

Abstract
With the improving survival of cystic fibrosis (CF) patients and the advent of highly effective cystic fibrosis transmembrane conductance regulator (CFTR) therapy, the clinical spectrum of this complex multi-system disease continues to evolve. One of the most important clinical events for patients with CF in the course of this disease is an acute pulmonary exacerbation. Clinical and microbial epidemiology studies of CF pulmonary exacerbations continue to provide important insight into the disease course, prognosis, and complications. This work has now led to a number of large scale clinical trials with the goal of improving the treatment paradigm for CF pulmonary exacerbation. The primary goal of this review is to provide a summary of the pathophysiology, the clinical epidemiology, microbial epidemiology, outcome and treatment of CF pulmonary exacerbation.
Keywords: FEV1, respiratory symptoms, pulmonary exacerbation, cystic fibrosis, treatment duration
Cystic fibrosis (CF) is the most common life shortening inherited disease in Caucasians and affects approximately 30,000 individuals in the U.S.1 Advances in care for individuals with CF have resulted in dramatic improvements in survival, but people with CF still have debilitating symptoms and die far too early.2,3 Acute pulmonary exacerbations are frequent and central events in the lives of individuals with CF. They may result in permanent loss of lung function, worse quality of life, and shortened survival.4–8 They are typically treated with multiple modalities including the use of antibiotics, given in various combinations of systemic and inhaled routes9. In this review, I will summarize what is known about the pathophysiology of exacerbations, the various definitions, clinical epidemiology and treatment approaches of PEx in CF.
Pathophysiology of CF Pulmonary Exacerbation
Despite our detailed understanding of CF at a cellular level, very little is known about the pathophysiology of recurrent episodes of increasing pulmonary symptoms termed exacerbations. Exacerbations of patients’ pulmonary disease are very common and present clinically with changes in cough, sputum production, dyspnea, decreased energy level and appetite, weight loss, and decreases in spirometry. These episodes are likely related to a complex relationship between host defense and airway microbiology that impact sputum production and airflow obstruction. Viral infections, including respiratory syncytia virus (RSV), may play a role in the initiation of these events10 although data regarding the impact of vaccination against viral infection is limited.11,12 In ecologic population studies, a portion of pulmonary exacerbations have been attributable to viral infections like RSV and influenza.13,14 Pulmonary exacerbations have also been associated with the acquisition of new organisms or with a change in the bacterial density of colonizing flora.15,16 Bacterial concentrations of Pseudomonas aeruginosa are high during an exacerbation and decrease with treatment; and treatment with antimicrobial agents reduces symptoms and improves lung function.15–17 More recent data suggest that the majority of exacerbations are not due to acquisition of new strains of pathogen (i.e. Pseudomonas), but potentially due to a clonal expansion of existing strains.18 Aaron and colleagues noted that among 80 individuals followed for 2 years with quarterly sputum cultures, 40 patients experienced a pulmonary exacerbation.18 Only 36 had isolates that could be genotyped and among those, only two subjects demonstrated acquisition during exacerbation of a new clone that had not been present during a period of clinical stability. Inflammatory response in the airway in conjunction with the increase in bacterial concentration and polymorphonucleocytes has been documented with increases in IL-8, IL-6, IL1β, TNF-α, leukotriene B4 (LBT4), and free neutrophil elastase; these inflammatory mediators have been noted to decrease with treatment of the pulmonary exacerbation.19–24 Treatment of inflammation in randomized controlled trials and observational studies have had variable impact on clinical outcomes, both positive and negative (lung function and exacerbation rates).25–30
Definitions/diagnosis
Despite calls for a consensus diagnosis of pulmonary exacerbations by a CF Outcomes Group in 1994, no consensus diagnostic criteria exist.31 A prior review highlighted prior definitions employed32 – to date most clinical trials have employed the definition developed for the rhDNase trials (Fuchs definition with or with treatment required).33 Other diagnostic tools include the Acute Respiratory Illness Checklist (ARIC)12 and the Respiratory and Systemic Symptoms Questionnaire (RSSQ©) used in a large scale phase 2 CF clinical trial for an LBT4 inhibitor34 but neither has been widely adopted. No definition has been formally validated. Definitions generally combine patient reported symptomatology, laboratory data (particularly spirometry) and a clinician based evaluation of the patient with the addition of a physician decision to treat the event. Components of these definitions have been examined to see which clinical characteristics best predict a pulmonary exacerbation.35–37 Symptoms rather than physical examination and laboratory values were found to be more predictive of a pulmonary exacerbation in these studies. The signs and symptoms that were most predictive of a pulmonary exacerbation in all of these studies were increased cough, change in sputum (volume or consistency), decreased appetite or decreased weight, change in respiratory exam and respiratory rate.38 Given the lack of a consensus definition of these events, it is not surprising that patterns of treatment of pulmonary exacerbation continue to be quite variable.39
These events have a broad spectrum of clinical presentation from cough without change in lung function to severe drops in lung function inducing hypoventilation, hypoxemia and potentially respiratory failure. Like the overall definition, no universally accepted gradation of exacerbation exists. What is clear is that severe events – those that could get a patient admitted to the intensive care unit, are associated with a high one year mortality (upwards of 50%).40,41 Multivariate predictors of mortality from such an event include annual decline in FEV1, simplified acute physiology score II, and the use of invasive mechanical ventilation. ICU mortality at single centers have been lower (32–55%).42,43 Improved outcomes have been recently noted using non-invasive oxygen and bi-level ventilation.44 More recent data suggests that the mortality for CF patients requiring mechanical ventilation remains high but is decreasing.45
CF Pulmonary Exacerbation as a predictor and outcome variable
The annual rate of CF pulmonary exacerbation has clearly been associated with 2 year and 5 year survival in two separate prediction models evaluating the odds of death during follow-up and recent models from the French and Canadian/UK registries.4,5,46–50 In a recent publication, the addition of pulmonary exacerbation in CF has added substantively to the lung allocation score in CF patients being listed for lung transplant in the US.51 CF pulmonary exacerbations requiring intravenous antibiotics have also been associated with later diminished lung function in children ages 1 to 6 years6, with CF related diabetes52, with sleep disturbances and health related quality of life.7,8 Lastly, pulmonary exacerbation rate has been an important marker of disease severity and as such has been used as an adjustment variable in studies looking at survival,53,54 a variable to consider as an inclusion criteria55 and an important outcome measure when assessing the impact of socioeconomic status and environmental exposure on CF.56,57 PEx rate has also been used as an important variable to assess novel outcome measures like high resolution computed tomography of the chest or cough frequency.58,59 Last, PEx has been central to drug agency approval of a number of key therapies.17,33,60–62
Epidemiology of CF exacerbation
The incidence of PEx appears to be relatively constant over the life of a CF patient, but antibiotic treatments change as patient airway infections become more complex and lung disease advances.63 In adolescents and adults, the proportions of PEx that are treated with intravenous (IV) antibiotics steadily increases.63,64 Several studies have shown that in approximately 25% of exacerbations, patients do not return to within 90% of their baseline lung function following treatment for the exacerbation (Figure 1).65,66 One factor associated with poor response to exacerbation treatment may be longer time from symptom onset to exacerbation treatment, suggesting that delayed treatment results in worse treatment outcomes. There is also evidence suggesting that CF centers that see patients more frequently and treat patients more aggressively, e.g. more antibiotic use, have better clinical outcomes.39,67 PEx are also associated with more rapid lung function decline, poorer quality of life, and increased healthcare costs.68
Figure 1.
