

Abstract
Herpes zoster is associated with an increased dementia and neovascular macular degeneration risk and a decline in glycemic control in diabetes mellitus. Because amyloid is present and pathogenic in these diseases, we quantified amyloid, Aβ40, Aβ42, and amylin in 14 zoster and 10 control plasmas. Compared to controls, zoster plasma had significantly elevated amyloid that correlated with Aβ42 and amylin levels; and increased amyloid aggregation with addition of exogenous Aβ42 or amylin. These results suggest that zoster plasma contains factor(s) that promote aggregation of amyloidogenic peptides, potentially contributing to the toxic amyloid burden and explaining accelerated disease progression following zoster.
Keywords: Herpes zoster, Varicella zoster virus, Amyloid, Aβ40, Aβ42, Amylin
Introduction
Varicella zoster virus (VZV) is latent in ganglionic neurons in >90% of the population (Mahalingam et al. 1990; Kennedy et al. 1998, 1999; Nagel et al. 2014). With aging or immunosuppression, VZV reactivates to produce herpes zoster (shingles). VZV can also spread centrally along immunoprivileged nerve fibers to infect end target organs where virus elicits a robust, often persistent, inflammatory response, resulting in a spectrum of other diseases, including cranial nerve palsies, transient ischemic attacks, stroke, myelopathy, and gastrointestinal disease (Gilden et al. 2013; Gershon et al. 2018), with or without zoster.
Recent studies have linked zoster to 3 amyloid-associated diseases of aging – Alzheimer’s disease (AD), neovascular age-related macular degeneration (“wet” AMD), and diabetes mellitus (DM). A retrospective cohort study showed that compared to healthy controls, herpes zoster ophthalmicus (HZO) subjects have a 2.97-fold greater risk of developing dementia over a 5-year period (p < 0.001) (Tsai et al. 2017); this risk was still significantly elevated, but to a lesser degree, if zoster occurred in any dermatome (1.11-fold; p < 0.0014); and antiviral therapy reduced the risk (Chen et al. 2018). Additionally, HZO subjects have a 4.62-fold increased risk of developing neovascular AMD compared to matched controls over 3 years (p < 0.001) (Ho et al. 2019) . Finally, several studies demonstrated that DM subjects are 20-30% more likely to develop zoster compared to non-diabetic controls (reviewed in Kawai and Yawn 2017); furthermore, following zoster, DM subjects experienced a deterioration of glycemic control (Munoz-Quiles et al. 2017).
The biological plausibility of VZV infection contributing to the pathogenesis of AD, AMD, and DM is supported by their shared clinicohistopathological features and established evidence that VZV can productively infect brain, retina, and pancreas (Gilden et al. 2013; Tran et al. 2003; Wang et al. 2014). Specifically, during productive VZV infection, inflammation, vascular involvement, and cell death are seen. These 3 features are also characteristic of (1) AD with neuroinflammation, cerebrovascular ischemia and neuronal death; (2) neovascular AMD with inflammation of the retina, neoangiogenesis, hemorrhage of retinal vessels, and retinal cell death; and (3) DM with infiltrating immune cells in pancreas, widespread vascular disease, and beta cell degeneration (Donath et al. 2008; Schaper et al. 2000; Hoppener et al. 2000, respectively); thus, a concurrent VZV infection in the central nervous system (CNS)/cerebrovasculature, eye, or pancreas, respectively, could accelerate disease progression. What is not known is whether VZV infection can contribute to the formation of the proinflammatory and cytotoxic amyloid seen in brains of AD, retina of AMD, and pancreatic islets of DM subjects. A recent study showed that VZV-infected primary human spinal astrocytes in vitro leads to accumulation of intracellular amyloidogenic proteins and amyloid, as well as an amyloid-promoting extracellular environment, in part through the production of amyloidogenic viral peptides (Bubak et al. 2019); these findings suggest that VZV infection in vivo could also produce an amyloidogenic environment, accelerating progression of amyloid-associated diseases. Thus, we determined if subjects with acute zoster produced an amyloidogenic environment by comparing levels of plasma amyloid and cellular peptides with the capacity to aggregate and form amyloid (amyloid-beta [Aβ]40, Aβ42, and amylin [islet amyloid polypeptide]) to plasma of non-zoster, control subjects.
