

Abstract
Germline pathogenic variants in CBL are associated with an autosomal dominant RASopathy and an increased risk for malignancies, particularly juvenile myelomonocytic leukemia. Herein, we describe a patient with clinical features of a Noonan-spectrum disorder who developed embryonal rhabdomyosarcoma of the bladder at age two years. Tumor analysis using the OncoKids® cancer panel revealed a CBL pathogenic variant: NM_005188.3:c.1100A>C (p.Gln367Pro). Sanger sequencing of peripheral blood DNA confirmed a de novo heterozygous germline variant. This is the first report of embryonal rhabdomyosarcoma in association with a germline CBL pathogenic variant, further broadening the CBL cancer predisposition spectrum.
Keywords: CBL, Cancer predisposition, RASopathy, Embryonal rhabdomyosarcoma
Introduction
CBL is a proto-oncogene encoding a RING finger E3 ubiquitin ligase , which primarily functions as a negative regulator of several receptor protein tyrosine kinase signaling transduction pathways, including the RAS-MAPK pathway, by targeting receptors for degradation [1]. Somatic mutations in CBL have primarily been identified in myeloid malignancies, especially juvenile myelomonocytic leukemia (JMML) [2,3], but not in embryonal rhabdomyosarcoma (ERMS). Germline pathogenic variants in CBL are associated with autosomal dominant RASopathy, a Noonan syndrome-like disorder with or without JMML [MIM: 613563] [4] that shows considerable phenotypic variation [5]. Patients display craniofacial features overlapping Noonan syndrome such as cardiac disease, ectodermal and musculoskeletal anomalies, and have variable cognitive deficits. Several RASopathies, including neurofibromatosis type 1, Costello syndrome, and Noonan syndrome, are considered cancer predisposition syndromes, with their associated mutations in NF1, HRAS, and PTPN11, resulting in enhanced RAS-MAPK pathway activation or dysregulated signaling [6]. Depending on the gene mutated, patients may have an increased risk for rhabdomyosarcoma, neuroblastoma or leukemia. Patients with germline CBL pathogenic variants have an increased risk for certain malignancies, particularly JMML. Although recent case reports suggest that CBL germline mutations may also predispose patients to teratoma, acute myelogenous leukemia and glioma [7–9], to date, germline CBL mutations have not been reported in patients with ERMS.
Little is known regarding the underlying genetic susceptibility to rhabdomyosarcoma; most cases appear to be sporadic. Genetic alterations in TP53 [10], DICER1 [11], NF1 [12], HRAS [13], sonic hedgehog pathway genes SUFU and PTCH1 [14], as well as imprinted genes IGF2, H19, and CDKN1C within the chromosome 11p15.5 region [15] have been reported in patients with rhabdomyosarcoma. RAS-MAPK pathway somatic mutations in NRAS, KRAS, HRAS, and NF1 have also been observed in ERMS, and are reported at higher frequencies in high-risk patients [16]. Here we report the first known patient with a germline CBL pathogenic variant and ERMS.
Case description
A 2-year old Hispanic male presented with an enlarging left sided lobulated intra-abdominal mass. Histology and immunostaining profiles of a biopsy from the abdominal mass were consistent with ERMS (Fig. 1). The patient was classified as intermediate risk, IRS Stage 3, Group II. He was treated with standard rhabdomyosarcoma therapy including systemic chemotherapy, surgery and radiation at the primary tumor site as per the Children’s Oncology Group Study D9803 for patients with intermediate risk rhabdomyosarcoma. The child’s past medical history was significant for developmental delay, a small atrial septal defect secundum, low-lying conus, splenomegaly, pancytopenia, and bilateral cryptorchidism. A subsequent dysmorphology examination documented down-slanting palpebral fissures, ptosis, low-set ears, five café au lait spots and widely-spaced nipples. The dysmorphic facial features, together with his congenital heart defect, cryptorchidism, café au lait spots, and widely-spaced nipples were suggestive of a Noonan-spectrum disorder.
Fig. 1.