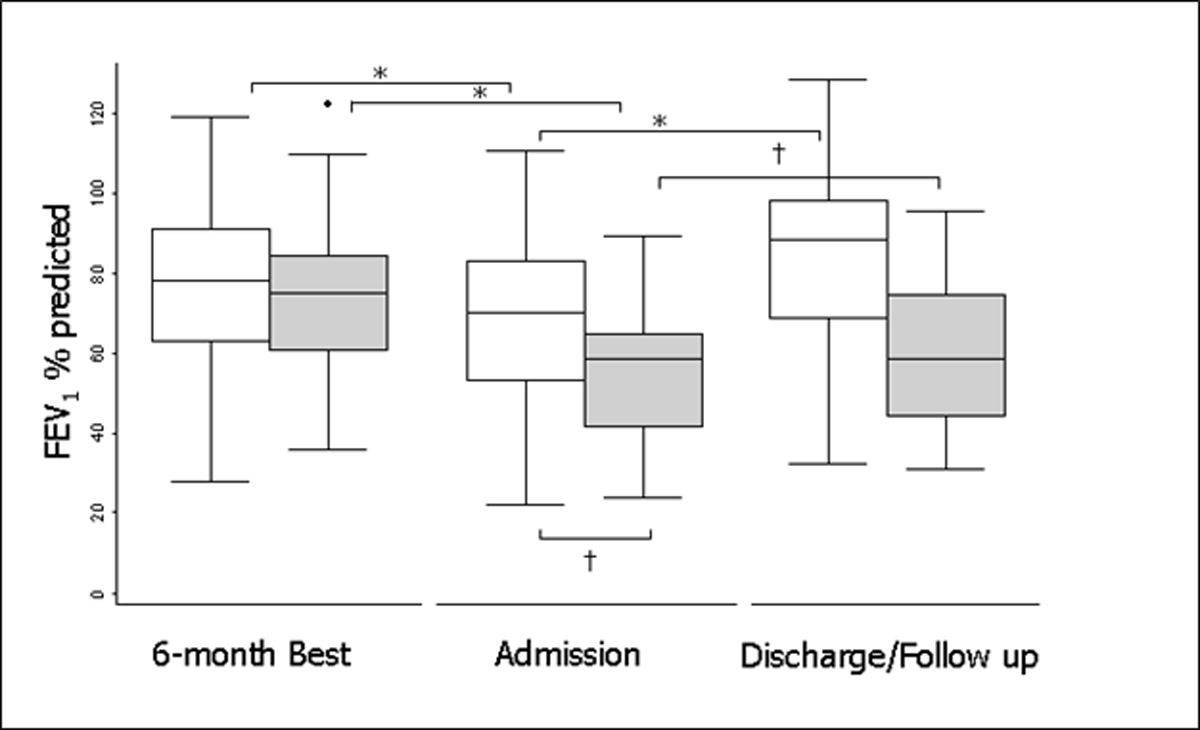
FEV1 for nonresponders and responders at baseline, treatment initiation, and the best FEV1 in the 3 months after treatment (n = 4,391). Responders are in white, nonresponders are in gray. The boxes represent the middle 50% of patients; the whiskers include all patients in each group. The horizontal line within the box represents the median FEV1. The x axis represents three time points: the best FEV1 in the 6 months before admission (baseline); FEV1 at treatment initiation (exacerbation); and the best FEV1 in the 3 months after treatment (follow-up). Reprinted with permission from the AJRCCM. * represent P<0.05
Pulmonary exacerbation in small children
PEx’s are also meaningful events in young children.69 Frequent PEx’s, especially in the first two years of life are associated with decreased spirometry (FEV1) at age 5; PEx’s treated with IV antibiotics in small children are associated with the presence of bronchiectasis on chest CT and decreased weight-for-age at age 5. The challenge in CF infants and toddlers with exacerbations is that the frequency of PEx’s is very similar to the frequency of viral upper respiratory tract infections that occur in healthy children without CF.70 Children with CF are more likely to have prolonged viral infections and of greater severity, so these event are intrinsically linked.71 The presence of rhinovirus (RV)72 and respiratory syncytial virus (RSV)73 may enable Pseudomonas to more easily infect airway epithelial cells from patients with CF. Preventing exacerbations early in life has become paramount. Of concern, the recent Infant Study of Inhaled Saline in Cystic Fibrosis study (ISIS) found that inhaled hypertonic saline failed to decrease the rate of PEx’s in children ages 4–60 months with CF.74 Importantly, a subset of patients performed infant pulmonary function testing, and patients who received inhaled hypertonic saline had a significantly larger mean improvement in forced expiratory volume in 0.5 seconds suggesting exacerbation may not be an ideal endpoint in pediatric studies.74 A recent study demonstrated hope at decreasing PEx’s in small children. Early introduction of azithromycin when added to inhaled tobramycin as part of a Pseudomonal eradication program did decrease PEx’s; the challenge with this study is that it was stopped early for efficacy and thus was underpowered to detect a change in Pseudomonas aeruginosa recurrence rate compared to placebo.75 Although this study met its key endpoint – PEx reduction, it left unanswered this key microbiologic question.
