

Abstract
Acute kidney injury (AKI) significantly increases the risk of development of chronic kidney disease (CKD). Recently, our laboratory generated a mouse model with the typical phenotypes of AKI to CKD transition in the unilateral kidney. However, AKI, CKD, and even the transition from AKI to CKD usually occur bilaterally rather than unilaterally in patients. Therefore, in the present study, we further modified the strategy and developed a new model of CKD transitioned from bilateral ischemia-reperfusion injury (IRI) in C57BL/6 mice. In this new model, unilateral severe IRI was performed in one kidney while the contralateral kidney was kept intact to maintain animal survival; then, following 14 days of recovery, when the renal function of the injured kidney restored above the survival threshold, the contralateral intact kidney was subjected to a similar IRI. Animals of these two-stage bilateral IRI models with pedicle clamping of 21 and 24 min at a body temperature of 37°C exhibited incomplete recovery from AKI and subsequent development of CKD with characteristics of progressive decline in glomerular filtration rate, increases in plasma creatinine, worsening of proteinuria, and deleterious histopathological changes, including interstitial fibrosis and glomerulosclerosis, in both kidneys. In conclusion, a new bilateral AKI to CKD transition animal model with a typical phenotype of CKD was generated in C57BL/6 mice.
Keywords: acute kidney injury, animal model, chronic kidney disease, ischemia-reperfusion injury
INTRODUCTION
Acute kidney injury (AKI), characterized by a sudden and dramatic loss of kidney function, is a common clinical syndrome with significant morbidity, mortality, and medical costs (1, 21, 26). A large number of studies have demonstrated or reported that AKI significantly promotes the development of and accelerates the progression of chronic kidney disease (CKD) (3, 4, 8, 15). Despite extensive experimental and clinical investigations, the underlying mechanisms for the transition of AKI to CKD have not been clarified.
Various animal models that mimic the transitional process of AKI to CKD have been generated and characterized, which significantly advanced the understanding of the post-AKI pathophysiological changes and molecular mechanisms (6). For example, repeated low-dose cisplatin is a well-established nephrotoxic model of inducing chronic renal pathologies such as tubulointerstitial fibrosis, glomerulosclerosis, and even the persistent decline of glomerular filtration rate (GFR) with a longer period of observation (6, 11, 22, 24). Beside nephrotoxin, renal ischemia-reperfusion injury (IRI) is another common cause of AKI occurring in a variety of clinical settings ranging from critical illness to kidney transplantation. However, unlike the outcomes of repeated administration of nephrotoxic drugs, the strategy of repeated IRI is not able to induce a progression to CKD but provides protective effect against the latter IRI (10, 23, 33).
Recently, several renal IRI models developed by our laboratory (30) and others (12, 13, 32, 34) with distinct strategies have achieved the transition from IRI-induced AKI to CKD in the unilateral kidney while retaining an intact contralateral kidney or using a delayed contralateral nephrectomy. The unilateral model may introduce some confounding factors, such as compensation in renal hemodynamics and suppression of the renin-angiotensin-aldosterone system, affecting the progression of kidney injury. In the present study, we developed a two-stage bilateral IRI (bIRI) model of the AKI to CKD transition in mice. A severe IRI was induced in the unilateral kidney while the contralateral kidney was kept intact and then subjected to a delayed but equivalent IRI at 14 days later. In this way, the bilateral successive IRIs induced a progression from ischemic AKI to CKD in both kidneys.
METHODS
Animals.
Animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all animal protocols were approved by the Institutional Animal Care and Use Committee of the University of South Florida College of Medicine. C57BL/6 mice (male, 8–10 wk old) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed individually at 23°C on a 12:12-h light-dark cycle while being maintained on water and food (Teklad Global Rodent Diet, no. 2918, 18% protein and 0.2% sodium, Envigo, Indianapolis, IN).
Surgery protocols.
At the beginning of stage I (day 1), unilateral IRI (uIRI) of the left kidney was performed as we have previously described (27, 30). Briefly, the mouse was anesthetized with pentobarbital (50 mg/mL ip), and body temperature was controlled at 36.8–37.2°C during surgery with a temperature-controlled operating table (Vestavia Scientific). The right kidney was kept intact. The left renal pedicle was carefully dissected and clamped with a silver clip (FE690K, AESCULAP) for 15, 18, 21, 24, 27, or 30 min, respectively. After release of the clip, the wound was sutured, and the animal was allowed to recover for 14 days. At the beginning of stage II (day 14), equivalent IRI was induced in the right kidney by clamping of the right renal pedicle for 15, 18, 21, 24, 27 or 30 min (two-stage bIRI 15-, 18-, 21-, 24-, 27-, and 30-min groups, respectively). Sham-operated animals without clamping at day 1 and day 14 served as controls (two-stage bIRI 0-min group).
