* indicates the beginning of the microscopy acquisition, corresponding roughly to 30 min after cell encapsulation and droplet entrapment within the microfluidic device. (D)–(F) Graphs summarizing the results of individual NK-92 cytotoxicity assays against single K562, Jurkat, or KG1a cells employing differently sized droplets. The time point 0* indicates the beginning of the microscopy acquisition, corresponding roughly to 30 min after cell encapsulation and droplet entrapment within the microfluidic device. Error bars display standard error of the mean (SEM). (G–I) Comparison of flow cytometry (white bars) and droplet-based (black bars) cytotoxic assays against K562, Jurkat, or KG1a target cells. White bars represent the mean ± standard deviation (SD) of three technical replicates. For black bars, errors depict SEM. The analyzed droplets (∼67 μm) contained just one NK-92 and one target cell each, and in the case of the flow cytometry assay, the target cells and NK-92 cells were co-cultured at a ratio of 1:1 inside a 96-well plate.
We also examined the influence of droplet confinement on the cytotoxic behavior of individual NK-92 cells against two and three target cells (see the Supporting Information, Figure S5). As expected, we observed slightly better performance in cytotoxicity dynamics for the confinements of ∼85 μm in size. This confinement size seems to provide a better balance between the amount of space necessary for cellular motility while ensuring killer-to-target proximity as well as transient scanning of the NK cell across the surface of the target cells.21,31,32 Low cell mortality was observed (see the Supporting Information, Figure S2) for effector and target cells encapsulated alone, precluding the risk of cell death due to prolonged confinement within the droplet.
Our experiments showed that close proximity as well as adequate physical space available for effector and target cell interactions are very important factors to be considered when designing confinement for NK-92 cell cytotoxicity assays. Another determinant of cytotoxic behavior is the ability of the effector cell to recognize a target, which greatly impacts the percentage of killing events observed within a 12 h period. To better understand the impact of recognition, we worked with three different target cell lines. Our data confirm that K562 cells, which proved to be a very effective target in NK-92 cytotoxicity assays,13,35 are around 10–15% more susceptible to NK-92 cytotoxicity than Jurkat cells (Figure 3E). KG1a cells, in comparison, are even less susceptible than Jurkat cells, with a maximum of only 14% killing events during coencapsulation with NK-92 cells (Figure 3F). Moreover, no impact of the confinement size on NK-92 cytotoxicity against KG1a target cells was observed. We assume that low cytotoxicity against KG1a cells is related to the fact that a subpopulation of these CD34+, CD38–, and CD54+ cells may be more resistant, similarly to leukemia stem cells (LSCs), which are the major cause of relapse in acute myeloid leukemia.35,36 It is important to mention here that in order to obtain mechanistic information regarding regulation pathways and ligands involved in triggering or eluding NK cell cytotoxicity (e.g., killer cell immunoglobulin-like receptors or human leukocyte antigens), the developed droplet-based assay must be merged with phenotype and cytokine secretome analyses of both effector and target cells.
Flow Cytometry NK-92 Cytotoxicity Assay
To evaluate the strength and sensitivity of our droplet-based cytotoxicity assay, we compared the assays performed in ∼67 μm droplets to flow cytometry measurements of bulk interactions between NK-92 and target cells (see the Supporting Information, Figure S6, for summarized data). Figure 3G–I depicts the percentage of droplet-based killing events (black bars) in comparison to the percentage of dead target cells recorded with a flow cytometry setup (white bars). Note that a 1:1 ratio of effector/target cells for the bulk assay was evaluated. For all three target cell lines, the percentage of killing events after 3 h of incubation in droplets is higher than in bulk flow cytometry measurements. In the case of K562 and Jurkat target cells, these values converge and, on average, level off over time. Only in the case of KG1a target cells, after 12 h, the number of killing events in bulk exceeds that in droplets. The obtained results revealed faster cytotoxicity dynamics inside the droplets in the early stage of the assay: the necessary assay time for revealing cytolytic activity in optimized confinements could be potentially halved compared to standard flow cytometry. Moreover, the ability to spatiotemporally investigate the behavior of unlabeled NK cells highlights another advantage of the droplet-based assay in comparison to flow cytometry.
Assessment of Early-Stage Serial Killing Behavior of NK-92 Cells inside Droplet-Based Confinements
To further investigate the effect of confinement on the dynamics of cytolytic activity of NK-92 cells toward multiple target cells, we evaluated droplets with diameters of ∼85 and 105 μm containing a single effector cell in addition to two or three target cells. We estimated that droplets of ∼85 μm diameter provide a better environment with sufficient space for the interaction of at least three cells (Figures 1 and S5). In accordance with our observations concerning the restricted ability of NK-92 to effectively recognize KG1a cells as a target for killing, we did not observe any serial killing behavior against KG1a cells. Note that since NK cells can kill up to seven target cells in a row, in this study, we focused our investigation on the early commencement of the serial killing behavior of NK-92 cells, hence evaluating their capability to serially kill up to two or three target cells.
