

Abstract
Background
Microsatellite instability (MSI) predicts oncological response to checkpoint blockade immunotherapies. Although microsatellite mutation is pathognomonic for the condition, loci have unequal diagnostic value for predicting MSI within and across cancer types.
Methods
To better inform molecular diagnosis of MSI, we examined 9438 tumor-normal exome pairs and 901 whole genome sequence pairs from 32 different cancer types and cataloged genome-wide microsatellite instability events. Using a statistical framework, we identified microsatellite mutations that were predictive of MSI within and across cancer types. The diagnostic accuracy of different subsets of maximally informative markers was estimated computationally using a dedicated validation set.
Results
Twenty-five cancer types exhibited hypermutated states consistent with MSI. Recurrently mutated microsatellites associated with MSI were identifiable in 15 cancer types, but were largely specific to individual cancer types. Cancer-specific microsatellite panels of 1 to 7 loci were needed to attain ≥95% diagnostic sensitivity and specificity for 11 cancer types, and in 8 of the cancer types, 100% sensitivity and specificity were achieved. Breast cancer required 800 loci to achieve comparable performance. We were unable to identify recurrent microsatellite mutations supporting reliable MSI diagnosis in ovarian tumors. Features associated with informative microsatellites were cataloged.
Conclusions
Most microsatellites informative for MSI are specific to particular cancer types, requiring the use of tissue-specific loci for optimal diagnosis. Limited numbers of markers are needed to provide accurate MSI diagnosis in most tumor types, but it is challenging to diagnose breast and ovarian cancers using predefined microsatellite locus panels.
Keywords: next-generation DNA sequencing, microsatellite instability, cancer, mismatch repair, screening, immunotherapy
Introduction
Microsatellite instability (MSI) is a molecular tumor phenotype that is indicative of genomic hypermutability, usually reflecting inactivation of the mismatch repair (MMR) system (1, 2). MSI is marked by spontaneous gains or losses of nucleotides from repetitive DNA tracts, resulting in new alleles of differing length that serve as the basis for its clinical diagnosis (2, 3). Although classically associated with colorectal and endometrial tumors (4–6), MSI has now been recognized in most cancer types with varying prevalence (7–10) and is accompanied by a generally increased rate of mutations genome-wide (11, 12). Testing for MSI has subsequently emerged as a pan-cancer biomarker of therapeutic response to PDL-1 and PD-1 immune checkpoint inhibitors (13–15), where the MSI positive (microsatellite high, or MSI-H) phenotype is believed to serve as an indicator of mutation-associated neoantigens that enable a more robust T lymphocyte response than for MSI negative (microsatellite stable, or MSS) cases (12, 13, 16).
Molecular diagnosis of MSI in clinical practice is most commonly achieved using multiplexed PCR of defined microsatellite loci, followed by capillary electrophoresis to detect new alleles (MSI-PCR) qualitatively (17, 18). Alternatively, we and others have developed quantitative next-generation sequencing (NGS) methods to identify MSI by assessing overall microsatellite mutation frequency at repetitive loci that are either directly targeted or incidentally captured by targeted gene enrichment oncology panels (7, 19–27). Nevertheless, both conventional and NGS approaches interrogate markedly limited subsets of the millions of microsatellite markers available in the human genome: only 5 loci are included in standard MSI-PCR (17, 18), whereas dozens to hundreds of sites are examined by typical NGS approaches (7, 19–26).
Recent studies have revealed tissue-specific signatures of microsatellite mutation, such that alterations in specific loci can occur with disparate frequencies in tumors from different tissues (7, 8). Consequently, microsatellites that are diagnostic for MSI potentially have unequal prognostic value within (3) and across tumor types (7), such that loci useful in one cancer type may not yield accurate diagnoses in others. Supporting this hypothesis, standard MSI-PCR markers were developed for use in colon cancers (2, 18) and can exhibit poor performance in other malignancies (23, 28–30).
The choice of markers is therefore critical to maximize sensitivity and specificity of molecular MSI diagnosis (3), regardless of whether testing is performed by conventional or NGS methods. Nevertheless, little effort to date has focused on using systematic, genome-scale analysis to identify optimal microsatellite loci for diagnosing MSI (7, 8). Here, we systematically evaluated tumor-normal pairs of exome and whole genome data from 32 different cancer types to ascertain the most informative microsatellites for predicting MSI.
Methods
Sequence Data and Identification of Microsatellite Loci
Genomic microsatellite loci were identified as previously (7), with some modifications. Briefly, microsatellites were defined in the human genome (GRCh37/hg19) as repeating subunits of 1–5 bp in length and comprising ≥5 repeats using MISA (31). Adjacent microsatellites within 10 base pairs of each other were termed ‘complex’ (c*) single loci if comprised of tracts with different repeating subunit lengths or ‘compound’ (c) single loci for those having the same repeat length. This analysis defined 19 035 602 loci, of which the 18 882 838 present on autosomes and chromosome X were retained. Repeat features were annotated using ANNOVAR (32) (24 February 2014 release).