Plasma and urine biochemistry.
Blood samples (~50 µL/each) were collected through the retroorbital venous sinus and centrifuged at 8,000 rpm for 5 min at 4°C to separate plasma (~25 µL/each). Phosphocreatine (PCr) was measured by HPLC at the O’Brien Center Core of the University of Alabama at Birmingham. Urine samples were collected in metabolic cages for 24 h. The urinary albumin concentration was measured with an ELISA kit (ab-108792, Abcam, Cambridge, MA), and the urinary creatinine concentration was also measured by HPLC at the O’Brien Center Core of the University of Alabama at Birmingham. The urine albumin-to-creatinine ratio was calculated by the urinary albumin concentration over urinary creatinine concentration, as we have previously described (29, 30).
GFR measurement.
GFR in conscious mice was measured by the clearance of plasma FITC-sinistrin with a single bolus injection, as we have previously described (16, 28, 37). Briefly, mice were lightly anesthetized with isoflurane and injected with the FITC-sinistrin (5.6 mg/100 g body wt) via the retroorbital venous sinus. Blood samples (~10 µL/each) were collected into heparinized capillary tubes from the tail vein at 3, 7, 10, 15, 35, 55, 75, and 90 min after injection. Blood samples were centrifuged at 8,000 rpm for 5 min at 4°C to separate plasma (~5 µL/each). The fluorescence of FITC-sinistrin in the plasma samples (1 µL/sample) was measured using a plate reader (Cytation3, BioTek, Winooski, VT) with 485-nm excitation and 538-nm emission. GFR was calculated with GraphPad Prism 8 (GraphPad Software, San Diego, CA).
Measurement of blood pressure with the radio telemetry system in conscious mice.
Mean arterial pressure (MAP) was measured with a radio telemetry system (PA-C10, Data Sciences International) in conscious mice, as we have previously described (16, 35, 36). Briefly, mice were anesthetized with inhaled isoflurane via vaporizer (Vaporizer Sales and Service, Rockmart, GA). Under aseptic conditions, a small incision was made in the middle of the neck to expose the left carotid artery. The pressure catheter of the transmitter was carefully inserted into the left carotid artery and further advanced down to the aortic arch. The body of the transmitter was then placed subcutaneously in the right ventral flank. Mice were allowed 7 days of recovery before data were collected. MAP was recorded for 10 s every 1 min for 4 h each day from 1 to 5 PM.
Anesthetized blood pressure measurement.
MAP was measured at the end of the experiment, as we have previously described (27, 30, 36). Mice were anesthetized with pentobarbital sodium (50 mg/mL ip), and body temperature was maintained at 36.8–37.2°C with a temperature-controlled operating table. Following a cannulation of the trachea, the carotid artery was catheterized for blood pressure measurement via a PowerLab system (AD Instruments, Boulder, CO).
Morphological evaluation with light microscopy.
The kidneys were harvested at the end of experiments, fixed in 4% paraformaldehyde solution overnight, and then embedded in paraffin, as we have previously described (29, 30). Kidney slices (2 µm) were cut and treated with Masson’s trichrome or periodic acid-Schiff stain. In the Masson’s trichrome-stained slices, tubular atrophy, interstitial fibrosis, and inflammatory cell invasion were scored on a scale of 0−3 using the following criteria: 0, affecting 0–5% of the renal area; 1, affecting 6–25% of the renal area; 2, affecting 26–50% of the renal area; and 3, affecting >50% of the renal area, as previously reported (9, 14, 30), at ×400 magnification. In the periodic acid-Schiff-stained slices, the glomerulosclerosis index was evaluated and scored on a scale of 0−4 using the following criteria: 0, normal glomeruli; 1, 1–25% sclerotic area; 2, 26–50% sclerotic area; 3, 51–75% sclerotic area; and 4, 76–100% sclerotic area, as previously reported (18, 31), and collapse of glomerular tufts as well as dilation of Bowman’s capsules were graded by their presence or absence, as previously reported (7, 9, 30), at ×1,000 magnification. All morphometric analysis was performed in a blinded manner.
Statistical analysis.