The heat maps in Figure 4A–H visualize the data compiled from the observation of serial killing events inside individual droplets of different sizes. Per condition, at least 75 and 21 droplets have been analyzed for 1:2 and 1:3 effector-to-target ratios, respectively. Note that the disparity in the analyzed number of droplets (n ≥ 21 and ≥75) is attributed to the Poisson distribution of cells within droplets, leading to a higher number of droplets containing three cells (i.e., one NK-92 cell and two targets) in comparison to droplets containing four cells (i.e., one NK-92 cell and three targets).
Figure 4.

Evaluation of the dynamics of NK-92 serial killing behavior via droplet-based microfluidics. (A, B) Heat maps summarizing the killing events by one NK-92 effector cell against two K562 target cells inside ∼105 and 85 μm sized droplets (n ≥ 75) over time (12 h). (C, D) Heat maps summarizing the killing events by one NK-92 effector cell against two Jurkat target cells inside ∼105 and 85 μm sized droplets (n ≥ 75) over time (12 h). (E, F) Heat maps summarizing the killing events by one NK-92 effector cell against three K562 target cells inside ∼105 and 85 μm sized droplets (n ≥ 21) over time (12 h). (G, H) Heat maps summarizing the killing events by one NK-92 cell against three Jurkat target cells inside ∼105 and 85 μm sized droplets (n ≥ 21) over time (12 h). Each row represents the timeline of cell killing events recorded in a single droplet over 12 h of coencapsulation. (I, J) Summary of the effect of confinement size on the ability of NK-92 effector cells to kill two (1:2) and three (1:3) K562 (I) or two (1:2) and three (1:3) Jurkat target cells (J) in series following 12 h of coencapsulation. (K, L) Summarized data of the effect of confinement size on the dynamics of NK-92 serial killing behavior against two K562 or two Jurkat target cells. NK-92 cells with the ability to kill their targets in a row were classified according to their killing speed and termed as fast serial killers (two targets in ≥1 h), mediocre serial killers (two targets in ≥3 h), or slow serial killers (two targets in ≥6 h).
We formatted the color scale according to the number of killing events per droplet: the darker the color, the more the killing events observed. Each row represents the timeline of the NK-92 cell killing events recorded in a single droplet. Columns correspond to time points during coencapsulation. Reducing the droplet diameter from ∼105 to 85 μm for droplets containing a 1:2 effector/target cell ratio led to an increase in the percentage of two consecutive killing events. This increase was from 37 to 55% (Figure 4A,B) and from 5 to 37% for K562 and Jurkat cells (Figure 4C,D), respectively. In droplets containing three target cells, confinement size reduction boosted the number of consecutive killing events from 28 to 37% in the case of K562 target cells (Figure 4E,F) and from 3 to 14% in the case of Jurkat target cells (Figure 4G,H). These results again underscored the significance of confinement on NK-92 serial killing behavior against both K562 and Jurkat target cells (see Figure 4I,J for summarized data).
To further advance our method of classifying NK-92 cells according to their killing efficiency, we also looked at the speed with which the NK-92 cells kill their target (for the purpose of this analysis, only droplets containing one serial killer NK-92 cell and two target cells were considered). With regard to killing speed, we differentiated between “fast serial killers” that kill two target cells in ≥1 h, “mediocre serial killers” referring to cells that can achieve this in ≥3 h, and “slow serial killer” that take longer than 6 h (Figure 4K,L). We then assessed how droplet size (i.e., ∼105 μm compared to 85 μm diameter) affects the speed with which serial killer cells are able to kill K562 or Jurkat target cells in droplets with two target cells. Our results show that in droplets containing two K562 target cells, around 13% of serial killer cells act fast and 54% are slow to kill the two targets in droplets of ∼105 μm diameter. The percentage of fast serial killer cells is higher in ∼85 μm sized droplets, where 50% of effector cells kill the two target cells in less than 3 h and only 16% take longer than 6 h to achieve this. All of the while, the percentage of mediocre serial killer cells is stable at around 30%; in other words, their level of killing activity is almost unaffected by the droplet size. In contrast, confinement has a different effect on the killing speed that NK-92 cells can achieve when interacting with Jurkat cells. In this case, an average of 22–25% fast serial killer cells were detected irrespective of the droplet diameter, whereas the percentage of mild and slow serial killer cells was affected by droplet size. However, the overall number of NK-92 serial killer cells increased from ∼105 to 85 μm droplets.