Sequence alignments of tumors and patient-matched normal specimens from exome and whole genome sequencing projects were obtained from The Cancer Genome Atlas (TCGA) Research Network (33). Alignments were standardized prior to analysis by converting alignments to FASTQ files using PICARD v1.98, re-aligning to GRCh37/hg19 using BWA-MEM v0.7.12, and indexing with SamTools v1.1.
Cataloging Microsatellite Instability Events
The process used for cataloging microsatellite instability events is diagrammed in Supplemental Fig. 1.
For each tumor and normal specimen we quantified the number of sequence reads supporting different tract lengths at each locus using mSINGS (Git Commit ID a7e9ea9) (19).
To identify instability events, we compared multinomial distributions of allele lengths for tumor at each locus to the joint multinomial distribution of allele lengths across tumor and normal at the site by:
where n refers to the number of reads at a site, p the proportion supporting each alternative allelic length, and Mult indicates sampling from a multinomial distribution with those parameters. “Unstable” microsatellites (those evidencing somatic mutations) were defined as those with nominally significant differences (P < 0.05) by likelihood ratio (G) tests without continuity correction. We estimated rates for calling false positive instability at this heuristic threshold as <3% at all sites having ≥2 reads in tumor and ≥1 read in its paired normal by simulating and comparing two distributions of 1000 normal sites with median observed multinomial distributions of allele lengths by:
To confidently define “stable” sites in tumor (those lacking somatic mutations), we simulated from empirically observed multinomial read distributions for highly unstable sites in cases having >30 read coverage in both tumor and normal by:
Down-sampling analyses estimated that ≥18 reads per site yielded 95% power for identifying an unstable locus, providing strong evidence to conclude that a site was “stable” if no difference in allelic distribution was observed at that coverage. Sites with 5–18 reads in both tumor and paired normal but no indication of instability (P > 0.05), and those covered by fewer than 5 reads in either tumor or normal samples were marked as “missing data.”
As a quality control measure, we excluded samples having ≥75% missing data, leaving 9438 tumor-normal exome pairs and 901 whole genome pairs for subsequent analysis. We similarly excluded individual loci for which ≥75% of specimens evidenced missing data.
Gaussian Mixture Model to Classify MSI Status
We quantified the overall frequency of microsatellite mutations for each tumor as the fraction of unstable sites over total callable sites, and given the skewed nature of the data, performed log10 transformation. For each cancer type, we then fit a Gaussian mixture model to these values using Mclust v5.4.5 with one or two mixture components with equal variance. If the two-component model could be validly applied to a cancer type, individual tumors were classified as MSS (lower mode), MSI-H (higher mode), or indeterminate (uncertainty value > 0.1). If distributions were instead consistent with a single component, all tumors of that cancer type were classified as MSS.
Identifying and Modeling Predictive Abilities of Informative Markers
After excluding “intermediate” MSI classifications, 80% of tumors in each cancer type were randomly assigned into training sets and 20% into validation sets. Using the training sets for each tissue type, we identified microsatellites that were most frequently mutated in MSI-H relative to MSS tumors by Fisher’s exact test. The proportions of stable and unstable sites between MSS and MSI-H tumors were compared (excluding missing data), allowing loci to be rank-ordered by P value. For each tissue type, we then selected subsets of the top n most informative loci (ranging from 1 to 2000 markers) and calculated the percentage of unstable loci for each sample (excluding missing data from both numerator and denominator). We used these values to calculate the area under the receiver operating characteristic (AUROC) using pROC v1.15.3. The optimal percentage on the receiver operating characteristic curve was identified by Youden’s J statistic. This value was subsequently used as the threshold to assign MSS or MSI-H classification to each sample based on the fraction of mutated markers identified from the simulated panel and to determine sensitivity and specificity for each tissue type.
To account for the possibility of more complex trans-genomic interactions between sites not captured by an additive model of instability, we explored machine learning approaches including random forest and boosted trees. Iterative reduction of marker features was performed using both Fisher exact test P values and Shapley feature importance values in the training dataset. However, these approaches did not meaningfully outperform the simpler additive model.
Identification of MSI-Associated Microsatellite Mutations Common to Multiple Tissue Types
The top 2000 loci most strongly associated with MSI-H status for each tissue type were examined for cross-performance across tissue types by hierarchical clustering of normalized P values. Normalization was required to account for differences in the uneven sample sizes, and was accomplished by log10-transformation of raw P values, followed by scaling on a per-tissue basis from a range of 0 (least significant) to 1 (most significant). A heatmap was generated using superheat v0.1.0, and pairwise and cophenetic distances between tissue types were subsequently calculated and compared using the base-R dist, cophenetic, and cor functions.
Features of Informative Microsatellites
We examined enrichment of particular locus features (annotated genic context of the repeat, repeat class, and number of repeat subunits in deciles) among individual microsatellite associations with MSI-H status using linear regression. Intergenic annotation, pentanucleotide repeats, and a length of 5–11 repeats, respectively, were arbitrarily selected as reference levels for these analyses. Whole genome data were used for this analysis, as they more comprehensively represented feature annotations across coding and noncoding regions than exome data.