The CF community has been evaluating newer outcome measures to identify progression of lung disease given that PEx as an endpoint may not be sensitive enough. Serial chest CT scans have demonstrated structural lung disease, such as bronchiectasis, is persistent and progressive even in young children with CF.76 The AREST-CF study suggests that at least some very young patients will have bronchiectasis very early.77 Additionally, the lung clearance index (LCI), an outcome measure using the multiple-breath washout (MBW) method has been shown to be repeatable, reproducible, and sensitive in detecting the presence of lung disease in children with CF as young as 4 months of age.78 LCI in 3–5 year-olds with CF is predictive of future LCI at 6–10 years of age.79 These tools may allow for closer clinical follow-up in these small children. These tools are essential because without spirometry it is difficult to assess whether young children ultimately recover following PEx’s.65
Microbiologic diagnosis
Chronic bacterial airway infections are characteristically seen in the majority of individuals with CF. These infections are commonly polymicrobial and rarely can be eradicated with antimicrobial therapy. Knowledge of the natural history of colonization and infection can be helpful in the management of CF PEx’s. Culture of respiratory tract specimens from individuals with CF can present challenges to microbiology laboratories unaccustomed to processing them, because of problems related to sample viscosity, the polymicrobial nature of infections, and slow bacterial growth. In addition, many of the available commercial systems for organism identification and antimicrobial susceptibility testing are inaccurate for CF pathogens.80–82
Given polymicrobial infections are the norm in CF airway infections, they can be problematic to culture since the organisms in the specimen may have very different growth requirements. Pseudomonas aeruginosa is often present and, because of its mucoid phenotype, frequently overgrows both Gram-positive bacteria such as Staphylococcus aureus and more fastidious or slower-growing Gram-negative organisms such as Haemophilus influenzae and Burkholderia cepacia complex. Selective media, which inhibits the growth of P. aeruginosa, is very useful for the isolation of S. aureus and H. influenzae and is mandatory for the isolation of B. cepacia complex.83–86 In addition, multiple subcultures may need to be performed in order to isolate pure bacterial cultures for identification and susceptibility testing. Slow bacterial growth also requires that culture plates receive prolonged incubation. Laboratories specializing in CF microbiology frequently use incubation times of 48 hours for cultures expected to yield P. aeruginosa, and up to 96 hours before reporting a culture negative for B. cepacia complex.85
Microbiologic sampling
Sampling of lower airway secretions is considered essential for determining the infectious etiology of PEx in CF. This is most readily accomplished using expectorated sputum. However, some individuals with CF are unable to expectorate. In addition, with the advent of highly effective CFTR modulators, expectoration has been noted to decrease with marked CF symptoms improvement.87 Bronchoalveolar lavage is an excellent way of sampling the lower airway in non-expectorating CF individuals, but this is too invasive for routine culturing.
Oropharyngeal swabs have served as a surrogate but may not be representative of lower airway infection88,89; oropharyngeal cultures for P. aeruginosa had a sensitivity of 44% and a specificity of 95%89. H. influenzae was similar, but the specificity was significantly lower for S. aureus. Oropharyngeal swabs obtained after chest physiotherapy were found to have increased sensitivity and specificity for the detection of both P. aeruginosa and S. aureus compared with swabs obtained prior to physiotherapy.90
Hypertonic saline induction of sputum has been reported to be a good surrogate for lower airway sampling in CF.91,92 Several studies suggest that induced sputum may be more sensitive in detecting bacteria in the lower airway compared with expectorated sputum and even bronchoalveolar lavage.92–94 Sputum induction has been used to monitor both inflammation and infection after intravenous antibiotic therapy for pulmonary exacerbations in CF.22 With the advent of highly effective CFTR therapy for over 90% of CF patients, patients may have marked reduction in expectoration, potentially increasing the role for sputum induction in CF management of PEx.
Antibiotic resistance and choice of antibiotics
Susceptibility testing of CF isolates of P. aeruginosa is difficult, for many of the same reasons that impact organism isolation and identification. Slow growth and mucoidy may impact the utility of automated systems for susceptibility testing of P. aeruginosa as well as for organism identification.82,95 When compared with broth microdilution methodology, agar diffusion methodologies including disk diffusion (Kirby-Bauer) and Etest performed well for the majority of antibiotics tested.96
Early infections with P. aeruginosa are commonly susceptible to anti-pseudomonal β-lactam antibiotics (including ticarcillin, piperacillin, ceftazidime, cefoperazone, and the penems), the aminoglycosides, and the fluoroquinolones. However, as patients age antibiotic resistance appears more frequently. At Danish CF centers, a significant increase in resistance to β-lactams was seen over two decades, but no correlation was found between increase in minimal inhibitory concentration and number of courses of anti-pseudomonal therapy.97 Multiple antibiotic resistance, defined as in vitro susceptibility to only a single class of antimicrobial agents, has been reported in up to 11.6% of P. aeruginosa isolates from individuals with CF in the United States and up to 17.4% in Italy.86,98 These multiple resistant isolates present important challenges in the care of CF patients, both because of concerns of patient to patient spread and antibiotic choices. Unfortunately, in patients with multiple resistant Pseudomonas isolates, synergy testing and multiple combination bactericidal testing (MCBT) have not been demonstrated to improve clinical or microbiological outcomes compared to usual susceptibility testing.99,100 Interestingly, even standard susceptibility testing has not been clearly demonstrated to improve patient outcome noted by evaluation of observational data.101 Not only has standard susceptibility testing failed to demonstrate benefit, more elaborate testing of P. aeruginosa grown in biofilms also failed to have superior impacts on lung function in two separate studies.102,103 These results and others have led to a recent systematic review on this topic.
Recently, Somayaji et. al. with the Antimicrobial Resistance in Cystic Fibrosis International Working Group published a systematic review of antimicrobial susceptibility testing (AST) in CF.104 The review addressed two key questions: 1) “For individuals with CF, is clinical response to antimicrobial treatment of bacterial airways infection predictable from antimicrobial susceptibility testing results available at treatment initiation?” and 2) “For individuals with CF, is clinical response to antimicrobial treatment of bacterial airways infection affected by the method used to guide antimicrobial selection?” Most of the studies involved P. aeruginosa with 13 specifically addressing pulmonary exacerbation. Two studies suggested antimicrobial susceptibility testing correlated with pulmonary exacerbation treatment response correlations, while 11 did not. The authors’ conclusions were that there is little evidence that antibiotic susceptibility testing “predicts the clinical outcome of CF antimicrobial treatment, suggesting a need for careful consideration of current AST use by the CF community.”
B. cepacia complex organisms are often highly antibiotic resistant. All are intrinsically resistant to the aminoglycosides105 and the rate of in vitro resistance to the β-lactam antibiotics, with the exception of meropenem, is also quite high.106,107 The quinolones have variable activity, but resistance can be readily induced.106 In vitro susceptibility testing suggests that there are combinations of antibiotics that act synergistically against B. cepacia complex using either synergy testing or MBCT.107,108 Synergy testing, using two drug combinations, found that for 57% of isolates tested, no active combination could be identified.108 The most active combinations were chloramphenicol plus minocycline (49% of isolates) and chloramphenicol plus ceftazidime (26% of isolates). MBCT testing using two or three drug combinations determined that at least one combination could be identified for all isolates tested.109 The majority of active combinations included meropenem. Unfortunately, it was not possible to predict for a given isolate whether a drug combination would be synergistic, additive or antagonistic.