Altogether, we would attribute the increase of “fast” serial killer cell nor to a change of phenotype of effector cells neither to the target cell type. Since we are investigating the same cell types coencapsulated together within different volumes, we believe that the major force driving this phenomenon is the physical confinement. The appropriate confinement (roughly 85 μm) that balanced the closest proximity possible between effector and target cells while providing enough space for cellular motility enhanced the dynamics of the serial killing.
Summary and Conclusions
In this study, we developed a droplet-based microfluidic assay for time-efficient evaluation of NK cell cytotoxic heterogeneity against hematological cancer cells. Toward this end, we generated cell-laden water-in-oil droplet compartments of different sizes with the goal of optimizing the identification of NK cells that have the ability to kill one or multiple targets. We demonstrated that the confinement size has an effect on the cytotoxic behavior of individual NK-92 cells against both K562 and Jurkat target cells. We, in fact, showed that to fastly observe NK cell cytolytic activity toward target cells, the optimal droplet size should provide a balance between the amount of space necessary for transient conjugation as a part of the cellular recognition process and ensuring killer-to-target proximity. The developed droplet-based cytotoxic assay enables us to assess the dynamics behind repeated cell killing behavior. Again, the size of the confinement had an important effect on the speed with which NK-92 cells can kill multiple targets in series. Overall, the assay time of the developed droplet-based cytotoxicity assay in optimized confinement conditions (∼67 and 85 μm) could potentially be halved in comparison with the flow cytometry-based cytotoxic assay.
Materials and Methods
Materials
Heat-inactivated fetal bovine serum (h.i. FBS), heat-inactivated horse serum (h.i. HS), penicillin/streptomycin solution, Gibco GlutaMAX (100x), Gibco 2-Mercaptoethanol, sodium pyruvate (100 mM), Molecular Probes Cell Tracker Green CMFDA dye, RPMI 1640 medium, CO2-independent medium, propidium iodide (PI), interleukin-15 (IL-15), and human recombinant were purchased from Thermo Fisher Scientific (Germany). Interleukin-2, human recombinant (hrIL-2), and minimum essential medium eagle-α modification (α-MEM) were purchased from Merck (Germany). Iscove’s modified Dulbecco’s medium (IMDM) was purchased from ATCC (Germany).
Cell Lines
NK-92 cells were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Germany) and cultured in α-MEM medium enriched with 10% (v/v) h.i. FBS, 10% (v/v) h.i. HS, 2 mM l-glutamate, 1 mM sodium pyruvate, 100 μM 2-mercaptoethanol, and IL-2 (10 ng/mL). Chronic myelogenous leukemia cells K562 were purchased from ATCC and cultured in IMDM supplemented with 10% (v/v) FBS. Acute T-cell leukemia Jurkat E6.1 and acute myelogenous leukemia KG1a cells were purchased from Cell Line Services (Germany) and cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS and in IMDM enriched with 10%(v/v) FBS and 2 mM l-glutamate, respectively. All cell lines used in this study were maintained in a suspension culture following the manufacturer’s recommendations in a controlled environment of 37 °C and 5% CO2. NK-92 cells were used as effector cells, whereas K562, Jurkat, and KG1a were used as target cells. Prior to cell encapsulation, the effector and target cells were resuspended in the CO2-independent medium containing 10% (v/v) h.i. FBS, 10% (v/v) h.i. HS, 2 mM l-glutamate, 1 mM sodium pyruvate, 100 μM 2-mercaptoethanol, IL-2 (10 ng/mL), IL-15 (30 ng/mL), and PI (1 μg/mL).