Results
Prevalence of Microsatellite Mutation and MSI-H Tumors Vary across Cancer Types
To ascertain the relative degree of genomic instability within and across cancer types, we assessed the overall frequency of microsatellite mutations within each of 32 cancer types using publicly available paired tumor-normal exome sequencing data. For each cancer type, we used Gaussian mixture modeling to circumscribe subpopulations of tumors having high burdens of microsatellite mutation, corresponding to MSI-H cases (7, 8, 19, 21, 34) (Fig. 1, Table 1, Supplemental Table 1). Comparison of MSI classifications established by this approach showed high agreement with determinations made by “gold-standard” MSI-PCR (accuracy 97.8%, 95% CI 95.5%–98.35%) or MOSAIC genome-scale analysis (7) (accuracy 98.8%, 95% CI 98.5%–99.1%), supporting their validity.
Fig. 1.
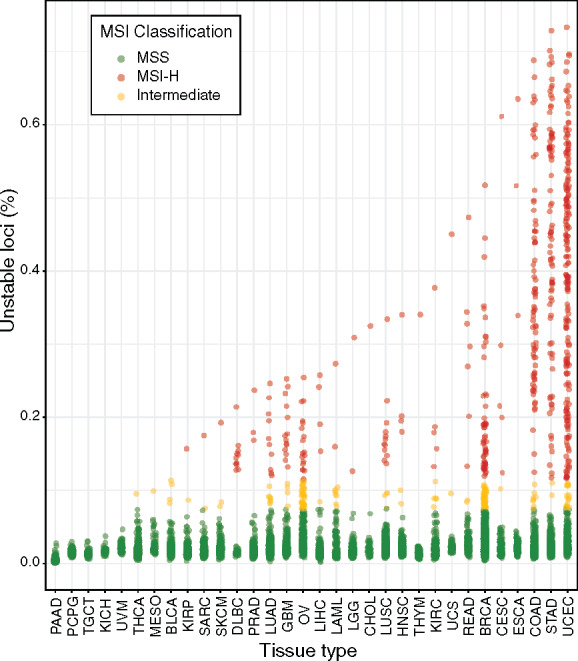
Relative burden of microsatellite instability events across cancer types. Percentage of total microsatellite loci found to be mutated are reported per tumor specimen, as stratified by cancer type. Each point corresponds to an individual tumor, and its coloration indicates the corresponding MSI classification. TCGA abbreviations for cancer types are as described in the Table 1 footnote.
Table 1.
Summary of inferred MSI diagnoses from tumor exome sequencing.
| Cancer type (TCGA code)a | Fraction of Cases (Count) |
||
|---|---|---|---|
| MSS | Intermediate | MSI-H | |
| BLCA | 0.99 (375) | 0.01 (4) | 0 (0) |
| BRCA | 0.9 (866) | 0.04 (43) | 0.06 (53) |
| CESC | 0.97 (206) | 0.005 (1) | 0.02 (5) |
| CHOL | 0.98 (49) | 0 (0) | 0.02 (1) |
| COAD | 0.79 (309) | 0.02 (8) | 0.19 (74) |
| DLBC | 0.77 (36) | 0 (0) | 0.23 (11) |
| ESCA | 0.98 (145) | 0 (0) | 0.02 (3) |
| GBM | 0.95 (356) | 0.01 (4) | 0.04 (14) |
| HNSC | 0.99 (506) | 0.004 (2) | 0.01 (4) |
| KICH | 1 (64) | 0 (0) | 0 (0) |
| KIRC | 0.97 (321) | 0.02 (5) | 0.02 (5) |
| KIRP | 0.99 (248) | 0.004 (1) | 0.004 (1) |
| LAML | 0.92 (113) | 0.07 (8) | 0.02 (2) |
| LGG | 1 (494) | 0 (0) | 0.004 (2) |
| LIHC | 0.99 (329) | 0.003 (1) | 0.01 (4) |
| LUAD | 0.97 (525) | 0.01 (7) | 0.02 (10) |
| LUSC | 0.96 (436) | 0.01 (4) | 0.03 (12) |
| MESO | 0.98 (59) | 0.02 (1) | 0 (0) |
| OV | 0.87 (339) | 0.09 (34) | 0.05 (18) |
| PAAD | 1 (174) | 0 (0) | 0 (0) |
| PCPG | 1 (184) | 0 (0) | 0 (0) |
| PRAD | 0.99 (455) | 0 (0) | 0.01 (3) |
| READ | 0.94 (133) | 0.01 (2) | 0.05 (7) |
| SARC | 0.99 (186) | 0.01 (1) | 0.01 (1) |
| SKCM | 0.99 (439) | 0.005 (2) | 0.002 (1) |
| STAD | 0.79 (328) | 0.005 (2) | 0.2 (84) |
| TGCT | 1 (156) | 0 (0) | 0 (0) |
| THCA | 1 (454) | 0.002 (1) | 0 (0) |
| THYM | 0.99 (118) | 0 (0) | 0.01 (1) |
| UCEC | 0.57 (255) | 0.04 (17) | 0.4 (179) |
| UCS | 0.96 (55) | 0.02 (1) | 0.02 (1) |
| UVM | 1 (80) | 0 (0) | 0 (0) |
BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma.