Materials and methods
Subjects
Whole blood was obtained from subjects with clinically-diagnosed zoster, de-identified, and analyzed (University of Texas Health Science Center’s Institutional Review Board protocol approval HSC-MS-14-0520). The zoster group consisted of 14 subjects (7 men and 7 women; mean age, 60.2 years (standard error of the mean (SEM), 3.86 years; range, 42-87 years) (Table 1). Blood was collected between 1-4 days after rash onset when subjects still exhibited a unilateral, dermatomal-distribution vesicular rash (days and distribution noted in Table 1); none of the subjects were on antiviral therapy at time of blood draws. De-identified, non-zoster control blood was obtained commercially from Vitalant (Denver, CO). The zoster-negative control group consisted of 10 subjects (5 men and 5 women; mean age, 50 years (SEM, 3.33 years; range, 40–67 years) (Table 1). Because samples were de-identified, additional clinical data, including history of zoster, zoster vaccination, diabetes, or inflammatory disease, was limited; however, at time of blood collection, control subjects did not exhibit rash and were not acutely ill. Furthermore, blood samples were tested and negative for human immunodeficiency virus; hepatitis B and C virus, and; cytomegalovirus.
Table 1.
Subjects examined for levels of amyloid, amylin, Aβ42 and Aβ40
| Analyte levels | ||||||||
|---|---|---|---|---|---|---|---|---|
| Raw values | Natural log Transformed values | |||||||
| Subject number | Age, years | Sex | Zoster distribution | Days from rash onseta | Amyloid (Thio-T, A.U.) | Amylin (pM) Aβ42 (pg/mL) Aβ40 (pg/mL) |
Amyloid | Amylin Aβ42 Aβ40 |
| Group 1 (healthy, non-zoster controls) | ||||||||
| 1 | 58 | M | NA | NA | 16891.67 | 36.12 20.68 121.58 |
9.73 | 3.59 3.03 4.80 |
| 2 | 60 | M | NA | NA | 18484.67 | 13.74 3.48 80.96 |
9.82 | 2.62 1.25 4.39 |
| 3 | 45 | F | NA | NA | 25691.00 | 9.64 2.53 92.34 |
10.15 | 2.27 0.93 4.53 |
| 4 | 67 | F | NA | NA | 23272.33 | 9.73 2.97 85.87 |
10.06 | 2.27 1.09 4.45 |
| 5 | 43 | F | NA | NA | 19603.00 | 99.56 6.57 89.45 |
9.88 | 4.60 1.88 4.49 |
| 6 | 62 | M | NA | NA | 26170.00 | 179.50 NM NM |
10.17 | 5.19 NM NM |
| 7 | 41 | M | NA | NA | 16579.00 | 239.79 9.56 111.68 |
9.72 | 5.48 2.26 4.72 |
| 8 | 40 | F | NA | NA | 23064.00 | 23.39 32.70 120.21 |
10.05 | 3.15 3.49 4.79 |
| 9 | 43 | F | NA | NA | 24202.67 | 26.55 26.70 116.73 |
10.09 | 3.28 3.28 4.76 |
| 10 | 40 | M | NA | NA | 22822.67 | 20.61 2.90 79.09 |
10.04 | 3.03 1.07 4.37 |
| Mean (SEM) | 50.00 (3.33) | 21678.00 (1115.00) | 65.90 (25.70) 12.00 (3.88) 99.80 (5.84) |
9.97 (0.05) | 3.55 (0.37) 2.03 (0.34) 4.59 (0.06) |
|||