Microfluidic Chip Design and Fabrication
Droplet-based microfluidic devices made of poly(dimethylsiloxane) (Dow Corning) were prepared by photolithography and soft lithography methods as described previously.37−39 Briefly, the geometry of the microfluidic device was drawn on QCAD-pro (Ribbon-Soft, Switzerland) and the respective photomasks were printed on chrome-coated soda lime glass (JD-Photodata, U.K.). For photolithography processes, a negative SU8-3050 photoresist (MicroChem) was spin-coated onto a silicon wafer at 1700 rpm to achieve an 80 μm thin uniform coating. Following SU8 coating, the wafer was subjected to a soft bake step at 65 °C on a hot plate for 5 min and another 15 min at 95 °C. Next, the wafer was moved to an MJB4 mask aligner (SÜSS MicroTec, Germany), aligned to the respective photomask, and exposed to UV light for 7.5 s. Two postexposure baking steps were performed, including 1 min on a hot plate at 65 °C and 5 min at 95 °C. Afterward, the remaining photoresist was removed using an mr-DEV 600 developer (MicroChemicals, Germany). Finally, the wafers were placed in the oven at 150 °C for 15 min. To produce microfluidic chips, poly(dimethylsiloxane) (PDMS) soft lithography was performed as previously described.38 Briefly, the PDMS oligomer was mixed with the polymerization catalyst at a 10:1 (w/w) ratio, degassed, and poured over the silicon master wafer. The PDMS-covered wafer was cured in an oven for 2 h at 65 °C. Afterward, the PDMS mold was peeled off from the wafer and a biopsy puncher with a diameter of 0.75 mm (World Precision Instruments) was used to generate the inlets and outlets. The PDMS device together with a 24 × 60 mm #1 coverslip (Carl Roth, Germany) were cleaned with ethanol and activated for 30 s with TePla 100 oxygen plasma at 0.4 mbar and 200 W (PVA TePla, Germany).40 After activation, the PDMS device and the coverslip were bonded together and placed in an oven at 65 °C for 2 h. The microchannel surfaces were finally rendered hydrophobic through passivation with Sigmacote (Sigma-Aldrich, Germany).
Surfactant Synthesis
The synthesis of a PFPE (2500 g/mol)–Jeffamine (600 g/mol)–PFPE (2500 g/mol) triblock copolymer surfactant was accomplished following a previously published protocol.20,41 Briefly, the synthesis of the triblock copolymer was carried out under an argon atmosphere in flame-dried glassware. First, Krytox 2500 (10 g, 4 mmol, 1.0 equiv; DuPont, Netherlands) was dried under reduced pressure and afterward dissolved in 30 ml of anhydrous HFE-7100 (IoLiTec—Ionic Liquids Technologies GmbH, Germany). One drop of anhydrous dimethylformamide (DMF) was added to the Krytox solution, and the mixture was cooled to 0 °C in an ice bath. Oxalylchloride (3 mL, 35 mM, 8.75 equiv; Sigma-Aldrich) was added to the solution dropwise over 1 h and stirred for an additional hour at 0 °C. Under continuous stirring, the solution was slowly heated to room temperature and stirred overnight. The next day, the solvent was removed under reduced pressure at 40 °C and the resulting acid chloride was again dissolved in 30 mL of anhydrous HFE-7100. In the second flame-dried flask, Jeffamine 600 (2.4 g, 4 mM, 1.0 equiv; Sigma-Aldrich) was dissolved in 12 mL of anhydrous HFE-7100 and 24 mL of anhydrous DCM. Triethylamine (1 mL, 7.2 mM, 1.8 equiv) was added to this solution, and the mixture was stirred for 30 min. Afterward, the before-prepared acid chloride solution was added and the resulting mixture was stirred again overnight. The crude product was obtained by removing the solvent under reduced pressure at 40 °C. Afterward, the crude product was again dissolved in 20 mL of HFE-7100 and 2 mL of Jeffamine 600 and 4 g of PEG 100 000 was added to remove unreacted Krytox. The mixture was stirred for 2 h and then centrifuged for 24 h (3000 rpm, 4 °C). After centrifugation, the product was filtered (0.22 μm syringe filters, TPP, Switzerland) and the solvent was removed under reduced pressure at 40 °C. The obtained product was a clear, highly viscous liquid (5 g, 1.07 mM, 26.8%).
Water-in-Oil Droplet Production
Stable droplets42 were generated using a medium containing different concentrations of effector and/or target cells (aqueous phase) and FC-40 oil (Acros Organics, Germany) containing 10 mM custom-made triblock PFPE–Jeffamine–PFPE copolymer surfactant (oil phase). Oil and aqueous phases were injected into the microfluidic chip via PTFE tubing (0.4–0.9 mm, Bola, Germany) connected to 1 mL microtube (Eppendorf) at one end and to the respective inlets of the microfluidic device at the other end. A flow controller system (ELVEFLOW-OB1 MK3, France) was used to control the flow rates. Droplets of different diameters were obtained by tuning the flow rates of the oil and aqueous phase inside the microfluidic device (Table 1). The droplet production quality was monitored with a high-speed camera (Phantom 7.2, Vision Research).
Cell Encapsulation and Distribution in Droplets
Prior to encapsulation, target cells (K562, Jurkat, and KG1a) were stained using Cell Tracker Green CMFDA dye (CTG; excitation/emission peak values, 492/517 nm) according to the manufacturer’s instructions and then resuspended in the CO2-independent medium supplemented with 10% (v/v) h.i. FBS, 10% (v/v) h.i. HS, 2 mM l-glutamate, 1 mM sodium pyruvate, 100 μM 2-mercaptoethanol, IL-2 (10 ng/mL), IL-15 (30 ng/mL), and PI (1 μg/mL). A spiral inlet channel43 connected to a T-junction (channel width ∼80 μm, see Figure 2A) was used to coencapsulate NK-92 cells and target cancer cells into droplets (Figure 2D). By adjusting the flow rates of the oil and aqueous phases as well as the concentration of the effector and target cells in the aqueous phase, droplets of a specific size containing NK-92 and target cells were achieved (Table 1). The efficiency of coencapsulation was assessed by bright-field and confocal fluorescence microscopy.