Other antibiotic resistant Gram-negative CF isolates include S. maltophilia and A. xylosoxidans. Treatment of these organisms is often complicated by resistance to the aminoglycosides and variable susceptibility to the β-lactams and quinolones. The most active single drugs in vitro against S. maltophilia are ticarcillin/clavulanate and trimethoprim/sulfamethoxazole; the most active combination in synergy studies is ticarcillin/clavulanate plus aztreonam.110 In a study of 106 CF isolates of A. xylosoxidans, the most active drugs were imipenem (59% susceptible), piperacillin/tazobactam (55%), meropenem (51%) and minocycline (51%).111 The most active additive or synergistic combinations were chloramphenicol plus minocycline, ciprofloxacin plus imipenem, and ciprofloxacin plus meropenem.
Overall approach to Treatment
A recent systematic review evaluated the literature regarding management of PEx. The authors performed a systematic review to develop treatment guidelines for the management of PEx and found insufficient evidence for most treatment decisions with the exception of treatment with antibiotics (including oral, inhaled and intravenous formulations in various combinations) which are considered standard of care.112 There has been no consensus as to how long to treat PEx with IV antibiotics and treatment durations can range from days to weeks.113 The number of antibiotics chosen (commonly two for Pseudomonas aeruginosa) was graded with insufficient data to make a recommendation after including 17 trials for review. The main challenge with these trials was an inability to demonstrate meaningful clinical improvement. Recent Cochrane reviews for the treatment of CF patients with PEx’s complicated Burkholderia cepacia complex infection found no quality evidence to guide treatment decisions.114CF consensus pulmonary guidelines for the treatment of PEx identified key questions for which additional studies were needed, including whether there was an optimal duration of IV antibiotic treatment for CF pulmonary exacerbation.112
Additional key aspects of PEx treatment approaches other than antibiotics include identifying and monitoring CF related diabetes, frequent airway clearance (four times a day), nutritional support and reducing risks of new organism acquisition if hospitalized. The role of oral steroids in pulmonary exacerbation remains unknown. In a recent US multicenter observational study of 220 CF adolescent and adults treated with IV antibiotics, 18.2% were treated with oral steroids for the exacerbation and 20% were treated with mucolytic agents.115 The only randomized controlled trial (RCT) of prednisone as an adjunct to treatment for PEx demonstrated a non-statistically significant improvement in lung function and no clear improvement in symptoms or sputum inflammatory markers compared to placebo.116 A large scale RCT is now underway in Canada to address the role of prednisone in patients with PEx who have not recovered to their baseline lung function with standard treatment by day 7 (NCT03070522).
Antibiotic selection for treatment remains challenging. Although studies have compared two versus one antibiotic to cover Pseudomonas, no consensus algorithm is available. As noted above, a systematic review could not come to a strong recommendation in regards to two anti-Pseudomonal agents to treat exacerbation. In one recent study of exacerbation, delineating the complexity of antibiotics used in a prospective multi-center cohort of 220 CF adolescents and adults, 51.8% received 2 IV antibiotics and 32.7 received 3 or more IV antibiotics.115 Intravenous tobramycin was most commonly used (in 59.5% of the cases) with meropenum, piperacillin/tazobatam/ceftazidime and vancomycin all prescribed in about 25% of the patients. Over 25% of patients also received an oral antibiotic with only 9% receiving an inhaled antibiotic. In regards to dosing interval, daily vs three times a day aminoglycosides have been studies in RCTs. The best known trial (the TOPIC study) reported equal efficacy between once and three-times daily tobramycin given with ceftazidime, with a trend to less nephrotoxicity in CF children.117 Based on this study, many centers now use one daily IV tobramycin. Our group noted higher rates of toxicity with 10 mg/kg (used in the TOPIC study) in adults with CF as others have reported, and we now start our dosing at 8 mg/kg. The use of IV gentamycin has markedly decreased since an observational study in the UK suggested increased toxicity compared to tobramycin.118 Another key topic of dosing of antibiotics in CF PEx has been the use of continuous infusions of beta lactams, combination penicillins/beta-lactamase inhibitors (i.e. piperacillin/tazobactam), or aminoglycosides.119–121 The rationale has been to optimize pharmacokinetic/pharmacodynamic (PK/PD) properties. Despite the PK/PD properties, studies to date have not been powered to demonstrate clinical superiority to routine dosing.
Treatment and treatment response
Several prospective cohort studies have clearly delineated the treatment response to IV antibiotics in CF PEx. These newer studies support earlier studies that note that an acute drop in pulmonary function is highly associated with the diagnosis and treatment of PEx37 and that treatment with IV antibiotics has been shown to result in improved lung function in CF patients experiencing PEx.15 Sagel et al. enrolled 103 patients and collected clinical data (symptom scores, spirometry) and inflammatory markers; 84% recovered at least 90% of their baseline FEV1 within 3 months of the exacerbation.20 Having higher IL-6, IL-8, and α1AT concentrations at the time of exacerbation were associated with an increased risk of being a non-responder. An extension of this cohort study (n=123) demonstrated that 33% experienced <10% relative improvement in FEV1 during treatment. Symptom improvement was observed but was not associated with subsequent lung function or time to next antibiotic therapy, which had a median recurrence time of 143 days.21 More recently, the Standard Treatment of Pulmonary exacerbation (STOP) study prospectively enrolled 220 CF subjects admitted to the hospital for treatment of a PEx to evaluate the variability of treatment durations and to identify the clinical outcome measures deemed most important to care-givers in determining treatment success.115,122–124 These two cohorts frequently assessed spirometry and used a symptom diary [Cystic Fibrosis Respiratory Symptom Diary Chronic Respiratory Infection Symptom Scale (CFRSD-CRISS)] to evaluate treatment outcome.125,126 Figures 1a and 1b describe the pattern of treatment response in the STOP trial in regards to spirometry and symptom score; these data show that respiratory symptoms as measured with the CFRSD-CRISS and average lung function typically improves with IV treatment but later declines. Of note, other patient reported outcome measures also can be used to assess response to treatment of a pulmonary exacerbation (the CFQ-R).127
Duration of treatment for a pulmonary exacerbation
The most common duration used to treat exacerbations is 14 days but there is considerable variance, including 10% of patients receiving >23 days. As noted above, systematic reviews of the literature found there was insufficient evidence upon which to develop guidelines for most treatment decisions, including antibiotic selection and duration128 and there is little evidence of additional clinical benefit for specific treatment durations in CF Foundation Patient Registry (CFFPR) analyses.129,130 Studies of PEx’s assessing response to treatment have shown that lung function improves up to day 8–10 with little additional improvement with longer treatments.113 A recent single-center retrospective study demonstrated that some patients continue to experience improvement in symptoms and FEV1 after cessation of treatment with longer antibiotic courses131, but it is not clear that this improvement was related to extending treatment, or that similar improvements would not have been observed with shorter treatment durations. Data show that average lung function typically declines after cessation of treatment. The total duration of IV antibiotic therapy for CF PEx has large implications on resource utilization, treatment burden, and potential for toxicity associated with extended treatment so remains a critical question in CF.