Statistical analysis was performed using Prism 8 (GraphPad Software). The effects of interest were tested using two-way ANOVA or repeated-measures two-way ANOVA followed by Sidak multiple-comparisons test when appropriate. Data are presented as means ± SE. P values of <0.05 were considered statistically significant.
RESULTS
Plasma creatinine.
PCr was monitored throughout the entire experiment. During stage I, because of the presence of an intact right kidney, PCr was not significantly changed in any group with uIRI of the left kidney. During stage II, after the original insults of the successive IRIs in both kidneys, PCr gradually increased from 0.19 ± 0.03 mg/dL (day 28) to 0.35 ± 0.04 mg/dL (day 98) in the two-stage bIRI 21-min group and from 0.28 ± 0.04 mg/dL (day 28) to 0.46 ± 0.06 mg/dL (day 98) in the two-stage bIRI 24-min group. PCr of the two-stage uIRI 15-min and 18-min groups returned to basal levels and maintained until the end of experiments (day 98; Fig. 1A).
Fig. 1.

Parameters of the two-stage bilateral ischemia-reperfusion injury (bIRI) model. A and B: phosphocreatine (PCr; A; n = 15 or 16 mice/group) and glomerular filtration rate (GFR; B; n = 15 or 16 mice/group) in two-stage bIRI models from day −1 to day 98. bIRI0, bIRI15, bIRI18, bIRI21, bIRI24, bIRI27, and bIRI30, two-stage bIRI 0-, 15-, 18-, 21-, 24-, 27-, and 30-min groups, respectively. C: GFR normalized to body weight (n = 15 or 16 mice/group) in two-stage bIRI models at day 98. *P < 0.01 vs. the bIRI0 group. D: survival rate (D; n = 15 or 16 mice/group) of two-stage bIRI models from day −1 to day 98. E: urine albumin-to-creatinine ratio (ACR; n = 9–11 mice/group) in two-stage bIRI models from day −1 to day 98. *P < 0.01 vs. the bIRI0 group. F: mean arterial pressure (MAP; n = 6–8 mice/group) in two-stage bIRI0 and bIRI24 groups from day −1 to day 28. *P < 0.01 vs. the bIRI0 group. For A, B, E, and F, statistical differences were calculated by repeated-measures two-way ANOVA followed by a Sidak multiple-comparisons test. For C, statistical differences were calculated by two-way ANOVA followed by a Sidak multiple-comparisons test.
Glomerular filtration rate.
GFR was monitored throughout the entire experiment. Changes of GFR were in a similar inverse pattern to PCr. After the original insults of the successive IRIs in both kidneys, GFR gradually declined from 166.85 ± 16.44 µL/min (day 28) to 107.47 ± 9.43 µL/min (day 98) in the two-stage bIRI 21-min group and from 143.84 ± 10.56 µL/min (day 28) to 79.92 ± 13.27 µL/min (day 98) in the two-stage bIRI 24-min group. GFR of the two-stage bIRI 15-min and 18-min groups restored to basal levels and maintained until the end of experiments (day 98; Fig. 1, B and C).
Survival rate.
All mice in the two-stage bIRI 15-min, 18-min, and 21-min groups as well as 91% of mice in the two-stage bIRI 24-min group survived until the end of experiments (day 98). All mice in the two-stage bIRI 27-min and 30-min groups died during the initial phase of stage II, within 7 days after IRI of the right kidneys (Fig. 1D).
Urine albumin-to-creatinine ratio.
The urine albumin-to-creatinine ratio was monitored throughout the entire experiment. The urine albumin-to-creatinine ratio gradually increased from a basal level of 21.7 ± 11.6 μg/mg (day −1) to 521.3 ± 173.5 μg/mg (day 98) in the two-stage bIRI 21-min group and from 15.8 ± 7.8 μg/mg (day −1) to 822.1 ± 216.8 μg/mg (day 98) in the two-stage bIRI 24-min group. In contrast, the urine albumin-to-creatinine ratio was not significantly changed in the two-stage bIRI 15-min or 18-min groups (Fig. 1E).
Blood pressure.
MAP was measured with a catheter at the end of experiments (day 98) under pentobarbital anesthesia. Anesthetized MAP was 89 ± 2.8, 93 ± 3.1, 91 ± 3.4, 88 ± 2.7, and 90 ± 3.3 mmHg in the two-stage 0-min, 15-min, 18-min, 21-min, and 24-min groups, respectively, without a significant difference among groups (Table 1). Moreover, MAP was also measured with a radio telemetry system in conscious mice of two-stage bIRI 0-min and bIRI 24-min groups from day −1 to day 28 (Fig. 1F). Consistent with the findings in anesthetized mice, conscious MAP was not significantly changed in the two-stage bIRI 24-min group compared with the two-stage bIRI 0-min group, implying that blood pressure may not play an important role in the progression of kidney injury in this two-stage bIRI model.