Droplet Trapping in the Microfluidic Observation Chamber
Following droplet generation at the T-junction, the droplets flowed into the microfluidic observation chamber (Figure 2A,B). The observation chamber consists of 80 channels, each containing 80 spherical traps with a diameter of dtrap ∼ 100 μm, thus yielding a total of 6400 observation traps. By instantly reducing the pressure to 0 bar, no new droplets were generated at the cross junction and the droplets flowing into the chamber were trapped inside the spherical trap. Once the observation chamber is filled with droplets, both the inlets and the outlet of the microfluidic device are sealed to avoid any further oil evaporation. Finally, the microfluidic chip is transferred into the incubation chamber of a laser-scanning confocal fluorescence microscope (LSM880, Carl Zeiss, Germany) for time-lapse analysis. The time point 0* (Figure 3) indicates the beginning of the microscopy acquisition, corresponding roughly to 30 min after cell encapsulation and droplet entrapment within the microfluidic device. Notably, both droplets trapped within and outside the observation chamber were analyzed.
Confocal Fluorescence Microscopy
An LSM880 confocal microscope (Carl Zeiss, Germany) with a 20x/0.8 Plan-APOCHROMAT air objective (∞/0.17 Carl Zeiss, Germany) was used for droplet analysis with a microfluidic observation chamber. Upon entrapment of the droplets in the observation chamber, fluorescence and bright-field microscopy images were automatically acquired on average every 20–25 min within the first 6 h and every 40–60 min for the following 6 h for a total period of 12 h (resolution of 512 × 512 pixels). Fluorophores were excited with an argon laser (λex = 488), and the detection filters chosen accordingly to Cell Tracker Green emission and propidium iodide (PI) emission.
Cell Viability and Cytotoxicity Assay in Droplets
For the NK-92 cells cytotoxicity assays inside the droplets, effector cells and fluorescently labeled target cells were encapsulated inside droplets containing 1 μg/mL PI. PI is an intercalating fluorescent agent used to distinguish between apoptotic and living cells based on membrane integrity.44 The viability of effector and target cells alone (see the Supporting Information, Figure S2) and the specific cytotoxicity of the effector cells inside droplet compartments were quantified by observing the intensity change in cellular fluorescence caused by PI accumulation in the cytosol over a period of 12 h.
Flow Cytometry
Fluorescently labeled (CTG stained) target cells were suspended in α-MEM medium supplemented with 10% (v/v) h.i. FBS, 10% (v/v) h.i. HS, 2 mM l-glutamate, 1 mM sodium pyruvate, 100 μM 2-mercaptoethanol, IL-2 (10 ng/mL), IL-15 (30 ng/mL), and PI (1 μg/mL) and seeded at a concentration of roughly 80,000 cells per well in a round-bottom 96-well plate at different ratios of NK-92/target cells (1:1, 1:3, 1:5, 3:1, 5:1 and 10:1). After 3, 6, or 12 h incubation time, 30 000 cells per condition were analyzed using a BD LSRFortessa X-20 cell analyzer (BD Biosciences) equipped with an HTS module. The different lasers and filters were used to record CTG and PI fluorescence: a violet laser with a 525/50 nm filter for CTG and a yellow/green laser with a 610/20 nm filter for PI. Unstained or single-fluorophore-stained target cells were used as controls to measure the fluorescence spillover and determine the thresholds for labeled target cells and dead target cells. The data were analyzed with FlowJo software. The gating strategy involved the removal of cell debris (FSC-A/SSC-A) and the selection of the target cell population (CTG fluorescence). The percentage of dead target cells was quantified using PI fluorescence.
Data Analysis
Image processing and analysis were carried out using the open-source platform ImageJ. Cell viability of target and effector cells as well as cell cytotoxicity of effector cells was assessed manually based on the fluorescent signal due to PI staining. Flow cytometry data were analyzed using the FlowJo software (TreeStar Software, Ashland, Oregon), and flow cytometry experiments were performed measuring three technical replicates for each condition. GraphPad Prism (GraphPad Software Inc.) was used for the graphical representation of the results.