Twenty-five of 32 cancer types evidenced one or more hypermutated tumors consistent with an MSI-H phenotype, ranging in incidence from 0.2% to 40%, similar to other reports (7–9). The total fraction of mutated microsatellites in MSI-H tumors varied considerably across cancer types, with the greatest microsatellite mutation burdens occurring in stomach, colon, and endometrial tumors (Fig. 1).
Parallel analyses were performed using whole genome data (Supplemental Fig. 2, Supplemental Table 2, Supplemental Table 3), which included intergenic regions but had lower sequencing read depths and were available for fewer cases and cancer types. Although data were sparser, results from whole genome analysis were consistent with those from exome data.
Informative Microsatellite Markers Differ between Cancer Types
We next sought to identify microsatellite mutations most predictive of MSI by cataloging events occurring significantly in MSI-H relative to MSS tumors of each cancer type. This analysis was restricted to cancers for which locus performance could be evaluated in both testing and validation sets, permitting examination of 15 cancer types by exome data alone (Supplemental Table 4).
Hierarchical clustering (Fig. 2) revealed that cancer types displayed markedly different, and largely non-overlapping subsets of informative microsatellites from across the genome. Correlation between measurements of pairwise (Supplemental Table 5) and cophenetic (online Supplemental Table 6) distance matrices of locus informativity was 0.82, indicating that clustering accurately represents a large degree of variation in patterns of MSI-associated mutation across cancers. Endometrial and colon tumors were most similar by these metrics, sharing 34.6% of sites (4767 loci) nominally associated with MSI-H diagnosis (at uncorrected P < 0.05), whereas esophageal carcinoma and brain lower grade glioma were most disparate, having only 5% informative microsatellites (79 loci) in common. Similarities in microsatellite mutation patterns were apparent among rectal, stomach, colon, and uterine corpus endometrial tumors, as well as between lung adenocarcinoma and squamous cell carcinoma (Fig. 2, Supplemental Table 4). However, most cancer types evidenced distinct profiles of microsatellite mutations.
Fig. 2.
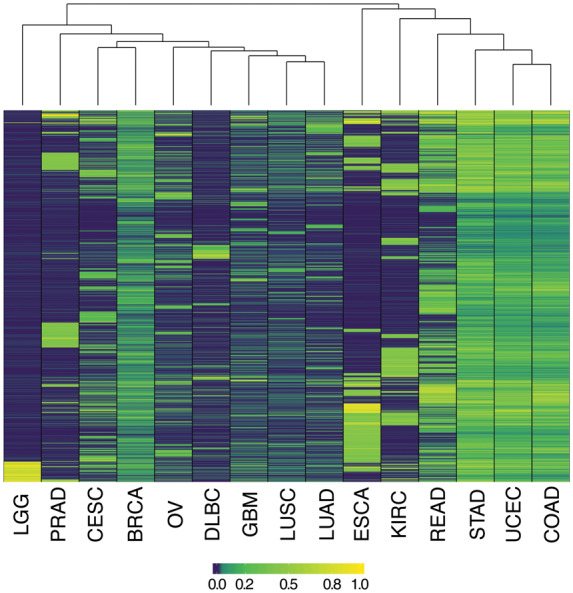
Informative markers for diagnosing MSI vary across cancer types. The 2000 most informative microsatellites (rows) for discriminating MSI positive from MSI negative tumors from each of the indicated cancer types (columns) are displayed. Heatmap coloration indicates the association of individual loci with the MSI-H phenotype, normalized on a per-cancer type basis from zero (least significant) to one (most significant). Hierarchical clustering of cancer types according to similarity in microsatellite mutation patterns is indicated at top. TCGA abbreviations for cancer types are as described in the Table 1 footnote.
We conclude that the great majority of loci whose mutation correlates with an MSI-H phenotype are specific to particular cancer types.
Predictive Value of Microsatellites for MSI Diagnosis Within and Across Cancer Types
Subsets of 1 to 2000 of the most highly informative loci identified per cancer type were used to computationally evaluate their performance for diagnosing MSI within their tumor type of origin, using an independent set of tumor samples for validation (Fig. 3). Specimens were classified as MSS or MSI-H based on the fraction of mutated microsatellites observed in a given panel, with optimal thresholds for discriminating these classifications determined empirically (Table 2). These classifications were compared against determinations made from exome-wide analysis (Supplemental Table 1) to assess their accuracy.
Fig. 3.