| Group 2 (zoster) | ||||||||
| 1 | 59 | F | Right, T8 | 2 | 27991.00 | 15.18 32.00 94.46 |
10.24 | 2.72 3.47 4.55 |
| 2 | 48 | F | Right, V1 | 1 | 27017.00 | NM NM 195.58 |
10.20 | NM NM 5.28 |
| 3 | 46 | F | Left, V1 | 4 | 28476.00 | 29.73 4.48 50.88 |
10.26 | 3.39 1.50 3.93 |
| 4 | 50 | M | Left, T5 | 3 | 24292.67 | 13.16 2.48 41.81 |
10.10 | 2.58 0.91 3.73 |
| 5 | 52 | M | Left, C6 | 2 | 23199.33 | 12.50 4.37 72.68 |
10.05 | 2.53 1.47 4.29 |
| 6 | 60 | F | Left, S2 | 4 | 26093.67 | 11.04 4.76 72.03 |
10.17 | 2.40 1.56 4.28 |
| 7 | 42 | M | Left, T6 | 1 | 28754.00 | 9.38 3.97 48.12 |
10.27 | 2.24 1.38 3.87 |
| 8 | 86 | F | Right, T5 | 3 | 26268.33 | 7.74 5.83 73.75 |
10.18 | 2.05 1.76 4.30 |
| 9 | 59 | F | Left, V1/V2 | 3 | 28837.00 | 19.22 46.40 94.22 |
10.27 | 2.96 3.84 4.55 |
| 10 | 70 | F | Right, S2 | 3 | 30617.67 | 73.87 27.34 118.47 |
10.33 | 4.30 3.31 4.77 |
| 11 | 87 | M | Left, C8 | 1 | 22082.00 | 9.22 4.73 95.90 |
10.00 | 2.22 1.55 4.56 |
| 12 | 74 | M | Left, T11 | 2 | 19757.67 | 9.24 2.90 75.43 |
9.89 | 2.22 1.03 4.32 |
| 13 | 63 | M | Right, V2 | 1 | 23484.00 | 37.87 7.10 65.38 |
10.06 | 3.63 1.96 4.18 |
| 14 | 47 | M | Right, T10 | 2 | 25940.33 | 11.07 NM 58.70 |
10.16 | 2.40 NM 4.07 |
| Mean (SEM) | 60.20 (3.86) | 25915.00 (810.00) | 19.90 (5.13) 12.20 (4.20) 82.70 (10.40) |
10.16 (0.03) | 2.74 (0.18) 1.98 (0.28) 4.33 (0.11) |
|||
Days since rash onset and serum collection
Thio-T Thioflavin-T, A.U. arbitrary units, M male, NA not applicable, F female, NM not measured, SEM standard error of the mean, T thoracic dermatome, V trigeminal distribution(s), C cervical dermatome
Thioflavin-T assay and peptide quantification
Blood was centrifuged at 1700 RPM for 10 min, plasma collected and frozen (−80°C) until use. Amyloid (comprised of Aβ and/or other amyloidogenic peptides) were quantified by adding 5 μL of plasma (assayed in triplicate) to 195 μL of nanopure water and 75 μL of Thioflavin-T (Thio-T; 13.5 μM in 50 mM glycine; MilliporeSigma, Burlington, MA) in a black 96-well plate (Corning, Corning, NY) that was incubated for 10 min in the dark. Thio-T detects β sheets within amyloid-like fibrillar structures (prefibrillar oligomers and amyloid fibrils). Similarly, for Aβ42 and amylin spiking experiments, 10 μL of plasma and 8 μL of either Aβ42 (4 μM) or amylin (10 μM) were combined with 182 μL of nanopure water and 75 μL of Thio-T, then incubated for 24 h in the dark. Fluorescence intensity was quantified (excitation, 440 nm; emission, 490 nm). Aβ40 and Aβ42 were quantified in duplicate using the V-Plex Plus Aβ peptide panel 1 ELISA (MesoScale Diagnostics, Rockville, MD); amylin was quantified by ELISA (MilliporeSigma).