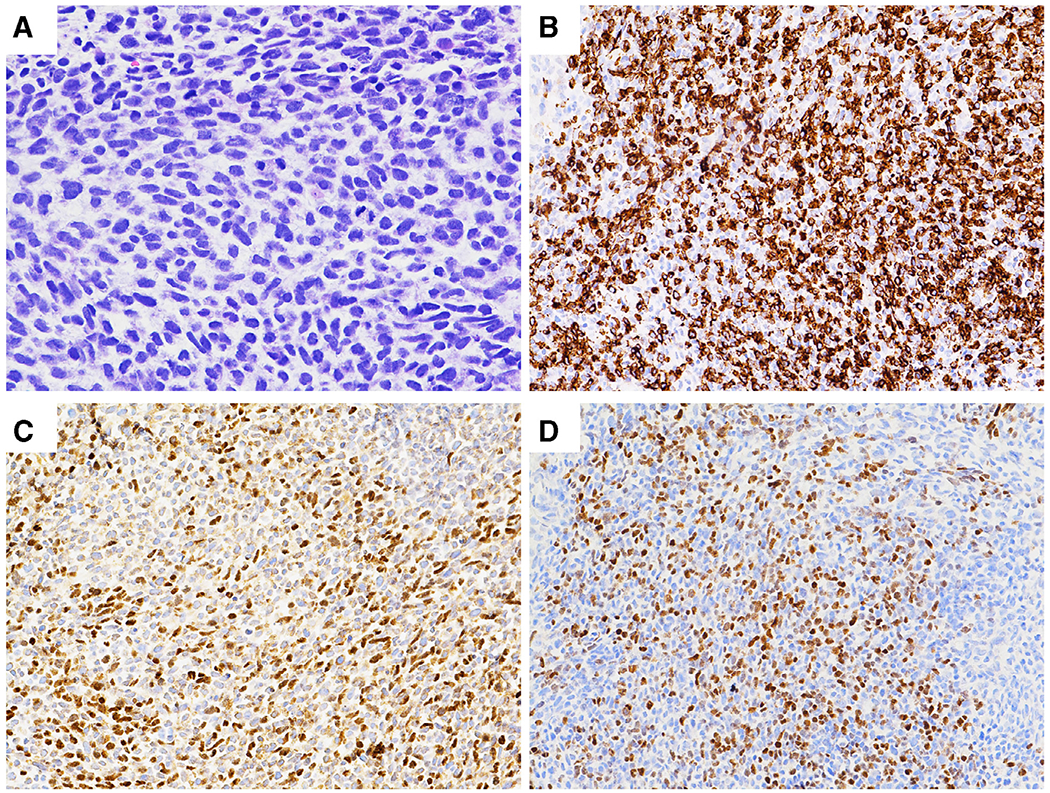
Histology and immunostaining results of the patient’s abdominal mass. (A). H&E: A hypercellular tumor with spindle shaped, hyperchromatic and pleomorphic nuclei, coarse chromatin, inconspicuous nucleoli and tapering eosinophilic cytoplasm. Frequent mitoses and karyorrhexis are present. (B). Desmin: Tumor cells were strongly and diffusely positive (cytoplasmic stain). (C). Myo-D: Approximately 35–40% of tumor cells were positive (nuclear stain). (D). Myogenin: Approximately 20% of tumor cells were positive (nuclear stain).
Materials and methods
DNA was extracted from frozen tumor tissue with the Qiagen Gentra Puregene Tissue Kit (Qiagen Inc., Valencia, CA). Chromosomal microarray analysis (CMA) was performed with the Affymetrix OncoScan platform following validated standard protocols (Thermo Fisher, Santa Clara, CA). CMA was performed on a peripheral blood sample from the patient using the Affymetrix CytoScan HD platform (Thermo Fisher, Santa Clara, CA).
RNA was extracted from frozen tumor with the Qiagen RNeasy Mini RNA Extraction Kit (Qiagen Inc. Valencia, CA) and DNA and RNA analysis was performed using the OncoKids® panel, a comprehensive next-generation sequencing panel for pediatric malignancies [17]. Sequence alterations were reported according to CAP/AMP/ASCO guidelines [18].
To determine whether the CBL variant was germline in origin, Sanger sequencing was performed on peripheral blood DNA from the patient and, subsequently, from both parents. Forward and reverse primer sequences used for CBL exon 8 (NM_005188.3) were 5′-ATGTGGTTTCACTTTAAACCCTG-3′ and 5′-TATCAGTAAAGGCTATATAATAC-3′, respectively.