In 2012, 35.1% of patients of all ages followed in the CFFPR were treated at least once with IV antibiotics for PEx, with the median number of IV antibiotic treatment days per PEx varying greatly by CF care center, from 3 days to 24.2 days (Figure 3).55,132 There are two clear peaks in duration of antibiotic duration – 14 and 21 days but very dramatic variation across US centers.55 The median IV antibiotic treatment duration was 13.5 days for children and 14.5 days for adults (≥ 18 years).132 Given that as many as a quarter of patients with PEx treated with IV antibiotics fail to return to even 90% of the lung function they had prior to exacerbation, we urgently need to determine appropriate treatment duration.66,133
Figure 3.
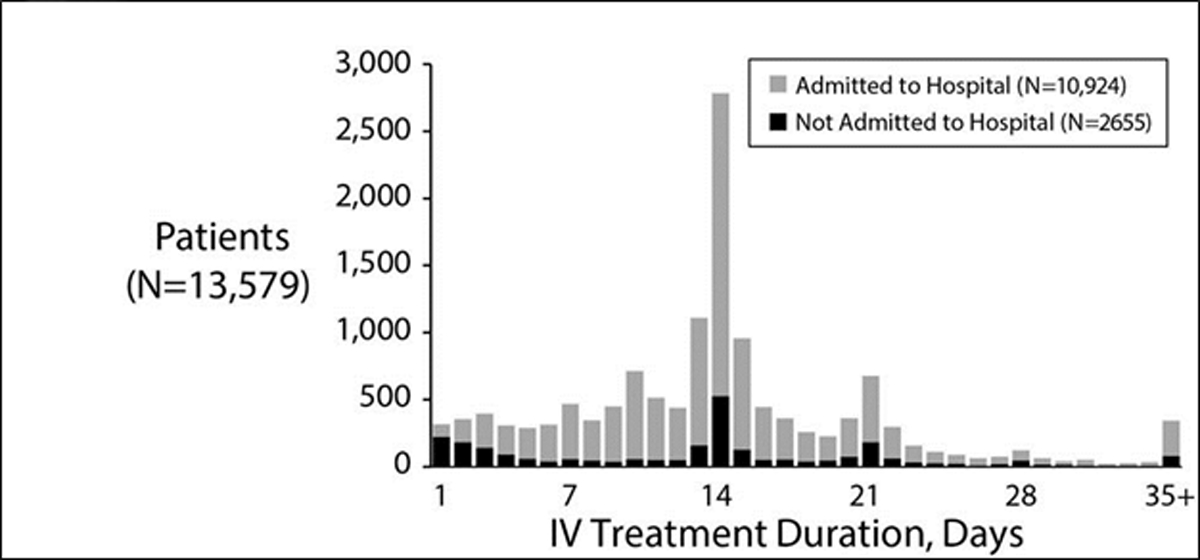
Distribution of IV antibiotic treatment durations in the US CFFPR. Grey bars reflect the number of exacerbations where patients were admitted to the hospital. Black bars show numbers of patients of a given treatment duration who were never admitted to hospital as part of their exacerbation treatment. Reprinted with permission of the Journal of Cystic Fibrosis.55
Studies that have focused on clinical outcomes after PEx have noted poorer short-term outcomes, including reduced recovery of lung function in a substantial fraction of events (up to 50%).65,134 It is possible that some PEx treatment decisions may account for poorer outcomes.55 For example, in the US, treatment with intravenous (IV) antibiotics for less than 9 days and treatment entirely as an outpatient have both been associated with an increased risk of retreatment with IV antibiotics within 30 days of PEx treatment completion, despite similar patient characteristics at IV antibiotic initiation.55
Given the strong evidence and successful implementation in other diseases, it is very possible for an optimal standard duration of IV antibiotic therapy to be defined in CF. For this reason, the STOP study (NCT02109822) was conducted to refine key clinical endpoints and variance for treatment response for an IV exacerbation in CF as noted above.115 This study led to the initiation of the STOP2 study (NCT02781610).124 The STOP2 trial is a divergent trial design that evaluates subjects’ interim improvement in lung function as measured by forced expiratory volume in one second (FEV1) and symptoms as measured by a CF specific respiratory symptom diary to tailor randomization to IV treatment duration (10 vs. 14 days for early responders, 14 vs. 21 days for delayed responders). Data from this large-scale randomized controlled trial (RCT) (STOP2 trial will have over 850 CF patients) will provide clear evidence regarding the optimal duration of IV antibiotics for adults with CF who present with a physician-defined PEx. Equally important, in conjunction with the RCT, there are ancillary studies including a detailed cost benefit analysis to fully understand the implications of the trial results.
Site of Care
As noted above, systematic reviews of the literature found there was insufficient evidence upon which to develop guidelines for most treatment decisions, including site of treatment (home or hospital).112 Only one comparative trial exists which demonstrated similar outcomes for the home and hospital setting.112,136 Observational data has suggested that optimal treatment responses may not be achieved by treating acute PEx’s in the outpatient setting. A recent analysis of a national data registry found a 10% greater likelihood of FEV1 returning to ≥90% of baseline comparing complete inpatient treatment with no inpatient treatment.137 The conclusions of this paper differed from another observational study using the CFFPR which found that site of treatment did not impact long-term outcomes of exacerbations.130 Thus, site of treatment remains a key unresolved question in the management of acute pulmonary exacerbations. In the CFFPR reports, the median treatment for PEx in adults was 12 days with a median hospitalization of 10 days,138 demonstrating that at least part of the treatment for PEx commonly occurs at home. The services to which patients have access are very different in the hospital compared to at home; differences may include nutritional support, regular chest physiotherapy and monitored antibiotic delivery. Given the costs of inpatient care, a clinical trial definitively addressing the best treatment setting for an IV exacerbation would be both ethically challenging and prohibitively costly.