Performance characteristics of variously sized tissue-specific panels for MSI diagnosis. Area under the receiver operating characteristic (AUROC) is shown as a function of the number of most highly informative markers examined for various cancer types. Results are shown for a) the training set from which informative microsatellites were initially identified and b) an independent validation set for each tumor type. TCGA abbreviations for cancer types are as described in the Table 1 footnote.
Table 2.
Performance characteristics of tissue-specific microsatellite panels for MSI diagnosis in validation set.
| Cancer Type (TCGA Code) | Smallest panel achieving sensitivity and specificity of ≥95% |
Smallest panel achieving AUROC of 0.9 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of markers | Minimum unstable markers for MSI-H classification (%) | Area under the curve | Sensitivity | Specificity | Number of markers | Minimum unstable markers for MSI-H classification (%) | Area under the curve | Sensitivity | Specificity | |
| COAD | 1 | 50.0% | 0.98 | 100.0% | 95.8% | 1 | 50.0% | 0.98 | 100.0% | 95.8% |
| ESCA | 1 | 50.0% | 1.00 | 100.0% | 100.0% | 1 | 50.0% | 1.00 | 100.0% | 100.0% |
| GBM | 1 | 50.0% | 1.00 | 100.0% | 100.0% | 1 | 50.0% | 1.00 | 100.0% | 100.0% |
| LUAD | 1 | 50.0% | 0.99 | 100.0% | 97.1% | 1 | 50.0% | 0.99 | 100.0% | 97.1% |
| LUSC | 1 | 50.0% | 0.99 | 100.0% | 97.1% | 1 | 50.0% | 0.99 | 100.0% | 97.1% |
| READ | 2 | 50.0% | 1.00 | 100.0% | 100.0% | 2 | 50.0% | 1.00 | 100.0% | 100.0% |
| STAD | 2 | 25.0% | 1.00 | 100.0% | 100.0% | 1 | 50.0% | 0.92 | 84.2% | 100.0% |
| LGG | 5 | 12.5% | 1.00 | 100.0% | 100.0% | 5 | 12.5% | 1.00 | 100.0% | 100.0% |
| PRAD | 5 | 16.7% | 1.00 | 100.0% | 100.0% | 5 | 16.7% | 1.00 | 100.0% | 100.0% |
| CESC | 7 | 45.8% | 1.00 | 100.0% | 100.0% | 7 | 45.8% | 1.00 | 100.0% | 100.0% |
| DLBC | 7 | 25.0% | 1.00 | 100.0% | 100.0% | 7 | 25.0% | 1.00 | 100.0% | 100.0% |
| UCEC | 20 | 12.9% | 0.98 | 97.0% | 97.9% | 2 | 25.0% | 0.95 | 90.3% | 97.2% |
| KIRC | 65 | 7.5% | 0.99 | 100.0% | 98.5% | 55 | 4.6% | 0.93 | 100.0% | 86.6% |
| BRCA | 800 | 3.6% | 1.00 | 100.0% | 99.5% | 50 | 4.3% | 0.92 | 100.0% | 81.1% |
| OVa | 2000 | 2.0% | 0.63 | 100.0% | 47.8% | 2000 | 2.0% | 0.63 | 100.0% | 47.8% |
Did not achieve cutoff for test-characteristic threshold with the maximum number of loci tested.
We initially determined the number of markers needed to achieve MSI diagnosis with at least 95% sensitivity and specificity (Fig. 3; Table 2). The number of requisite markers varied considerably across tumor types. For 11 of the 15 cancer types, 7 or fewer markers provided the requisite performance characteristics, and in 8 cancers, MSI could be diagnosed with 100% specificity and sensitivity. Diagnosis of MSI in endometrial tumors required 20 markers, while 65 were needed for classification of kidney renal clear cell cancers. In contrast, MSI determination in breast cancer required 800 markers to achieve comparable performance. MSI diagnosis in ovarian tumors, while favorable for the training set (Fig. 3A), did not enable reliable diagnosis in the validation set using any number of markers considered (Fig. 3B;Table 2).
The desired balance of sensitivity and specificity may vary by clinical application, and relates to the number of markers examined. We therefore additionally determined the predictive capacity of various numbers of markers as measured by the AUROC and the number of makers required to achieve an area under the curve (AUC) of 0.9 or greater (Table 2). Although the number of markers required for most cancers by this metric remained similar, decreases for breast (from 800 to 50), kidney renal clear cell (65 to 55), endometrial (20 to 2), and stomach (2 to 1) were observed.
A small number of loci were informative for MSI across multiple cancer types. We therefore examined informative microsatellites in endometrial, colon, rectal, and stomach cancers, which collectively showed closely related mutational patterns, to determine whether a common marker panel could be used to diagnose MSI in those tumors. After Bonferroni correction, 37 shared microsatellites were independently associated with MSI-H status in each of those four cancer types (Supplemental Table 7). The 37-marker panel demonstrated favorable performance characteristics for the 4 specific cancer types (0.98 AUC, sensitivity 94.3%, specificity 97.7%), but did not outperform respective tissue-type specific marker panels (Table 2) and functioned poorly when applied to other cancers. For example, AUC was 0.45 when the panel was applied to lung squamous cell carcinoma and lung adenocarcinoma, compared with AUC 0.95 for a similarly sized panel specific to those cancer types.