Table 1.
Characteristics of the two-stage bIRI model
| Anesthetized Mean Arterial Pressure, mmHg | Body Weight, g | Right Kidney Weight, g | Left Kidney Weight, g | |
|---|---|---|---|---|
| Two-stage bIRI 0-min group | 89 ± 2.8 | 32.9 ± 1.5 | 0.170 ± 0.018 | 0.165 ± 0.015 |
| Two-stage bIRI 15-min group | 93 ± 3.1 | 33.1 ± 1.6 | 0.169 ± 0.021 | 0.161 ± 0.019 |
| Two-stage bIRI 18-min group | 91 ± 3.4 | 31.5 ± 1.9 | 0.152 ± 0.017 | 0.146 ± 0.020 |
| Two-stage bIRI 21-min group | 88 ± 2.7 | 29.2 ± 2.1 | 0.091 ± 0.024* | 0.084 ± 0.021* |
| Two-stage bIRI 24-min group | 90 ± 3.3 | 26.8 ± 1.7 | 0.079 ± 0.029* | 0.060 ± 0.018* |
Values are means ± SE. There was no significant difference in anesthetized mean arterial pressure among all groups at the end of experiments (n = 9–10 mice/group). Compared with the two-stage bilateral ischemia-reperfusion injury (bIRI) 0-min group, body weight and kidney weight were lower in the two-stage bIRI 21-min and two-stage bIRI 24-min groups but not significantly different in the two-stage bIRI 15-min or two-stage bIRI 18-min groups (n = 9 or 10 mice/group).
P < 0.001 vs. the two-stage bIRI 0-min group.
Kidney size and weight.
Kidney size and weight were assessed at the end of experiments (day 98). Compared with the two-stage bIRI 0-min group, the kidneys were significantly smaller and lighter, with a granular appearance in the two-stage bIRI 21-min and 24-min groups, whereas they were not significantly different in the two-stage uIRI 15-min or 18-min groups (Fig. 2A and Table 1).
Fig. 2.


Histopathological analyses of the two-stage bilateral ischemia-reperfusion injury (bIRI) model. A: representative images of the left and right kidneys from each two-stage bIRI group. B and C: kidney slices with Masson’s trichrome (B) or periodic acid-Schiff staining (C) showed the typical histopathological features of chronic kidney disease, including tubular atrophy (black stars), interstitial fibrosis (yellow triangles), inflammatory cell infiltration (yellow stars), glomerulosclerosis (green triangle), collapse of glomerular tufts (green star), and dilated Bowman’s capsules (red stars).
Histopathological analysis.
Histological analysis with Masson’s trichrome stain (Fig. 2B) and periodic acid-Schiff stain (Fig. 2C) showed the typical morphological changes of CKD, including tubular atrophy, interstitial fibrosis, inflammatory cell infiltration, glomerulosclerosis, collapse of glomerular tufts, and dilated Bowman’s capsule, in the kidney slices of the two-stage bIRI 21-min and 24-min groups but not in the kidney slices of the two-stage bIRI 15-min or 18-min groups (Table 2).
Table 2.