Results
The pathologic findings from the patient’s tumor biopsy are shown in Fig. 1.
OncoKids® analysis of the tumor DNA revealed a missense NM_005188.3: c.1100A > C (p.Gln367Pro) variant of strong clinical significance in the CBL gene at a variant allele fraction of 98% (tumor percentage > 95%). In addition, variants of unknown clinical significance were detected in KMT2D (NM_003482.3: c.2204G > T (p.Cys735Phe) and RUNX1 (NM_001754.4: c.824C > T (p.Pro275Leu)) (Supplemental Table 1). There were no other clinically significant sequence alterations, gene amplification or RNA fusions detected.
CMA of the tumor demonstrated copy number alterations and regions of loss of heterozygosity (LOH) consistent with ERMS (Fig. 2A), including gains of most of chromosomes 2, 7, 8 (5 copies), 11, 12, 13, 14 and 19. A biallelic deletion of CDKN2A and CDKN2B in 9p21.3 was present. There were four copies of most of the short arm of chromosome 11; however, there was also LOH for the entire chromosome 11 (Supplemental Table 2), which included the CBL gene. This LOH accounted for the high variant allele frequency detected in CBL with OncoKids®. CMA of the patient’s peripheral blood DNA showed no evidence for LOH of chromosome 11, confirming that this was a somatic event.
Fig. 2.
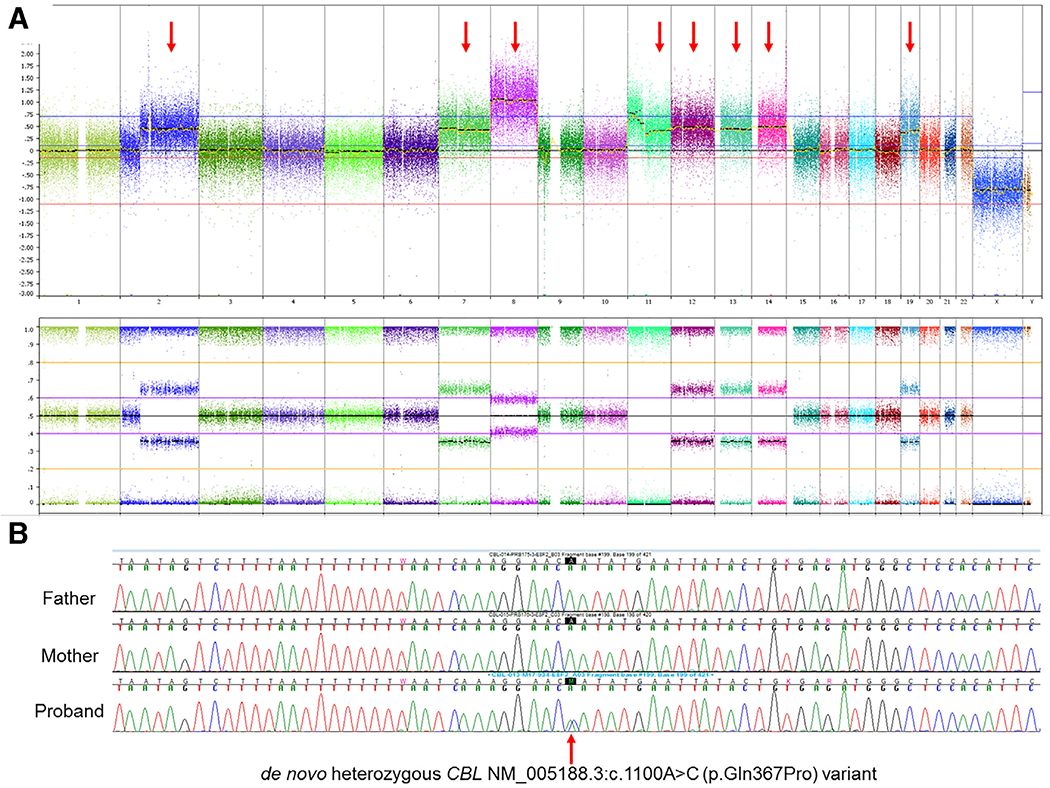
A. Chromosomal microarray whole genome view of the tumor. Copy number alterations and regions of loss of heterozygosity of this tumor sample were consistent with the clinical diagnosis of embryonal rhabdomyosarcoma. These included gains of most of chromosomes 2, 7, 8 (5 copies), 11,12,13,14 and 19, as shown by red arrows. There was also a homozygous deletion of CDKN2A/B in 9p21.3. LOH was observed for chromosome 11, including the CBL gene locus. Fig. 2B Follow-up targeted Sanger sequencing of the peripheral blood samples for the CBL variant initially identified in the tumor sample. The patient is heterozygous for the NM_005188.3:c.1100A > C (p.Gln367Pro) variant in the CBL gene. This patient’s biologic parents do not harbor the CBL variant, confirming that it is a de novo germline variant.