Statistical analyses
Although amyloid was normally distributed in both control and zoster groups, the data was natural log (In) transformed to retain the same scale as the amyloidogenic peptides that were not normally distributed. Means and standard deviations in the original scale of the biomarkers were calculated to aid interpretation. Exact two-sample Wilcoxon tests were used to determine statistically significant differences between amyloid, Aβ40, Aβ42 and amylin in zoster samples compared to controls, as well as for differences in fold-changes following exogenous Aβ42 and amylin applications. Due to small sample sizes, we avoided the asymptotic normality assumption in these analyses. We first determined whether the biomarker measures were distributed similarly in the 2 groups. Here, we used the exact Kolmogorov-Smirnov Test (K-S) and calculated the exact permutation-based P-value for the statistic. The result produces the maximum absolute value of the differences between two sample cumulative distribution functions. Based on results of the K-S tests, linear regression analyses with permutation-based P-values were used to determine the association of amyloid with Aβ40, Aβ42 and amylin, stratified by zoster status. We tested for differences by age and sex, as well as potential confounders. Results were considered significant at alpha = 0.05; all p-values were two-sided. Analyses were conducted in SAS 9.4 (SAS Institute INC, Cary, NC), R and Prism (GraphPad 8.1).
Results
Compared to control plasma, zoster plasma contained significantly higher levels of amyloid (p < 0.01; Fig. 1a); no significant differences were detected in levels of Aβ40, Aβ42, or amylin between zoster and control samples (Fig. 1b), as well as Aβ42/Aβ40 ratios (data not shown). Amyloid levels were significantly higher in females (p = 0.02), but no differences were seen by sex for Aβ40, Aβ42 or amylin. All raw values are presented in Table 1.
Fig. 1.
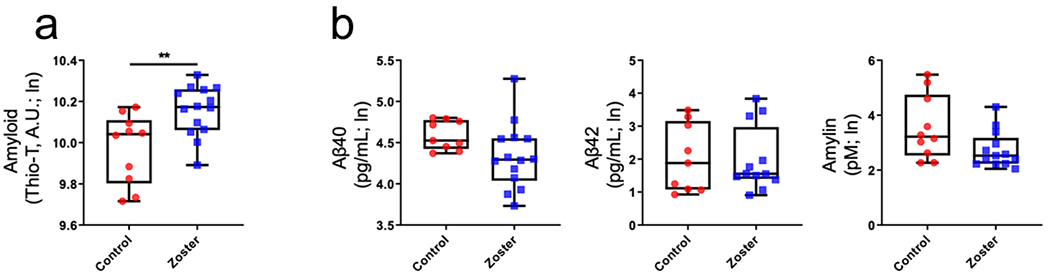
Levels of amyloid and cellular amyloidogenic peptides from plasma of zoster and control subjects. (a) Plasma amyloid levels were significantly higher in zoster compared to non-zoster, control subjects (**p < 0.01). (b) No statistically significant differences in concentrations of Aβ40, Aβ42 or amylin were detected between zoster and control subjects (p > 0.05). (In; natural log).
A cumulative distribution analysis was applied to determine if underlying processes leading to amyloid formation were different between control and zoster subjects. We found significant differences in amyloid distribution probabilities (Fig. 2a; p < 0.05; K-S test), without overlap, suggesting that different underlying mechanisms account for the increased amyloid burden seen in zoster compared to controls. The maximum distance between amyloid levels (Fig. 2a; vertical red dashed line) was ~10.05 arbitrary units (A.U.; Thio-T; amyloid), indicating ~70% of control subjects had a probability of containing at or below this amount of amyloid while only ~20% of zoster subjects contained this probability, i.e., ~80% of zoster subjects were predicted to contain more than 10.05 A.U. of amyloid while only ~30% of control subjects were predicted to have more than 10.05 A.U. of amyloid. This significant observation is remarkable to find in such a small sample size and demonstrates that the analysis is fully powered with only 24 subjects (14 zoster and 10 controls).
Fig. 2.