Histopathological analysis of the two-stage bIRI model
| Compartment | Parameter | Two-Stage bIRI 0-min Group | Two-Stage bIRI 15-min Group | Two-Stage bIRI 18-min Group | Two-Stage bIRI 21-min Group | Two-Stage bIRI 24-min Group |
|---|---|---|---|---|---|---|
| Tubule and interstitium | Tubular atrophy (0–3) | 0 | 0 | 0 | 2 | 2 |
| Interstitial fibrosis (0–3) | 0 | 0 | 0 | 1 | 2 | |
| Inflammatory cell invasion (0–3) | 0 | 0 | 0 | 1 | 3 | |
| Glomeruli | Glomerulosclerosis index (0–4) | 0 ± 0 | 0 ± 0 | 0.02 ± 0.01 | 2.39 ± 0.23 | 3.02 ± 0.20 |
| Collapse of glomerular tufts (present/absent) | 0/100 | 0/100 | 0/100 | 16/84 | 56/44 | |
| Dilatation of Bowman’s capsules (present/absent) | 0/100 | 0/100 | 1/99 | 21/79 | 62/38 |
Tubular atrophy, interstitial fibrosis, inflammatory cell infiltration, glomerulosclerosis, collapse of glomerular tufts, and dilated Bowman’s capsules were observed in the two-stage bilateral ischemia-reperfusion injury (bIRI) 21-min and two-stage bIRI 24-min groups but not in the two-stage bIRI 15-min, two-stage bIRI 18-min, or two-stage bIRI 0-min groups. Tubular atrophy, interstitial fibrosis, and inflammatory cell invasion were scored on a scale of 0−3 according to the following criteria: 0, affecting 0–5% of the renal area; 1, affecting 6–25% of the renal area; 2, affecting 26–50% of the renal area; and 3, affecting >50% of the renal area (n = 10 fields/mouse, 9 or 10 mice/group). The glomerulosclerosis index was scored on a scale of 0−4 using the following criteria: 0, normal glomeruli; 1, 1–25% of the sclerotic area; 2, 26–50% of the sclerotic area; 3, 51–75% of the sclerotic area; and 4, 76–100% of the sclerotic area; collapse of glomerular tufts as well as dilatation of Bowman’s capsules were graded by their presence or absence (n = 10 glomeruli/mouse, 10 mice/group).
DISCUSSION
In the present study, we developed a new model of CKD transitioned from bilateral AKI in C57BL/6 mice. In these animals, two-stage successive severe IRI induced an incomplete recovery from ischemic AKI and subsequent progression of CKD with the typical characteristics, including the gradual decline in kidney function, continuous worsening of proteinuria, and deleterious histopathological changes, in both kidneys.
The classical view of AKI as a transient, self-limited, and reversible symptom has been challenged by recent epidemiological studies. A growing body of compelling evidence has demonstrated that AKI significantly increases the risk of development of CKD, and the subsequent progression of CKD is associated with the severity of AKI (3, 4, 8, 15). The animal model of uIRI without contralateral nephrectomy has been used in experimental studies of the long-term post-AKI sequelae (5, 6, 20, 34). In this model, on one side, owing to the existence of an intact contralateral kidney, animals can survive from the unilateral life-threatening AKI and develop the chronic pathological changes in the injured kidney; on the other side, however, systemic biomarkers of renal function, including PCr and GFR, are normal due to the intact contralateral kidney. We speculate, as shown in Fig. 3, that even though GFR of the unilateral kidney with severe IRI would decline below the survival threshold, the intact contralateral kidney could maintain animal survival, functioning like dialysis in patients with severe AKI; then, after a period of recovery, when GFR of the injured kidney restores above the survival threshold to keep the animal alive, the intact contralateral kidney would be subjected to a delayed, but equivalent, IRI. In this way, both kidneys of the animal would undergo partial recovery followed by a subsequent decline in GFR.
Fig. 3.
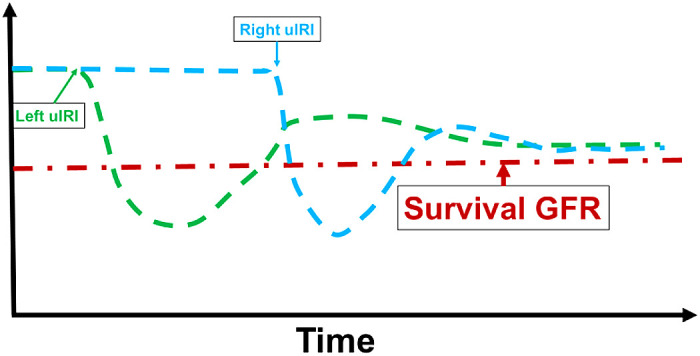
Rationale for the acute kidney injury to chronic kidney disease transition in the two-stage bilateral ischemia-reperfusion injury (bIRI) model. The red line suggests the threshold of the glomerular filtration rate (GFR) for the animal to survive. The green and blue lines represent the projected GFR changes for the left and right kidneys. In the two-stage bIRI model, severe IRI is induced in the left kidney while the right kidney is kept intact. Even though GFR of the injured left kidney decreases below the survival threshold, the nonischemic right kidney could still maintain the animal to survive. After GFR of the injured left kidney recovers above the survival threshold, the intact right kidney is subjected to delayed, but equivalent, IRI. In this way, both kidneys of the animal would successively undergo severe IRI with only partial recovery followed by a continuous decline in GFR.