Use of home monitoring for exacerbation
Very little has been published on self-monitoring for individuals with CF, and it has never been widely embraced by CF care providers. Investigators and clinicians at the University of Minnesota reported on their experiences with home monitoring in CF in the late 1980’s and early 1990’s.50;51 For a two year period 111 individuals with CF maintained daily diaries in which they recorded vital capacity, body weight, respiratory rate, pulse, and symptoms. The daily participation rate was maintained at approximately 80%.52 Subsequently, these investigators carried out a non-concurrent cohort study on 50 individuals with CF.51 Twenty-five participants were selected randomly from the group that had used home monitoring and were matched to 25 participants that had not done home monitoring. The groups were matched on age and gender. There were no specific protocols for treating an exacerbations noted in either arm of the trial. Participants were followed for four years. FEV1 declined from 73.1% predicted to 70.1% predicted in the home monitoring group (N.S.) and declined from 72.3% predicted to 60.8% (p<0.001) in the control group. While this was a small, retrospective study, it demonstrated that home monitoring was embraced by patients, did not adversely affect patients, and may result in beneficial health care behaviors. There was generally strong agreement that the feedback from self-monitoring was relevant, would allow patients to have more control of their health, and could play an important role in chronic disease management. Additional groups have integrating devices in the home to evaluate other aspects of CF care centered in Australia.139–141 Despite encouraging results from earlier studies in CF, a large scale randomized controlled trial of home monitoring in the US did not demonstrate clinical efficacy.142 Adherence with the monitoring arm of the protocol in this trial was lower than desired, with only 19% of participants in the intervention arm transmitting their home data twice weekly 80% of the time or greater for the year duration of the study. However, adherence with once weekly data transmission was 50% using the device at least once per week on more than 80% of their follow-up weeks and generated a total of 524 alarms among 97 participants (72%) in the monitoring arm. These alarms led to a shorter time to diagnosis of a protocol defined PEx and antibiotic prescription. The treatments of exacerbations in the monitoring arm were more likely solely oral antibiotics. Currently, optimal care of an acute PEx is not known [i.e. number of antibiotics, duration of antibiotics and location of delivery of antibiotics (home or inpatient)].128 Thus, identifying PEx’s alone may not be enough; one needs to optimize management of these events.
Exacerbations in an era of highly effective modulator therapy