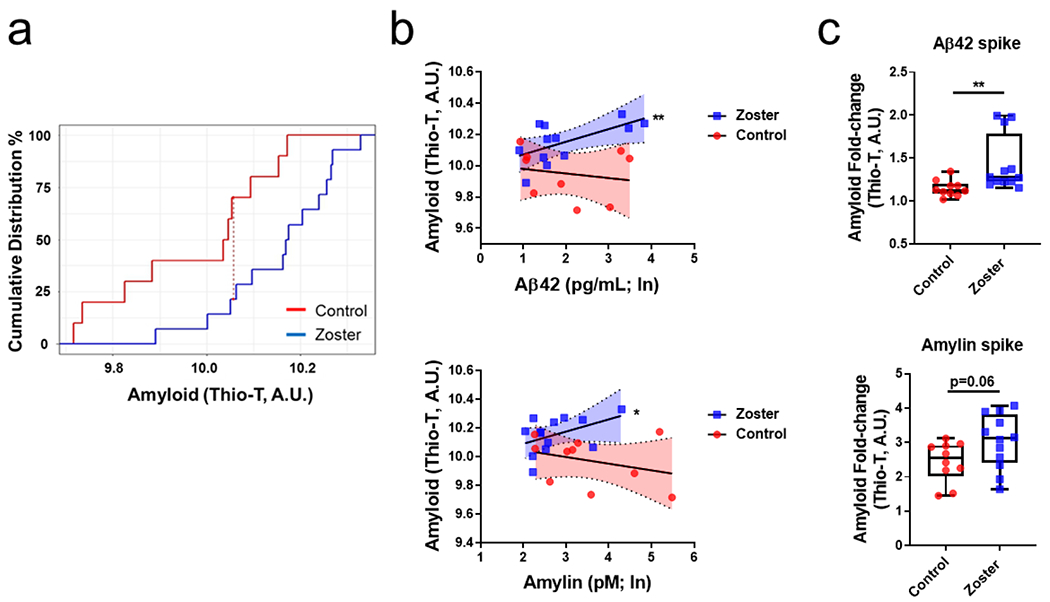
Amylin and Aβ42 were positive predictors for amyloid formation in zoster subjects. (a) Cumulative distribution analysis of amyloid between non-zoster control and zoster subjects revealed a significant increase in the distribution of amyloid probability in zoster subjects (p < 0.05; Kolmogorov-Smirnov test). The red dashed line represents the maximum distance between amyloid levels in control and zoster subjects such that at ~10.05 amyloid levels (Thio-T, A.U.), ~70% of control subjects (red line) are at or below this level of amyloid and 30% of controls have amyloid levels >10.05; in contrast, ~20% of zoster subjects (blue line) are at or below this level of amyloid and 80% of zoster subjects have amyloid levels >10.05. These results indicate that the accumulation of amyloid in the zoster group is occurring via a different mechanism than that in controls. (b) Aβ42 and amyloid were positively correlated in zoster subjects (p < 0.01) but no relationship was seen with control subjects (p > 0.05). Similarly, amylin and amyloid were positively correlated in zoster subjects (p < 0.05) but not in control subjects (p > 0.05). (In; natural log). Shaded region; 95% confidence intervals. (c) Zoster plasma had a significantly higher fold-change (spiked/unspiked; p < 0.01) in amyloid levels when spiked with Aβ42 compared to the control plasma fold-change (spiked/unspiked; top). Similarly, zoster plasma spiked with amylin had a marginally statistically significant fold-change (spiked/unspiked; p = 0.06) increase in amyloid levels compared to the control plasma fold-change (spiked/unspiked; bottom).