Thus, in the present study, we developed a two-stage bIRI model, in which uIRI was performed in the left kidney by clamping of the renal pedicle while the right kidney was kept intact for 14 days and then subjected to a similar IRI. To figure out the optimal condition that initiates the progression to CKD, a series of two-stage bIRI models with gradient severity in AKI were conducted by adjusting the ischemic duration at a body temperature of 37°C. We found that all mice of the two-stage bIRI 27-min and 30-min models died within a few days following the contralateral IRI, suggesting that >27-min ischemia-induced renal injury was too severe to recover over the survival threshold, whereas all mice of the two-stage bIRI 15-min and 18-min models survived well and had almost completely restored kidney function and renal histopathology, suggesting that <18 min of ischemia-induced AKI was not able to cause the progression to CKD within 98 days. Only animals of the two-stage bIRI 21-min and 24-min models developed into CKD with the characteristic features, including a continuous decline in GFR, elevation in PCr, and exacerbation in proteinuria. Moreover, both of the bilateral kidneys exhibited the typical morphological changes of CKD, including contracted size with granular appearance, tubulointerstitial fibrosis, and glomerular and tubular damage. These phenotypes demonstrated that the two-stage bIRI 21-min and 24-min models resulted in an incomplete recovery from ischemic AKI and subsequent transition to CKD.
In the present two-stage bIRI model, both kidneys essentially underwent two serial hits. In addition to the pedicle clamping-induced IRI on its own, each kidney also experienced an episode of uremic milieu that was induced by the contralateral renal IRI. Even though several previous studies (13, 25, 34) have shown that the contralateral kidney retains normal histology without apparent structural alterations in mouse models of uIRI, we speculate that the uremic milieu per se beyond the IRI in this model may cause oxidative stress as well as inflammation, which promote the progression of kidney injury.
Compared with the two-stage uIRI models, as described in our previous study (30), the optimal renal ischemic times that achieve the transition from AKI to CKD are similar, indicating that the severity of AKI is the key factor that determines whether AKI progresses to CKD regardless of unilateral or bilateral kidneys. However, the rates of the decline in GFR, rise in PCr, and aggravation in proteinuria during the progression of CKD are slower in the bilateral models than unilateral models, which is consistent with a consensus in both clinical and experimental studies that nephrectomy or reduced nephron number is a significant risk factor of the progression of CKD (2, 17, 29).
In addition, a recent study by Matsushita et al. (19) described the AKI to CKD transition in a C57BL/6 mouse cardiac arrest and cardiopulmonary resuscitation (CA/CPR) model of acute cardiorenal syndrome. In their model, CA/CPR resulted in near-zero GFR and oliguria at 24 h. Even though 63% of animals died in 5 days, all other animals that survived over day 5 survived to the predetermined end point of the experiment and exhibited a full recovery in GFR at 2 wk but had some typical features of CKD, including declined GFR and elevated blood urea nitrogen and albuminuria at 7 wk. The results from this CA/CPR model also support our contention above that a rapid recovery of GFR over the survival threshold is the key to success in animal models of AKI to CKD transition.
In conclusion, we created and characterized a new model of CKD transitioned from bilateral AKI in C57BL/6 mice. In this model, two-stage successive bilateral renal IRI resulted in incomplete recovery from severe AKI and subsequent development of CKD in both kidneys. This newly generated animal model is of more clinical relevance and is expected to provide an additional tool in the investigation of the underlying mechanisms and exploration of the potential therapeutic targets for the AKI to CKD transition.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.Z., X.W., and R.L. conceived and designed research; J.Z. and J.W. performed experiments; J.Z., X.W., J.W., L.X., and F.C. analyzed data; J.Z. and X.W. interpreted results of experiments; J.Z., X.W., and K.Y. prepared figures; J.Z. and X.W. drafted manuscript; J.Z., X.W., J.W., L.W., S.J., L.Q., L.F., J.B., and R.L. edited and revised manuscript; J.Z., X.W., J.W., L.W., S.J., L.X., L.Q., K.Y., L.F., J.B., F.C., and R.L. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of X. Wang: Shandong Provincial Hospital affiliated to Shandong University, Jinan, Shandong, China.