With the advent of highly effective modulators like ivacaftor60 and now ivacaftor/tezacaftor/ elexacaftor,143 clinicians do not know how the landscape of PEx will change. Based on clinical trial data, ivacaftor decreased exacerbation rates by 55% over 48 weeks.60 In post-marketing data from the Irish CF Registry, ivacaftor has the potential to reduce exacerbation rates treated with intravenous antibiotic courses by 46% (95% CI: −62.5% to −23.3%) and oral antibiotics by 49%( (95% CI, −61.1% to −32.1%).144 We do not yet have published data about the impact of the new triple combination therapy (ivacaftor/tezacaftor/ elexacaftor) on PEx rate, however, it is likely to be greater than ivacaftor in patients with G551D mutations.143 Even further data suggests that the future may be very bright for our patients; in a recent publication supporting earlier work, ivacaftor appears to reduce the incidence of new Pseudomonas aeruginosa acquisition.145,146 Despite the potential for radical changes in PEx rates, the majority of CF patients will still have moderate to extensive bronchiectasis complicated by lower airway infection and like non-CF bronchiectasis, will continue to have PEx’s managed by CF physicians.
Conclusions
Research regarding pulmonary exacerbations in cystic fibrosis continues to evolve improving our understanding of the natural history of disease of CF patients. Pulmonary exacerbations continue to significantly impact the lives of both children and adults with CF. Improving our understanding of these events will have implications for both basic research and clinical research in CF. We are now entering a new era of large scale clinical trials to provide high quality evidence to treatment of PEx. We are also about to enter an era when >90% of our patients will likely have highly effective CFTR therapy that could alter the severity and the rates of PEx in CF. Despite these advances, many questions remain about basic aspects of pathophysiology and treatment of PEx that will likely persist. More work is desperately needed to further the science of PEx.
Figure 2a and 2b.
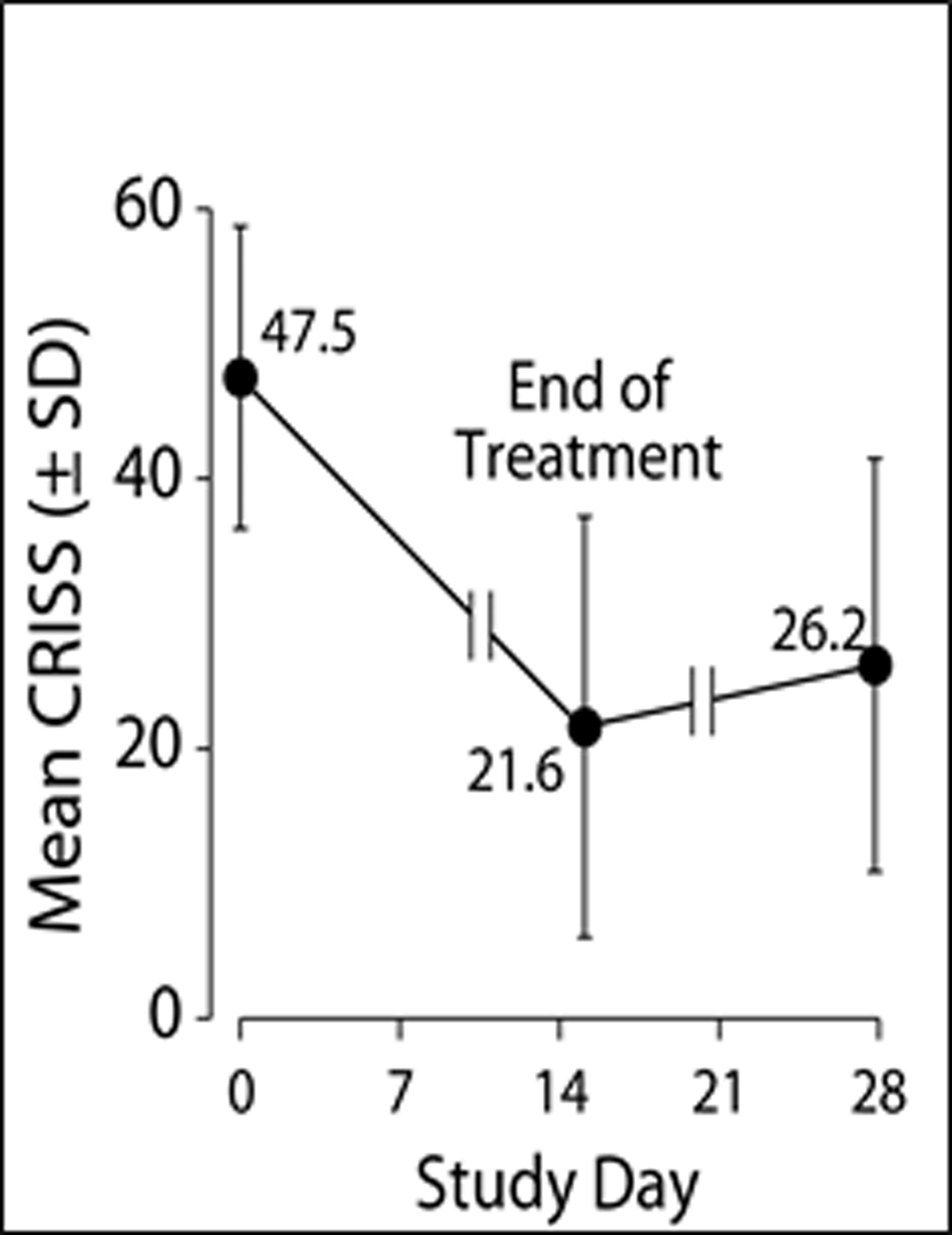
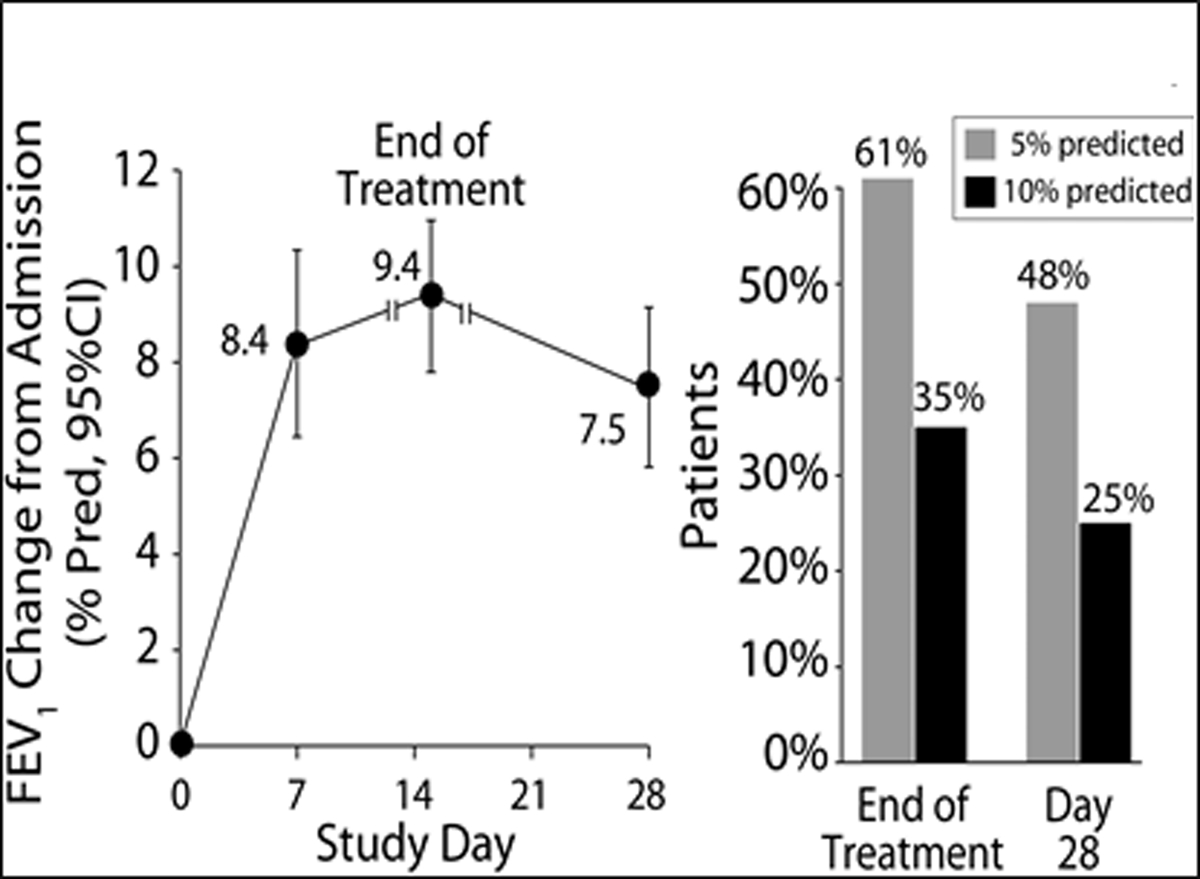
2a: Patient’s mean the Chronic Respiratory Infection Symptom Score (CRISS) +/− SD by day of treatment with day 0 the beginning of IV treatment. The CRISS ranges from 0–100 with 100 being the worst possible respiratory symptoms. 2b: notes the FEV1 % predicted change from admission for IV antibiotics by days of treatment. The bar graph notes those with an absolute change of 5% predicted and 10% predicted by end of IV treatment and them by day 28.
Funding:
Dr. Goss’ research time is supported by the Cystic Fibrosis Foundation Therapeutics (GOSS13A0) and NIH (P30 DK089507, UL1TR000423). There was no role of funding sources in writing of this manuscript, or the decision to submit for publication.