Targeted Sanger sequencing analysis of peripheral blood samples from the patient and his parents demonstrated the presence of the c.1100A > C (p.Gln367Pro) variant, identical to the tumor sample (Fig. 2B). Absence of the variant in either parent confirmed that it was de novo in the patient’s germline. This particular variant has been reported previously in association with an autosomal dominant Noonan syndrome-like phenotype [MIM: 613563] [4,7,19], and is consistent with the clinical RASopathy features observed in this patient. Additionally, the variant is absent from the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/). This substitution occurs at a highly conserved position in the linker region of CBL, which connects the RING finger domain and TKB domain. Both germline and somatic mutations have been described in this region in patients with hematological malignancies. There is a moderate physicochemical difference between glutamic acid (Gln) and proline (Pro) (Grantham dist.: 76 [0–215]). Multiple in silico algorithms (Align GVGD, SIFT, Polyphen2, MutationTaster) all suggest that the variant may be damaging to CBL protein structure and function. Based on the published literature, its de novo nature, rarity, computational predictions, and the patient’s clinical presentation, we considered this variant to be pathogenic for a CBL-related RASopathy.
The other detected variants, KMT2D p.Cys735Phe and RUNX1 p.Pro275Leu, have not been reported as somatic alterations in the COSMIC database, but have been observed in the general population at low frequencies (minor allele frequency < 0.01%). The significance of these variants, if any, is uncertain due to the overall paucity of clinical and functional evidence.
Our patient’s cancer diagnosis preceded the molecular diagnosis of his constitutional RASopathy. The finding of the germline pathogenic CBL variant did not change the risk assessment or therapy for this patient. He was treated as per the Children’s Oncology Group Study D9803 for patients with intermediate risk rhabdomyosarcoma and has responded well to therapy.
Discussion
We report a patient with clinical features of a RASopathy, and a de novo germline CBL variant who presented with ERMS. The CBL heterozygous variant was present at a variant allele fraction of 98% in our patient’s ERMS sample due to the acquired LOH of chromosome 11 (Fig. 2A). Interestingly, paternal uniparental disomy of 11p is a hallmark of syndromic and sporadic ERMS [15,20]. The chromosome 11 LOH that includes 11p15 may have been a secondary alteration relevant to the development of the ERMS in this patient. Molecular analysis of solid tumors in other patients with germline CBL mutations may help to clarify this.
While considerable phenotypic variability is reported with germline CBL pathogenic variants, the clinical presentation of this patient was consistent with previous reports [4,5]. The detailed comparison of clinical features for a total of five patients who carry the same germline CBL c.1100A > C (p.Gln367Pro) variant are listed in Supplemental Table 3. Among these patients, one developed JMML at age 3 weeks and another developed an ovarian mixed germ cell tumor at age 12.5 years [7,19]. It is known that patients with germline mutations in PTPN11, KRAS and NF1, and in CBL, are at increased risk for malignancies, particularly JMML [4]. A causal contribution of c-Cbl to the onset of myeloid malignancies has been supported by previous mouse model studies [2]. Solid tumors, including rhabdomyosarcoma, are described in patients with germline mutations in RAS-MAPK pathway genes [21,22]. Many of these genes, including CBL are widely expressed in most tissues in humans. Somatic or germline mutations in these RAS-MAPK pathway genes have been observed in ERMS, although infrequently [21,22]. Their presence does however suggest that alterations in these genes could contribute to the development of ERMS. Our current case further supports this and that specifically, CBL mutations may be involved in the pathogenesis of ERMS.