Due to evidence of differing distributions of amyloid, but largely unchanged Aβ peptides and amylin concentrations between the 2 groups, we investigated the relationship between amyloid and individual amyloidogenic peptides. In zoster samples, regression analyses revealed significant, positive associations between Aβ42 and amyloid (Fig. 2b, top, blue squares; p < 0.01), as well as between amylin and amyloid (Fig. 2b, bottom, blue squares; p < 0.05); in contrast, in control samples, no statistically significant relationship was observed for Aβ42 and amyloid (Fig. 2b, top, red circles; p > 0.05) or amylin and amyloid (Fig. 2b, bottom, red circles; p > 0.05). No significant relationship was seen between Aβ40 and amyloid in zoster (p > 0.05) or control (p > 0.05) subjects. Despite zoster subjects being older than controls, neither age nor sex were confounders in these associations nor were they statistically significant in the regression models.
To further test for a heightened amyloidogenic environment, plasma from control and zoster subjects were spiked with either Aβ42 (4 μM) or amylin (10 μM) and amyloid formation measured by a Thio-T fluorescence assay. Zoster plasma spiked with Aβ42 had a significantly higher fold-change (spiked/unspiked) in fluorescence intensity compared to control plasma (Fig. 2c, top; p < 0.01). Similarly, zoster plasma spiked with amylin was marginally statistically higher compared to control plasma spiked with amylin (Fig. 2c, bottom; p = 0.06).
Discussion
To date, more than 20 proteins have been shown to have the capacity to assemble into oligomers and insoluble amyloid fibrils that deposit extracellularly and have a pathogenic effect corresponding to sites of accumulation (reviewed in Benson et al. 2018). In AD, amyloid comprised of Aβ and amylin are present in senile plaques (Jackson et al. 2013); in DM, amylin forms amyloid within the islets of Langerhans (Westermark et al. 1996); and in AMD, drusen contain Aβ oligomers (Luibl et al. 2006). Amyloid formation is multifactorial and complex yet, herein, we showed that zoster plasma contains factor(s) that overall promoted amyloid fibrillization. Specifically, compared to healthy non-zoster controls, zoster subjects had significantly more amyloid in plasma; there were no significant differences in levels of Aβ40, Aβ42, Aβ42/Aβ40 ratios or amylin. Further statistical analyses revealed that in the zoster group, the amylin or Aβ42 levels were positive predictors of total amyloid levels. In contrast, in controls, there was no relationship observed between these cellular amyloidogenic peptides and amyloid. The positive correlation between Aβ42 and amyloid, as well as amylin and amyloid, present in zoster plasma, but not in control plasma, suggests that zoster plasma contains a factor(s) that may promote amyloid fibrillization of Aβ42 or amylin. Alternatively, or in addition, the zoster plasma may be lacking an inhibitory factor(s) that prevents Aβ42 or amylin from forming amyloid. These in vivo findings corroborate our in vitro study showing that direct VZV infection of primary human spinal astrocytes leads to intracellular accumulation of prefibrillar oligomers and/or amyloid fibrils detected by Thio-T and production of an extracellular amyloidogenic environment (Bubak etal. 2019).
The amyloidogenic environment seen in zoster plasma recapitulates the environment seen in AD plasma. An and colleagues (2017) found that compared to control plasma, AD plasma contained more oligomers following addition of Aβ42. Their study also demonstrates that a peripheral amyloidogenic environment can be detected in a CNS amyloid disease. We found similar results when adding exogenous Aβ42 or amylin into the plasma samples; the zoster group had an increase in amyloid compared to control samples indicating a heightened amyloidogenic environment. This was corroborated by regression analyses clearly demonstrating that as endogenous Aβ42 or amylin levels increased between zoster subjects, amyloid concentrations increased. Amyloid levels did not increase in control plasma when concentrations of these cellular amyloidogenic peptides increased or even surpassed levels measured in zoster plasma. Therefore, it was not the total concentration of cellular amyloidogenic proteins that solely determined amyloid formation; rather, there was a pro-amyloidogenic factor(s), lack of an inhibitory factor, or balance of both in zoster plasma that triggered fibrillization of Aβ42 and amylin. Precedence for such factors are evident in multiple reports, including a study describing antichymotrypsin as a pathological chaperone that binds Aβ and facilitates polymerization to amyloid filaments (Ma et al. 1994), and a study demonstrating that insulin-degrading enzyme hypofunction decreases degradation of Aβ (Farris et al. 2003).