Properties of Informative Microsatellites
We examined the sequence composition and genic feature annotations that were enriched in microsatellites having globally high informativity for MSI-H tumors (Fig. 4, Supplemental Table 8). Whole genome data were examined in order to allow exploration of coding and noncoding regions but showed agreement with analyses of exome sequencing where direct comparisons were possible (not shown). Of the different types of repeat sequence classes examined (Fig. 4A), mononucleotide microsatellite mutations correlated most highly with MSI-H, followed by complex repeats (which themselves may be composed of multiple mononucleotide repeats). MSI-informative markers were most significantly enriched in splice sites (Fig. 4B), and conversely, intergenic regions and noncoding intronic annotations contained the fewest informative microsatellite elements. Notably, we found a nonlinear correlation between the length of mononucleotide markers and their informativity (Fig. 4, C and D). Tracts comprised of 12 to 13 repeats proved most informative, with longer or shorter loci showing a decrease in informativity proportional to their distance from this maximum.
Fig. 4.
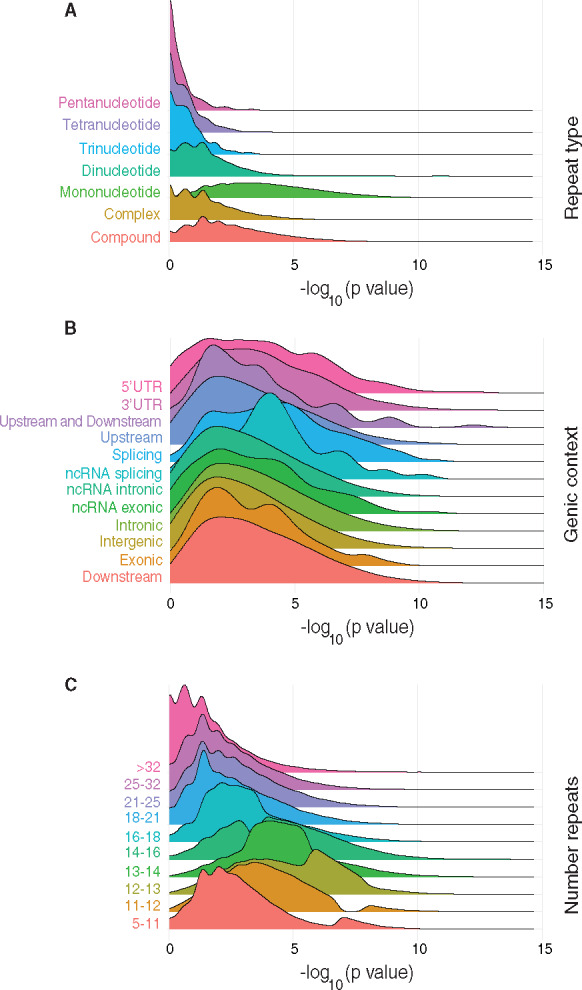
Properties of informative microsatellites. Ridgeline plots showing density of -log10 transformed P values for specified features among microsatellites that are informative for diagnosing MSI. A shift to the right in the placement of distributions corresponds to increased association with locus instability in MSI. a) Repeat type. b) Genomic context. c) Number of repeats in a microsatellite.
Conclusions
Here we used genomic analyses to prioritize microsatellite markers that are most informative for diagnosing MSI by molecular methods. Whereas prior work has cataloged loci that are frequently mutated in MSI-H tumors from the 3 (8) or 4 (7) cancer types where the phenotype occurs most often, here we have more broadly examined microsatellite mutation occurrence across cancer types and have also evaluated the performance of variously sized marker subsets for classifying MSI-H tumors in clinical practice.
We previously reported that cancer types exhibit distinct patterns of microsatellite mutation overall (7), and in this work similarly found that microsatellites that are informative for diagnosing MSI also vary across cancer types (Fig. 2). Our results strongly argue that MSI diagnostic panels developed for particular tumor types should not be considered generally applicable across all cancers (23, 28–30). Although a small subset of markers were cross-informative across a limited group of cancer types that shared similar mutational profiles (Table 2), microsatellites tailored to a specific cancer type provided maximal diagnostic accuracy. This phenomenon potentially reflects different selective pressures underlying tumor evolution, wherein mutation of specific microsatellites may alter gene expression or gene function and are therefore recurrently subjected to positive or negative selection in cancer types for which those changes are relevant (7).
Accordingly, in determining markers with the highest diagnostic utility (Fig. 4), we observed that microsatellite mutations within functional elements including splice sites and exons were significantly enriched, further supporting the notion that biological pressures are involved in selecting microsatellite mutations associated with MSI (7). We also observed that mononucleotide microsatellites, loci occurring in splicing regions, and microsatellites comprised of 12 to 13 repeat subunits are expected to provide the greatest diagnostic benefit, with microsatellites of 18 or greater repeats being significantly associated with stability. This latter finding argues that, unlike in in vitro systems (35), MSI phenotypes in vivo preferentially involve tracts of specific lengths.