This is the first report of ERMS in a patient with a germline pathogenic variant in CBL, further broadening the CBL cancer predisposition spectrum. While CBL associated cancer risks besides JMML have not yet been defined, systematic surveillance for solid malignancies may be warranted in addition to monitoring patients for a hematologic disorder.
Supplementary Material
Acknowledgment
We thank Jennifer Han, Dolores Estrine, Cindy Fong, and Vandana Mehta for their technical assistance.
Footnotes
Conflict of interest
None declared.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cancergen.2018.12.006.
References
- [1].Joazeiro CA , Wing SS , Huang H , Leverson JD , Hunter T, Liu YC . The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 1999;286:309–12 [DOI] [PubMed] [Google Scholar]
- [2].Katzav S, Schmitz ML. Mutations of c-Cbl in myeloid malignancies. Oncotarget 2015;6:10689–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Niemeyer CM , Kang MW, Shin DH , Furlan I , Erlacher M , Bunin NJ , Bunda S , Finklestein JZ, Gorr TA , Mehta P , Schmid I, Kropshofer G, Corbacioglu S, Lang PJ , Klein C, Schlegel PG , Heinzmann A, Schneider M , Stary J, van den Heuvel-Eibrink MM, Hasle H , Locatelli F , Sakai D , Archambeault S , Chen L , Russell RC , Sybingco SS , Ohh M , Braun BS , Flotho C , Loh ML , Germline CBL mutations cause developmental abnormalities and predispose to j uvenile myelomonocytic l eukemia. Nat Genet 2010;42:794–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Martinelli S , De Luca A , Stellacci E , Rossi C , Checquolo S , Lepri F , Caputo V , Silvano M , Buscherini F , Consoli F , Ferrara G , Digilio MC , Cavaliere ML , van Hagen JM , Zampino G , van der Burgt I , Ferrero GB , Mazzanti L , Screpanti I , Yntema HG , Nillesen WM , Savarirayan R , Zenker M , Dallapiccola B , Gelb BD , Tartaglia M , Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet 2010;87:250–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martinelli S, Stellacci E, Pannone L, D’Agostino D, Consoli F, Lissewski C, Silvano M, Cencelli G, Lepri F, Maitz S, Pauli S, Rauch A, Zampino G, Selicorni A, Melancon S, Digilio MC, Gelb BD, De Luca A, Dallapiccola B, Zenker M, Tartaglia M. Molecular diversity and associated phenotypic spectrum of germline CBL mutations. Hum Mutat 2015;36:787–96 [DOI] [PubMed] [Google Scholar]
- [6].Villani A, Greer MC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW, Pfister SM, Walsh MF, Wasserman JD, Zelley K, Kratz CP, Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res 2017;23:e83–90 [DOI] [PubMed] [Google Scholar]
- [7].Hanson HL, Wilson MJ, Short JP, Chioza BA, Crosby AH, Nash RM, Marks KJ, Mansour S, Germline CBL mutation associated with a noonan-like syndrome with primary lymphedema and teratoma associated with acquired uniparental isodisomy of chromosome 11q23. Am J Med Genet A 2014;164A:1003–9 [DOI] [PubMed] [Google Scholar]
- [8].Becker H, Yoshida K, Blagitko-Dorfs N, Claus R, Pantic M, Abdelkarim M, Niemoller C, Greil C, Hackanson B, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Dohner K, Schnittger S, Henneke P, Niemeyer CM, Flotho C, Pfeifer D , Ogawa S, Lubbert M, Tracing the development of acute myeloid leukemia in CBL syndrome. Blood 2014;123:1883–6 [DOI] [PubMed] [Google Scholar]