A limitation of our study was the de-identified nature of the samples. Specifically, we had limited clinical information on the non-zoster control samples, including history of zoster or zoster vaccination, pre-diabetes/diabetes (which could account for amylin variability) and inflammatory diseases (due to autoimmunity or unscreened pathogens). Thus, it is essential in future prospective studies to examine contributions of other clinical diseases in amyloid accumulation, as well as establish a larger, well-defined non-zoster inflammatory control population.
This pilot study of acute zoster subjects opens new avenues of research. To our knowledge, this is the first study demonstrating both elevated amyloid and an amyloid-promoting peripheral environment during infection in vivo. Thus, aside from collecting additional clinical information noted above, it is essential to determine if other pathogens can produce similar results. With regards to future zoster studies, it is important to determine how long the amyloidogenic environment conferred by zoster persists since the risk of dementia and neovascular AMD is seen up to 5 years post-zoster. The composition of the circulating amyloid needs to be dissected and the amyloid-promoting and/or inhibitory factors identified. The mechanisms through which an amyloidogenic environment in the periphery confers increased risk of dementia in the CNS needs to be investigated. Finally, the ability of antiviral therapy or zoster vaccination to reduce the amyloidogenic environment remains to be determined since these therapies may attenuate risk and improve disease outcomes.
In conclusion, shared clinicohistopathological features of inflammation, vascular disease, and cell death exist between VZV infection and the 3 major amyloid-associated diseases of aging: AD, DM, and neovascular AMD. These shared features, the capacity of VZV to infect the associated organs, and the amyloidogenic peripheral environment in zoster subjects described herein, provide a potential mechanism to explain how zoster is epidemiologically associated with an increased risk of AD or neovascular AMD, or with the observation that zoster contributes to deterioration of glycemic control in DM, through acceleration of amyloid pathology. Furthermore, if other environmental factors or genetic variants contribute to increased levels of amyloid or cellular amyloidogenic peptides (e.g. decreased amyloid degradation/clearance in aging or hyperamylinemia in DM), more amyloid generated during zoster may “tip the scales” and accelerate disease progression.
Acknowledgments
Funding This study was funded by National Institutes of Health (NIH) NIA P01 AG032958; and NIH NINDS R01 NS093716 and R01 NS082228.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest The authors declare that there are no conflicts of interest.
References
- An SSA, Lee BS, Yu JS, Lim K, Kim GJ, Lee R, Kim S, Kang S, Park YH, Wang MJ, Yang YS, Youn YC, Kim S (2017) Dynamic changes of oligomeric amyloid β levels in plasma induced by spiked synthetic Aβ42. Alzheimers Res Ther 17:86. doi: 10.1186/s13195-017-0310-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, Sipe JD, Westermark P (2018) Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 25:215–219. doi: 10.1080/13506129.2018.1549825 [DOI] [PubMed] [Google Scholar]
- Bubak AN, Como CN, Coughlan CM, Johnson NR, Hassell JE Jr, Mescher T, Niemeyer CS, Mahalingam R, Cohrs RJ, Boyd TD, Potter H, Russ H, Nagel MA (2019) Varicella zoster virus infection of primary human spinal astrocytes produces intracellular amylin, amyloid-beta, and an amyloidogenic extracellular environment. J Infect Dis, pii: jiz560 doi: 1093/infdis/jiz560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VC, Wu SI, Huang KY, Yang YH, Kuo TY, Liang HY, Huang KO, Gossop M (2018) Herpes zoster and dementia: a nationwide population-based cohort study. J Clin Psychiatry 79:16m11312. doi: 10.4088/JCP.16m11312 [DOI] [PubMed] [Google Scholar]
- Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA (2008) Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 31(Suppl 2), S161–S164. doi: 10.2337/dc08-s243 [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100:4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon M, Gershon A (2018) Varicella-zoster virus and the enteric nervous system. J Infect Dis 218 (Suppl 2):113–119. doi: 10.1093/infdis/jiy407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilden D, Nagel MA, Cohrs RJ, Mahalingam R (2013) The variegate neurological manifestations of varicella zoster virus infection. Curr Neurol Neurosci Rep 13:374. doi: 10.1007/s11910-013-0374-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JD, Lin HC, Kao LT (2019) Increased risk of neovascular age-related macular degeneration in patients with herpes zoster ophthalmicus: a retrospective cohort study. Acta Ophthalmol 97:e321–e322. doi: 10.1111/aos.13924 [DOI] [PubMed] [Google Scholar]
- Höppener JW, Ahrén B, Lips CJ (2000) Islet amyloid and type 2 diabetes mellitus. N Engl J Med 343:411–419. [DOI] [PubMed] [Google Scholar]
- Jackson K, Barisone GA, Diaz E, Jin LW, DeCarli C, Despa F (2013) Amylin deposition in the brain: a second amyloid in Alzheimer disease? Ann Neurol 74:517–526. doi: 10.1002/ana.23956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai K, Yawn BP (2017) Risk factors for herpes zoster: a systematic review and meta-analysis. Mayo Clin Proc 92:1806–1821. doi: 10.1016/j.mayocp.2017.10.009 [DOI] [PubMed] [Google Scholar]
- Kennedy PG, Grinfeld E, Gow JW (1998) Latent varicella-zoster virus is located predominantly in neurons in human trigeminal ganglia. Proc Natl Acad Sci U S A 95:4658–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PG, Grinfeld E, Gow JW (1999) Latent varicella-zoster virus in human dorsal root ganglia. Virology 258:451–454. [DOI] [PubMed] [Google Scholar]
- Luibl V, Isas JM, Kayed R, Glabe CG, Langen R, Chen J (2006) Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J Clin Invest 116:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer HB Jr, Das S, Potter H (1994) Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 372:92–94. [DOI] [PubMed] [Google Scholar]
- Mahalingam R, Wellish M, Wolf W, Dueland AN, Cohrs R, Vafai A, Gilden D (1990) Latent varicella-zoster viral DNA in human trigeminal and thoracic ganglia. N Engl J Med 323:627–631. [DOI] [PubMed] [Google Scholar]
- Munoz-Quiles C, López-Lacort M, Ampudia-Blasco FJ, Diez-Domingo J (2017) Risk and impact of herpes zoster on patients with diabetes: A population-based study, 2009-2014. Hum Vaccin Immunother 13:2606–2611. doi: 10.1080/21645515.2017.1368600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel MA, Rempel A, Huntington J, Kim F, Choe A, Gilden D (2014) Frequency and abundance of alphaherpesvirus DNA in human thoracic sympathetic ganglia. J Virol 88:8189–8192. doi: 10.1128/JVI.01070-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper NC, Nabuurs-Franssen MH, Huijberts MS (2000) Peripheral vascular disease and type 2 diabetes mellitus. Diabetes Metab Res Rev 16 (Suppl 1), S11–S15. [DOI] [PubMed] [Google Scholar]
- Tran TH, Rozenberg F, Cassoux N, Rao NA, LeHoang P, Bodaghi B (2003) Polymerase chain reaction analysis of aqueous humour samples in necrotizing retinitis. Br J Ophthalmol 87:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Cheng WL, Sheu JJ, Huang CC, Shia BC, Kao LT, Lin HC (2017) Increased risk of dementia following herpes zoster ophthalmicus. PLoS One 12:e0188490. doi: 10.1371/journal.pone.0188490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ye J, Han YH (2014) Acute pancreatitis associated with herpes zoster: case report and literature review. World J Gastroenterol 20:18053–18056. doi: 10.3748/wjg.v20.i47.18053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P, Li ZC, Westermark GT, Leckstrom A, Steiner DF (1996) Effects of beta cell granule components on human islet amyloid polypeptide fibril formation. FEBS Lett 379:203–206. [DOI] [PubMed] [Google Scholar]