REFERENCES
- 1.Bellomo R, Kellum JA, Bagshaw SM. Normotensive ischemic acute renal failure. N Engl J Med 357: 2205–2206, 2007. [PubMed] [Google Scholar]
- 2.Bertram JF, Douglas-Denton RN, Diouf B, Hughson MD, Hoy WE. Human nephron number: implications for health and disease. Pediatr Nephrol 26: 1529–1533, 2011. doi: 10.1007/s00467-011-1843-8. [DOI] [PubMed] [Google Scholar]
- 3.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 371: 58–66, 2014. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int 82: 516–524, 2012. doi: 10.1038/ki.2012.208. [DOI] [PubMed] [Google Scholar]
- 5.Colombaro V, Jadot I, Declèves AE, Voisin V, Giordano L, Habsch I, Malaisse J, Flamion B, Caron N. Lack of hyaluronidases exacerbates renal post-ischemic injury, inflammation, and fibrosis. Kidney Int 88: 61–71, 2015. doi: 10.1038/ki.2015.53. [DOI] [PubMed] [Google Scholar]
- 6.Fu Y, Tang C, Cai J, Chen G, Zhang D, Dong Z. Rodent models of AKI-CKD transition. Am J Physiol Renal Physiol 315: F1098–F1106, 2018. doi: 10.1152/ajprenal.00199.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodgin JB, Bitzer M, Wickman L, Afshinnia F, Wang SQ, O’Connor C, Yang Y, Meadowbrooke C, Chowdhury M, Kikuchi M, Wiggins JE, Wiggins RC. Glomerular aging and focal global glomerulosclerosis: a podometric perspective. J Am Soc Nephrol 26: 3162–3178, 2015. doi: 10.1681/ASN.2014080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu RK, Hsu CY. The role of acute kidney injury in chronic kidney disease. Semin Nephrol 36: 283–292, 2016. doi: 10.1016/j.semnephrol.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia T, Olauson H, Lindberg K, Amin R, Edvardsson K, Lindholm B, Andersson G, Wernerson A, Sabbagh Y, Schiavi S, Larsson TE. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol 14: 116, 2013. doi: 10.1186/1471-2369-14-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapitsinou PP, Haase VH. Molecular mechanisms of ischemic preconditioning in the kidney. Am J Physiol Renal Physiol 309: F821–F834, 2015. doi: 10.1152/ajprenal.00224.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katagiri D, Hamasaki Y, Doi K, Negishi K, Sugaya T, Nangaku M, Noiri E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int 89: 374–385, 2016. doi: 10.1038/ki.2015.327. [DOI] [PubMed] [Google Scholar]
- 12.Le Clef N, Verhulst A, D’Haese PC, Vervaet BA. Unilateral renal ischemia-reperfusion as a robust model for acute to chronic kidney injury in mice. PLoS One 11: e0152153, 2016. doi: 10.1371/journal.pone.0152153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lech M, Römmele C, Gröbmayr R, Eka Susanti H, Kulkarni OP, Wang S, Gröne HJ, Uhl B, Reichel C, Krombach F, Garlanda C, Mantovani A, Anders HJ. Endogenous and exogenous pentraxin-3 limits postischemic acute and chronic kidney injury. Kidney Int 83: 647–661, 2013. doi: 10.1038/ki.2012.463. [DOI] [PubMed] [Google Scholar]
- 14.Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12: 976–998, 2016. doi: 10.1080/15548627.2016.1166317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordoñez JD, Hsu CY. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int 76: 893–899, 2009. doi: 10.1038/ki.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu Y, Wei J, Stec DE, Roman RJ, Ge Y, Cheng L, Liu EY, Zhang J, Hansen PB, Fan F, Juncos LA, Wang L, Pollock J, Huang PL, Fu Y, Wang S, Liu R. Macula densa nitric oxide synthase 1β protects against salt-sensitive hypertension. J Am Soc Nephrol 27: 2346–2356, 2016. doi: 10.1681/ASN.2015050515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luyckx VA, Brenner BM. The clinical importance of nephron mass. J Am Soc Nephrol 21: 898–910, 2010. doi: 10.1681/ASN.2009121248. [DOI] [PubMed] [Google Scholar]
- 18.Maric C, Sandberg K, Hinojosa-Laborde C. Glomerulosclerosis and tubulointerstitial fibrosis are attenuated with 17beta-estradiol in the aging Dahl salt-sensitive rat. J Am Soc Nephrol 15: 1546–1556, 2004. doi: 10.1097/01.ASN.0000128219.65330.EA. [DOI] [PubMed] [Google Scholar]