- [9].Seong MW, Ka SH, Park JH, Park JH, Yoo HM, Yang SW, Park JM, Park D, Lee ST, Seol JH, Chung CH. Deleterious c-Cbl Exon skipping contributes to human Glioma. Neoplasia 2015;17:518–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hettmer S , Archer NM , Somers GR , Novokmet A , Wagers AJ , Diller L, Rodriguez-Galindo C, Teot LA, Malkin D. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer 2014;120:1068–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Doros L, Yang J , Dehner L , Rossi CT , Skiver K , Jarzembowski JA, Messinger Y, Schultz KA, Williams G, Andre N, Hill DA, DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer 2012;59:558–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crucis A, Richer W, Brugieres L, Bergeron C, Marie-Cardine A, Stephan JL, Girard P, Corradini N, Munzer M, Lacour B, Minard-Colin V, Sarnacki S, Ranchere-Vince D, Orbach D, Bourdeaut F, Rhabdomyosarcomas in children with neurofibromatosis type I: a national historical cohort. Pediatr Blood Cancer 2015;62:1733–8 [DOI] [PubMed] [Google Scholar]
- [13].Menke J, Pauli S, Sigler M, Kuhnle I, Shoukier M, Zoll B, Ganster C, Salinas-Riester G, Schaefer IM, Uniparental trisomy of a mutated HRAS proto-oncogene in embryonal rhabdomyosarcoma of a patient with costello syndrome. J Clin Oncol 2015;33:e62–5 [DOI] [PubMed] [Google Scholar]
- [14].Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol 2006;208:17–25 [DOI] [PubMed] [Google Scholar]
- [15].Gripp KW, Robbins KM, Sheffield BS, Lee AF, Patel MS, Yip S, Doyle D, Stabley D, Sol-Church K. Paternal uniparental disomy 11p15.5 in the pancreatic nodule of an infant with Costello syndrome: shared mechanism for hyperinsulinemic hypoglycemia in neonates with Costello and Beckwith-Wiedemann syndrome and somatic loss of heterozygosity in Costello syndrome driving clonal expansion. Am J Med Genet A 2016;170:559–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, Wu G, Bradley C, McEvoy J, Pappo A, Spunt S, Valentine MB, Valentine V, Krafcik F, Lang WH, Wierdl M, Tsurkan L, Tolleman V, Federico SM, Morton C, Lu C, Ding L, Easton J, Rusch M, Nagahawatte P, Wang J, Parker M, Wei L, Hedlund E, Finkelstein D, Edmonson M, Shurtleff S, Boggs K, Mulder H, Yergeau D, Skapek S, Hawkins DS, Ramirez N, Potter PM, Sandoval JA, Davidoff AM, Mardis ER, Wilson RK, Zhang J, Downing JR, Dyer MA, St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome P. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 2013;24:710–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hiemenz MC, Ostrow DG, Busse TM, Buckley J, Maglinte DT, Bootwalla M, Done J, Ji J, Raca G, Ryutov A, Xu X, Zhen CJ, Conroy JM, Hazard FK, Deignan JL, Rogers B, Treece AL, Parham DM, Gai X, Judkins AR, Triche TJ, Biegel JA, OncoKids(SM): a comprehensive next-generation sequencing panel for pediatric malignancies. J Mol Diagn 2018 [DOI] [PubMed] [Google Scholar]
- [18].Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, Nikiforova MN, Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19:4–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bulow L, Lissewski C, Bressel R, Rauch A, Stark Z, Zenker M, Bartsch O, Hydrops, fetal pleural effusions and chylothorax in three patients with CBL mutations. Am J Med Genet A 2015;167A:394–9 [DOI] [PubMed] [Google Scholar]
- [20].Robbins KM, Stabley DL, Holbrook J, Sahraoui R, Sadreameli A, Conard K, Baker L, Gripp KW, Sol-Church K. Paternal uniparental disomy with segmental loss of heterozygosity of chromosome 11 are hallmark characteristics of syndromic and sporadic embryonal rhabdomyosarcoma. Am J Med Genet A 2016;170:3197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Denayer E, Devriendt K, de Ravel T, Van Buggenhout G, Smeets E, Francois I, Sznajer Y, Craen M, Leventopoulos G, Mutesa L, Vandecasseye W, Massa G, Kayserili H, Sciot R, Fryns JP, Legius E, Tumor spectrum in children with Noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer 2010;49:242–52 [DOI] [PubMed] [Google Scholar]
- [22].Jongmans MC, Hoogerbrugge PM, Hilkens L, Flucke U, van der Burgt I, Noordam K, Ruiterkamp-Versteeg M, Yntema HG, Nillesen WM, Ligtenberg MJ, van Kessel AG, Kuiper RP, Hoogerbrugge N. Noonan syndrome, the SOS1 gene and embryonal rhabdomyosarcoma. Genes Chromosomes Cancer 2010;49:635–41 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.