Acknowledgments
The authors acknowledge the funding from the MaxSynBio Consortium, which is jointly funded by the German Federal Ministry of Education and Research and the Max Planck Society. They also acknowledge the support from the SFB 1129 (Project P1 and P15) of the German Research Foundation and the Volkswagenstiftung (priority call “Life?”). J.P.S. is the Weston Visiting Professor at the Weizmann Institute of Science and part of the excellence cluster CellNetworks at the University of Heidelberg. The authors thank Dr. Jan-Willi Janiesch and Ms. Désirée Sauter for their support with the synthesis of block copolymer surfactants and Dr. Corentin Gondrand for his assistance with flow cytometry. The Max Planck Society is appreciated for its general support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03264.
Evaluation of the droplet diameters for the NK-92 cytotoxicity assay, assessment of droplet sizes, characterization of cell mortality in droplets, characterization of effector/target cell distribution ratios in droplets, representative confocal fluorescence microscopy images, droplet-based microfluidic assessment of NK-92 cytotoxicity against two and three K562/Jurkat target cells, and flow cytometry measurements of NK-92 cell cytotoxicity (PDF)
video1_Nk-k562 bulk.avi (AVI)
The authors declare no competing financial interest.
Supplementary Material
References
- Farag S. S.; Caligiuri M. A. Human natural killer cell development and biology. Blood Rev. 2006, 20, 123–137. 10.1016/j.blre.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Klingemann H. Are natural killer cells superior CAR drivers?. Oncoimmunology 2014, 3, e28147. 10.4161/onci.28147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A.; Viswanathan C. Natural killer cells: In health and disease. Hematol./Oncol. Stem Cell Ther. 2015, 8, 47–55. 10.1016/j.hemonc.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Mehta R. S.; Rezvani K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. 10.3389/fimmu.2018.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegler E. L.; Zhu Y.; Wang P.; Yang L. Off-the-Shelf CAR-NK Cells for Cancer Immunotherapy. Cell Stem Cell 2018, 23, 160–161. 10.1016/j.stem.2018.07.007. [DOI] [PubMed] [Google Scholar]
- Bhat R.; Watzl C. Serial killing of tumor cells by human natural killer cells--enhancement by therapeutic antibodies. PLoS One 2007, 2, e326. 10.1371/journal.pone.0000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldevall K.; Brandt L.; Forslund E.; Olofsson K.; Frisk T. W.; Olofsson P. E.; Gustafsson K.; Manneberg O.; Vanherberghen B.; Brismar H.; Kärre K.; Uhlin M.; Önfelt B. Microchip Screening Platform for Single Cell Assessment of NK Cell Cytotoxicity. Front. Immunol. 2016, 7, 119 10.3389/fimmu.2016.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanherberghen B.; Olofsson P. E.; Forslund E.; Sternberg-Simon M.; Khorshidi M. A.; Pacouret S.; Guldevall K.; Enqvist M.; Malmberg K.-J.; Mehr R.; Önfelt B. Classification of human natural killer cells based on migration behavior and cytotoxic response. Blood 2013, 121, 1326. 10.1182/blood-2012-06-439851. [DOI] [PubMed] [Google Scholar]
- Miller R. G.; Dunkley M. Quantitative analysis of the 51Cr release cytotoxicity assay for cytotoxic lymphocytes. Cell. Immunol. 1974, 14, 284–302. 10.1016/0008-8749(74)90212-3. [DOI] [PubMed] [Google Scholar]
- Papadopoulos N. G.; Dedoussis G. V. Z.; Spanakos G.; Gritzapis A. D.; Baxevanis C. N.; Papamichail M. An improved fluorescence assay for the determination of lymphocyte-mediated cytotoxicity using flow cytometry. J. Immunol. Methods 1994, 177, 101–111. 10.1016/0022-1759(94)90147-3. [DOI] [PubMed] [Google Scholar]
- Jung T.; Schauer U.; Heusser C.; Neumann C.; Rieger C. Detection of intracellular cytokines by flow cytometry. J. Immunol. Methods 1993, 159, 197–207. 10.1016/0022-1759(93)90158-4. [DOI] [PubMed] [Google Scholar]