We evaluated the performance of variously sized subsets of the most informative loci per cancer type in data sets withheld from those used to identify informative loci (Fig. 3; Table 2). We note that due to the degree of missing microsatellite calls inherent to our dataset, existing performance estimates are likely to be conservative. Even so, 8 of the 15 cancer types achieved 100% sensitivity and specificity using 7 or fewer loci, and in 2 of those cancer types only a single marker was required. These modest testing requirements are compatible with MSI diagnosis by highly focused methods including conventional MSI-PCR or targeted NGS. Endometrial and kidney renal clear cell tumors required slightly more markers (20 and 65, respectively). In 2 extreme cases, breast cancer required 800 markers, whereas reliable MSI diagnosis could not be achieved in ovarian cancer using predefined subsets of microsatellite markers. These findings are consistent with microsatellites following less stereotyped patterns of mutation for breast and ovarian cancers, possibly owing to instability events having more neutral fitness effects for breast and ovarian tumors and subsequently resulting in fewer recurrently mutated loci. In these cancer types, and possibly others, a measurement of overall genome-wide microsatellite mutation burden may be required to establish MSI diagnosis reliably (7) (Fig. 1).
Although MSI status is currently the diagnostic marker approved by the United States Food and Drug Administration to indicate eligibility for PDL-1 and PD-1 inhibitor treatments (13, 14), alternative methods for testing tumor susceptibility to those immunotherapies are now available. The most widely used of these alternative approaches is tumor mutation burden (TMB), a biomarker based on estimating the total substitution and indel lesions present in a cancer genome (36). Nevertheless, TMB determination requires sequencing large gene panels (37) and is of contested clinical utility (36, 38–41). Because immunotherapy response is particularly associated with insertion-deletion mutational load (12), MSI is considered a more reliable positive predictor of treatment outcomes even though it is unable to identify all cancers for which a favorable response can be achieved (11). Although MSI and TMB determinations frequently overlap, they provide distinct information (11). Given these considerations, MSI and TMB can be considered complementary (11, 12), and we envision that dedicated testing for MSI will continue to provide utility as an inexpensive, primary screening method for immunotherapy response.
Our analyses define useful tissue-specific diagnostic panels for MSI, but their power and utility could be improved by future efforts. Although most cancer types include some fraction of cases that are distinguishable as MSI-H [Fig. 1 and (7–10)], existing data do not encompass enough MSI-H representatives to enable identification of informative loci from all cancer types, and in other cases the paucity of such specimens may limit the robustness analysis. Focused efforts to sequence MSI-H tumors from these cancer types would enable their more thorough characterization. Separately, extant sequence data provides variable coverage across genomic regions and among tumors, such that the instability of specific microsatellites cannot consistently be assessed within or across tumor types. This results in “missing” data that negatively affects the statistical power of our analyses and potentially obscure the identification of otherwise diagnostically useful markers, but could be remedied by greater read depths. Relatedly, we were unable to directly compare the tissue-specific diagnostic performance of loci identified in this study to microsatellite loci commonly included in clinical MSI assays (17, 18) due to both the unknown sensitivity of NGS relative to MSI-PCR for detecting microsatellite mutations and inadequate read depths for those markers in both exome and whole genome data. It is noteworthy that the most informative markers identified by our analyses do not overlap with those utilized in standard clinical assays for MSI (17, 18). Lastly, although informative microsatellites are enriched in splice sites and other transcribed features (Fig. 4) that are largely recovered by exome sequencing, the availability of high-depth, whole genome sequence data from MSI-H tumors would enable a more comprehensive search for rare, diagnostically useful loci present in noncoding regions. As additional higher quality sequencing data are generated that meet these needs, they will enable further refinement of optimally informative MSI markers across different malignancies.
Supplementary Material
Nonstandard Abbreviations
MSI, microsatellite instability: MMR, mismatch repair; MSI-H, microsatellite instability high; MSS, microsatellite stable; NGS, Next-Generation DNA sequencing; TMB, tumor mutation burden; AUC, area under the curve; AUROC, area under the receiver operating characteristic.
Human Genes: PD-1, programmed cell death 1; PDL-1, programmed cell death 1 ligand 1.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 4 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; (c) final approval of the published article; and (d) agreement to be accountable for all aspects of the article thus ensuring that questions related to the accuracy or integrity of any part of the article are appropriately investigated and resolved.
D.R. Long, statistical analysis; V.P. Panicker, statistical analysis; R.J. Hause, statistical analysis.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: None declared.
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Research Funding: D.R. Long, T32 GM086270-11 from National Institute of General Medical Sciences; S.J. Salipante, grant R33CA222344 from the National Cancer Institute.
Expert Testimony: None declared.