- 19.Matsushita K, Saritas T, Eiwaz MB, McClellan N, Coe I, Zhu W, Ferdaus MZ, Sakai LY, McCormick JA, Hutchens MP. The acute kidney injury to chronic kidney disease transition in a mouse model of acute cardiorenal syndrome emphasizes the role of inflammation. Kidney Int 97: 95–105, 2020. doi: 10.1016/j.kint.2019.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthijsen RA, Huugen D, Hoebers NT, de Vries B, Peutz-Kootstra CJ, Aratani Y, Daha MR, Tervaert JWC, Buurman WA, Heeringa P. Myeloperoxidase is critically involved in the induction of organ damage after renal ischemia reperfusion. Am J Pathol 171: 1743–1752, 2007. doi: 10.2353/ajpath.2007.070184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A; Acute Kidney Injury Network . Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11: R31, 2007. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, Dong Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest 121: 2709–2722, 2011. doi: 10.1172/JCI45586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem 276: 11870–11876, 2001. doi: 10.1074/jbc.M007518200. [DOI] [PubMed] [Google Scholar]
- 24.Sharp CN, Doll MA, Dupre TV, Shah PP, Subathra M, Siow D, Arteel GE, Megyesi J, Beverly LJ, Siskind LJ. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 310: F560–F568, 2016. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tantawy MN, Jiang R, Wang F, Takahashi K, Peterson TE, Zemel D, Hao CM, Fujita H, Harris RC, Quarles CC, Takahashi T. Assessment of renal function in mice with unilateral ureteral obstruction using 99mTc-MAG3 dynamic scintigraphy. BMC Nephrol 13: 168, 2012. doi: 10.1186/1471-2369-13-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 334: 1448–1460, 1996. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 27.Wei J, Song J, Jiang S, Zhang G, Wheeler D, Zhang J, Wang S, Lai EY, Wang L, Buggs J, Liu R. Role of intratubular pressure during the ischemic phase in acute kidney injury. Am J Physiol Renal Physiol 312: F1158–F1165, 2017. doi: 10.1152/ajprenal.00527.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei J, Zhang J, Jiang S, Wang L, Persson AEG, Liu R. High-protein diet-induced glomerular hyperfiltration is dependent on neuronal nitric oxide synthase β in the macula densa via tubuloglomerular feedback response. Hypertension 74: 864–871, 2019. doi: 10.1161/HYPERTENSIONAHA.119.13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei J, Zhang J, Wang L, Cha BJ, Jiang S, Liu R. A new low-nephron CKD model with hypertension, progressive decline of renal function, and enhanced inflammation in C57BL/6 mice. Am J Physiol Renal Physiol 314: F1008–F1019, 2018. doi: 10.1152/ajprenal.00574.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei J, Zhang J, Wang L, Jiang S, Fu L, Buggs J, Liu R. New mouse model of chronic kidney disease transitioned from ischemic acute kidney injury. Am J Physiol Renal Physiol 317: F286–F295, 2019. doi: 10.1152/ajprenal.00021.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yacov N, Feldman B, Volkov A, Ishai E, Breitbart E, Mendel I. Treatment with lecinoxoids attenuates focal and segmental glomerulosclerosis development in nephrectomized rats. Basic Clin Pharmacol Toxicol 124: 131–143, 2019. doi: 10.1111/bcpt.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zager RA, Baltes LA, Sharma HM, Jurkowitz MS. Responses of the ischemic acute renal failure kidney to additional ischemic events. Kidney Int 26: 689–700, 1984. doi: 10.1038/ki.1984.204. [DOI] [PubMed] [Google Scholar]
- 34.Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol 301: F1334–F1345, 2011. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Chandrashekar K, Lu Y, Duan Y, Qu P, Wei J, Juncos LA, Liu R. Enhanced expression and activity of Nox2 and Nox4 in the macula densa in ANG II-induced hypertensive mice. Am J Physiol Renal Physiol 306: F344–F350, 2014. doi: 10.1152/ajprenal.00515.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Qu HY, Song J, Wei J, Jiang S, Wang L, Wang L, Buggs J, Liu R. Enhanced hemodynamic responses to angiotensin II in diabetes are associated with increased expression and activity of AT1 receptors in the afferent arteriole. Physiol Genomics 49: 531–540, 2017. doi: 10.1152/physiolgenomics.00025.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Wei J, Jiang S, Xu L, Wang L, Cheng F, Buggs J, Koepsell H, Vallon V, Liu R. Macula densa SGLT1-NOS1-tubuloglomerular feedback pathway, a new mechanism for glomerular hyperfiltration during hyperglycemia. J Am Soc Nephrol 30: 578–593, 2019. doi: 10.1681/ASN.2018080844. [DOI] [PMC free article] [PubMed] [Google Scholar]