- Park K.-H.; Park H.; Kim M.; Kim Y.; Han K.; Oh E.-J. Evaluation of NK cell function by flowcytometric measurement and impedance based assay using real-time cell electronic sensing system. Biomed. Res. Int. 2013, 2013, 210726. 10.1155/2013/210726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanchi S. S.; McCulley K. J.; Somanchi A.; Chan L. L.; Lee D. A. A Novel Method for Assessment of Natural Killer Cell Cytotoxicity Using Image Cytometry. PLoS One 2015, 10, e0141074. 10.1371/journal.pone.0141074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausell-Tormos J.; Lieber D.; Baret J.-C.; El-Harrak A.; Miller O. J.; Frenz L.; Blouwolff J.; Humphry K. J.; Köster S.; Duan H.; Holtze C.; Weitz D. A.; Griffiths A. D.; Merten C. A. Droplet-Based Microfluidic Platforms for the Encapsulation and Screening of Mammalian Cells and Multicellular Organisms. Chem. Biol. 2008, 15, 427–437. 10.1016/j.chembiol.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Chokkalingam V.; Tel J.; Wimmers F.; Liu X.; Semenov S.; Thiele J.; Figdor C. G.; Huck W. T. S. Probing cellular heterogeneity in cytokine-secreting immune cells using droplet-based microfluidics. Lab Chip 2013, 13, 4740–4744. 10.1039/c3lc50945a. [DOI] [PubMed] [Google Scholar]
- El Debs B.; Utharala R.; Balyasnikova I. V.; Griffiths A. D.; Merten C. A. Functional single-cell hybridoma screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 11570–11575. 10.1073/pnas.1204514109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyer K.; Doineau R. C. L.; Castrillon C. E.; Briseño-Roa L.; Menrath V.; Mottet G.; England P.; Godina A.; Brient-Litzler E.; Nizak C.; Jensen A.; Griffiths A. D.; Bibette J.; Bruhns P.; Baudry J. Single-cell deep phenotyping of IgG-secreting cells for high-resolution immune monitoring. Nat. Biotechnol. 2017, 35, 977. 10.1038/nbt.3964. [DOI] [PubMed] [Google Scholar]
- Klein A. M.; Mazutis L.; Akartuna I.; Tallapragada N.; Veres A.; Li V.; Peshkin L.; Weitz D. A.; Kirschner M. W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konry T.; Dominguez-Villar M.; Baecher-Allan C.; Hafler D. A.; Yarmush M. L. Droplet-based microfluidic platforms for single T cell secretion analysis of IL-10 cytokine. Biosens. Bioelectron 2011, 26, 2707–2710. 10.1016/j.bios.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzman I.; Janiesch J.-W.; Spatz J. P. Synthesis of Nanostructured and Biofunctionalized Water-in-Oil Droplets as Tools for Homing T Cells. J. Am. Chem. Soc. 2013, 135, 3339–3342. 10.1021/ja311588c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S.; McKenney S.; Sabhachandani P.; Adler J.; Hu X.; Stroopinksy D.; Rosenblatt J.; Avigan D.; Konry T. Anti-myeloma activity and molecular logic operation by Natural Killer cells in microfluidic droplets. Sens. Actuators, B 2019, 282, 580–589. 10.1016/j.snb.2018.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A. H.-H.; Li H.; Jia Y.; Mak P.-I.; Martins R. P. dS.; Liu Y.; Vong C. M.; Wong H. C.; Wong P. K.; Wang H.; Sun H.; Deng C.-X. Drug screening of cancer cell lines and human primary tumors using droplet microfluidics. Sci. Rep. 2017, 7, 9109 10.1038/s41598-017-08831-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abate A. R.; Hung T.; Mary P.; Agresti J. J.; Weitz D. A. High-throughput injection with microfluidics using picoinjectors. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 19163–19166. 10.1073/pnas.1006888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang J.; Feng L.; Lin T. Fluid mixing in droplet-based microfluidics with a serpentine microchannel. RSC Adv. 2015, 5, 104138–104144. 10.1039/C5RA21181F. [DOI] [Google Scholar]
- Caen O.; Schütz S.; Jammalamadaka M. S. S.; Vrignon J.; Nizard P.; Schneider T. M.; Baret J.-C.; Taly V. High-throughput multiplexed fluorescence-activated droplet sorting. Microsyst. Nanoeng. 2018, 4, 33 10.1038/s41378-018-0033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzman I.; Janiesch J. W.; Matić J.; Spatz J. P. Artificial antigen-presenting interfaces in the service of immunology. Isr. J. Chem. 2013, 53, 655–669. 10.1002/ijch.201300060. [DOI] [Google Scholar]