Patents: D.R. Long, S.J. Salipante, R.J. Hause, and A. Waalkes have applied for a provisional patent based on this work.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, preparation of manuscript, or final approval of manuscript.
References
- 1. Vilar E, Gruber SB.. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 2010;7:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murphy KM, Zhang S, Geiger T, Hafez MJ, Bacher J, Berg KD, et al. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn 2006;8:305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baudrin LG, Deleuze J-F, How-Kit A.. Molecular and computational methods for the detection of microsatellite instability in cancer. Front Oncol 2018;8:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J Mol Diagn 2008;10:301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beamer LC, Grant ML, Espenschied CR, Blazer KR, Hampel HL, Weitzel JN, et al. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol 2012;30:1058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hause RJ, Pritchard CC, Shendure J, Salipante SJ.. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 2016;22:1342–50. [DOI] [PubMed] [Google Scholar]
- 8. Cortes-Ciriano I, Lee S, Park W-Y, Kim T-M, Park PJ.. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 2017;8:15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen H-Z, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol 2017;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maruvka YE, Mouw KW, Karlic R, Parasuraman P, Kamburov A, Polak P, et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat Biotechnol 2017;35:951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J.. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 2018;7:746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019;364:485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang L, Chang M, Chang HM, Chang F.. Microsatellite instability: a predictive biomarker for cancer immunotherapy. Appl Immunohistochem Mol Morphol 2017;26:e15–21. [DOI] [PubMed] [Google Scholar]
- 16. Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017;551:517–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de la Chapelle A, Hampel H.. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol 2010;28:3380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB, et al. Development of a fluorescent multiplex assay for detection of MSI-high tumors. Dis Markers 2004;20:237–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC.. Microsatellite instability detection by next generation sequencing. Clin Chem 2014;60:1192–9. [DOI] [PubMed] [Google Scholar]
- 20. Huang MN, McPherson JR, Cutcutache I, Teh BT, Tan P, Rozen SG.. MSIseq: software for assessing microsatellite instability from catalogs of somatic mutations. Sci Rep 2015;5:13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kautto EA, Bonneville R, Miya J, Yu L, Krook MA, Reeser JW, et al. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017;8:7452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, et al. Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis Oncol 2017;1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waalkes A, Smith N, Penewit K, Hempelmann J, Konnick EQ, Hause RJ, et al. Accurate pan-cancer molecular diagnosis of microsatellite instability by single-molecule molecular inversion probe capture and high-throughput sequencing. Clin Chem 2018;64:950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gray PN, Tsai P, Chen D, Wu S, Hoo J, Mu W, et al. TumorNext-Lynch-MMR: a comprehensive next generation sequencing assay for the detection of germline and somatic mutations in genes associated with mismatch repair deficiency and Lynch syndrome. Oncotarget 2018;9:20304–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hempelmann JA, Scroggins SM, Pritchard CC, Salipante SJ.. MSIplus: integrated colorectal cancer molecular testing by next-generation sequencing. J Mol Diagn 2015;17:705–14. [DOI] [PubMed] [Google Scholar]
- 26. Willis J, Lefterova MI, Artyomenko A, Kasi PM, Nakamura Y, Mody K, et al. Validation of microsatellite instability detection using a comprehensive plasma-based genotyping panel. Clin Cancer Res 2019;25:7035–45. [DOI] [PubMed] [Google Scholar]
- 27. Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, et al. A novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J Mol Diagn 2019;21:1053–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 2006;66:7810–7. [DOI] [PubMed] [Google Scholar]
- 29. Faulkner RD, Seedhouse CH, Das-Gupta EP, Russell NH.. BAT-25 and BAT-26, two mononucleotide microsatellites, are not sensitive markers of microsatellite instability in acute myeloid leukaemia. Br J Haematol 2004;124:160–5. [DOI] [PubMed] [Google Scholar]
- 30.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009;11:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beier S, Thiel T, Münch T, Scholz U, Mascher M.. MISA-web: a web server for microsatellite prediction. Bioinformatics 2017;33:2583–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang K, Li M, Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Cancer Genome Atlas Program. https://www.cancer.gov/tcga (Accessed July 2020).
- 34. Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014;30:1015–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koole W, Schäfer HS, Agami R, van Haaften G, Tijsterman M.. A versatile microsatellite instability reporter system in human cells. Nucleic Acids Res 2013;41:e158–e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagahashi M, Wakai T, Shimada Y, Ichikawa H, Kameyama H, Kobayashi T, et al. Genomic landscape of colorectal cancer in Japan: clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med 2016;8:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Danilova L, Wang H, Sunshine J, Kaunitz GJ, Cottrell TR, Xu H, et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc Natl Acad Sci U S A 2016;113:E7769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014;24:743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gonzalez-Cao M, Viteri S, Karachaliou N, Aguilar A, García-Mosquera JJ, Rosell R.. Tumor mutational burden as predictive factor of response to immunotherapy. Transl Lung Cancer Res 2018;7:S358–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.