- Sarkar S.; Motwani V.; Sabhachandani P.; Cohen N.; Konry T. T Cell Dynamic Activation and Functional Analysis in Nanoliter Droplet Microarray. J. Clin. Cell. Immunol. 2015, 6, 334 10.4172/2155-9899.1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segaliny A. I.; Li G.; Kong L.; Ren C.; Chen X.; Wang J. K.; Baltimore D.; Wu G.; Zhao W. Functional TCR T cell screening using single-cell droplet microfluidics. Lab Chip 2018, 18, 3733–3749. 10.1039/C8LC00818C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S.; Sabhachandani P.; Ravi D.; Potdar S.; Purvey S.; Beheshti A.; Evens A. M.; Konry T. Dynamic Analysis of Human Natural Killer Cell Response at Single-Cell Resolution in B-Cell Non-Hodgkin Lymphoma. Front. Immunol. 2017, 8, 1736. 10.3389/fimmu.2017.01736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-E.; Kim H.; Doh J. Single cell arrays of hematological cancer cells for assessment of lymphocyte cytotoxicity dynamics, serial killing, and extracellular molecules. Lab Chip 2019, 19, 2009–2018. 10.1039/C9LC00133F. [DOI] [PubMed] [Google Scholar]
- Khorshidi M. A.; Vanherberghen B.; Kowalewski J. M.; Garrod K. R.; Lindström S.; Andersson-Svahn H.; Brismar H.; Cahalan M. D.; Önfelt B. Analysis of transient migration behavior of natural killer cells imaged in situ and in vitro. Integr. Biol. 2011, 3, 770–778. 10.1039/c1ib00007a. [DOI] [PubMed] [Google Scholar]
- Olofsson P. E.; Forslund E.; Vanherberghen B.; Chechet K.; Mickelin O.; Ahlin A. R.; Everhorn T.; Onfelt B. Distinct Migration and Contact Dynamics of Resting and IL-2-Activated Human Natural Killer Cells. Front. Immunol. 2014, 5, 80. 10.3389/fimmu.2014.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitard L.; Cottinet D.; Kleinschmitt C.; Bremond N.; Baudry J.; Yvert G.; Bibette J. Monitoring single-cell bioenergetics via the coarsening of emulsion droplets. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 7181–7186. 10.1073/pnas.1200894109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouzes E.; Medkova M.; Savenelli N.; Marran D.; Twardowski M.; Hutchison J. B.; Rothberg J. M.; Link D. R.; Perrimon N.; Samuels M. L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 14195. 10.1073/pnas.0903542106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B. A.; Dief B.; Wang X.-H.; Keating A. NK-92 Preferentially Targets Acute Myeloid Leukemia Stem Cells. Blood 2010, 116, 4300. 10.1182/blood.V116.21.4300.4300. [DOI] [Google Scholar]
- Greenfield K.; Mokhtari S.; Haug M.; Porada C. D.; Almeida-Porada G. Identification and Phenotypic Characterization of a Subpopulation of Acute Myelogenous Leukemia (AML) Cells with Increased Plastic Adherence. Blood 2012, 120, 2556. 10.1182/blood.V120.21.2556.2556. [DOI] [Google Scholar]
- Xia Y.; Whitesides G. M. Soft Lithography. Angew. Chem., Int. Ed. 1998, 37, 550–575. 10.1002/(sici)1521-3773(19980316)37:53.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Duffy D. C.; McDonald J. C.; Schueller O. J. A.; Whitesides G. M. Rapid Prototyping of Microfluidic Systems in Poly(dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- Zamir E.; Frey C.; Weiss M.; Antona S.; Frohnmayer J. P.; Janiesch J.-W.; Platzman I.; Spatz J. P. Reconceptualizing Fluorescence Correlation Spectroscopy for Monitoring and Analyzing Periodically Passing Objects. Anal. Chem. 2017, 89, 11672–11678. 10.1021/acs.analchem.7b03108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisensky G. C.; Campbell D. J.; Beckman K. J.; Calderon C. E.; Doolan P. W.; Ottosen R. M.; Ellis A. B. Replication and Compression of Surface Structures with Polydimethylsiloxane Elastomer. J. Chem. Educ. 1999, 76, 537. 10.1021/ed076p537. [DOI] [Google Scholar]
- Holtze C.; Rowat A. C.; Agresti J. J.; Hutchison J. B.; Angilè F. E.; Schmitz C. H. J.; Köster S.; Duan H.; Humphry K. J.; Scanga R. A.; Johnson J. S.; Pisignano D.; Weitz D. A. Biocompatible surfactants for water-in-fluorocarbon emulsions. Lab Chip 2008, 8, 1632–1639. 10.1039/b806706f. [DOI] [PubMed] [Google Scholar]
- Baret J.-C. Surfactants in droplet-based microfluidics. Lab Chip 2012, 12, 422–433. 10.1039/C1LC20582J. [DOI] [PubMed] [Google Scholar]
- Kemna E. W. M.; Schoeman R. M.; Wolbers F.; Vermes I.; Weitz D. A.; van den Berg A. High-yield cell ordering and deterministic cell-in-droplet encapsulation using Dean flow in a curved microchannel. Lab Chip 2012, 12, 2881–2887. 10.1039/c2lc00013j. [DOI] [PubMed] [Google Scholar]
- Riccardi C.; Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.