References:
- 1.Knapp EA, Fink AK, Goss CH, et al. The Cystic Fibrosis Foundation Patient Registry. Design and Methods of a National Observational Disease Registry. Ann Am Thorac Soc 2016;13:1173–9. [DOI] [PubMed] [Google Scholar]
- 2.Jain M, Goss CH. Update in cystic fibrosis 2013. Am J Respir Crit Care Med 2014;189:1181–6. [DOI] [PubMed] [Google Scholar]
- 3.MacKenzie T, Gifford AH, Sabadosa KA, et al. Longevity of Patients With Cystic Fibrosis in 2000 to 2010 and Beyond: Survival Analysis of the Cystic Fibrosis Foundation Patient Registry. Ann Intern Med 2014;161:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liou T, Adler F, Fitzsimmons S, Cahill B, Hibbs J, Marshall B. Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol 2001;153:345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liou TG, Adler FR, Huang D. Use of lung transplantation survival models to refine patient selection in cystic fibrosis. Am J Respir Crit Care Med 2005;171:1053–9. [DOI] [PubMed] [Google Scholar]
- 6.Emerson J, Rosenfield M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 2002;34:91–100. [DOI] [PubMed] [Google Scholar]
- 7.Britto M, Kotagal U, Hornung R, Atherton H, Tsevat J, Wilmott R. Impact of recent pulmonary exacerbations on quality of life in patients with cystic fibrosis. Chest 2002;121:64–72. [DOI] [PubMed] [Google Scholar]
- 8.Dobbin CJ, Bartlett D, Melehan K, Grunstein RR, Bye PT. The effect of infective exacerbations on sleep and neurobehavioral function in cystic fibrosis. Am J Respir Crit Care Med 2005;172:99–104. [DOI] [PubMed] [Google Scholar]
- 9.Wagener JS, Rasouliyan L, VanDevanter DR, et al. Oral, inhaled, and intravenous antibiotic choice for treating pulmonary exacerbations in cystic fibrosis. Pediatr Pulmonol 2013;48:666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiatt PW, Grace SC, Kozinetz CA, et al. Effects of viral lower respiratory tract infection on lung function in infants with cystic fibrosis. Pediatrics 1999;103:619–26. [DOI] [PubMed] [Google Scholar]
- 11.Piedra PA, Grace S, Jewell A, et al. Purified fusion protein vaccine protects against lower respiratory tract illness during respiratory syncytial virus season in children with cystic fibrosis. Pediatr Infect Dis J 1996;15:23–31. [DOI] [PubMed] [Google Scholar]
- 12.Piedra PA, Cron SG, Jewell A, et al. Immunogenicity of a new purified fusion protein vaccine to respiratory syncytial virus: a multi-center trial in children with cystic fibrosis. Vaccine 2003;21:2448–60. [DOI] [PubMed] [Google Scholar]
- 13.Ortiz JR, Neuzil KM, Victor JC, Wald A, Aitken ML, Goss CH. Influenza-associated cystic fibrosis pulmonary exacerbations. Chest 2010;137:852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Somayaji R, Goss CH, Khan U, Neradilek M, Neuzil KM, Ortiz JR. Cystic Fibrosis Pulmonary Exacerbations Attributable to Respiratory Syncytial Virus and Influenza: A Population-Based Study. Clin Infect Dis 2017;64:1760–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regelmann WE, Elliott GR, Warwick WJ, Clawson CC. Reduction of sputum Pseudomonas aeruginosa density by antibiotics improves lung function in cystic fibrosis more than do bronchodilators and chest physiotherapy alone. Am Rev Respir Dis 1990;141:914–21. [DOI] [PubMed] [Google Scholar]
- 16.Smith AL, Redding G, Doershuk C, et al. Sputum changes associated with therapy for endobronchial exacerbation in cystic fibrosis. J Pediatr 1988;112:547–54. [DOI] [PubMed] [Google Scholar]
- 17.Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999;340:23–30. [DOI] [PubMed] [Google Scholar]
- 18.Aaron SD, Ramotar K, Ferris W, et al. Adult cystic fibrosis exacerbations and new strains of Pseudomonas aeruginosa. Am J Respir Crit Care Med 2004;169:811–5. [DOI] [PubMed] [Google Scholar]
- 19.Mayer-Hamblett N, Aitken ML, Accurso FJ, et al. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med 2007;175:822–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sagel SD, Thompson V, Chmiel JF, et al. Effect of treatment of cystic fibrosis pulmonary exacerbations on systemic inflammation. Ann Am Thorac Soc 2015;12:708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heltshe SL, Goss CH, Thompson V, et al. Short-term and long-term response to pulmonary exacerbation treatment in cystic fibrosis. Thorax 2016;71:223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ordonez CL, Henig NR, Mayer-Hamblett N, et al. Inflammatory and microbiologic markers in induced sputum after intravenous antibiotics in cystic fibrosis. Am J Respir Crit Care Med 2003;168:1471–5. [DOI] [PubMed] [Google Scholar]
- 23.Colombo C, Costantini D, Rocchi A, et al. Cytokine levels in sputum of cystic fibrosis patients before and after antibiotic therapy. Pediatr Pulmonol 2005;40:15–21. [DOI] [PubMed] [Google Scholar]
- 24.Waters VJ, Stanojevic S, Sonneveld N, et al. Factors associated with response to treatment of pulmonary exacerbations in cystic fibrosis patients. J Cyst Fibros 2015;14:755–62. [DOI] [PubMed] [Google Scholar]
- 25.Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med 1995;332:848–54. [DOI] [PubMed] [Google Scholar]
- 26.Lands LC, Milner R, Cantin AM, Manson D, Corey M. High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. J Pediatr 2007;151:249–54. [DOI] [PubMed] [Google Scholar]
- 27.Conrad C, Lymp J, Thompson V, et al. Long-term treatment with oral N-acetylcysteine: affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J Cyst Fibros 2015;14:219–27. [DOI] [PubMed] [Google Scholar]
- 28.Ren CL, Pasta DJ, Rasouliyan L, et al. Relationship between inhaled corticosteroid therapy and rate of lung function decline in children with cystic fibrosis. J Pediatr 2008;153:746–51. [DOI] [PubMed] [Google Scholar]
- 29.Chmiel JF, Konstan MW, Accurso FJ, et al. Use of ibuprofen to assess inflammatory biomarkers in induced sputum: Implications for clinical trials in cystic fibrosis. J Cyst Fibros 2015;14:720–6. [DOI] [PubMed] [Google Scholar]
- 30.Konstan MW, VanDevanter DR, Sawicki GS, et al. Association of High-Dose Ibuprofen Use, Lung Function Decline, and Long-Term Survival in Children with Cystic Fibrosis. Ann Am Thorac Soc 2018;15:485–93. [DOI] [PubMed] [Google Scholar]
- 31.Ramsey BW, Boat TF. Outcome measures for clinical trials in cystic fibrosis. Summary of a Cystic Fibrosis Foundation consensus conference. J Pediatr 1994;124:177–92. [DOI] [PubMed] [Google Scholar]
- 32.Goss CH, Burns JL. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax 2007;62:360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994;331:637–42. [DOI] [PubMed] [Google Scholar]
- 34.Konstan MW, Doring G, Heltshe SL, et al. A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. J Cyst Fibros 2014;13:148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dakin C, Henry RL, Field P, Morton J. Defining an exacerbation of pulmonary disease in cystic fibrosis. Pediatr Pulmonol 2001;31:436–42. [DOI] [PubMed] [Google Scholar]
- 36.Rosenfeld M, Emerson J, Williams-Warren J, et al. Defining a pulmonary exacerbation in cystic fibrosis. J Pediatr 2001;139:359–65. [DOI] [PubMed] [Google Scholar]
- 37.Rabin HR, Butler SM, Wohl ME, et al. Pulmonary exacerbations in cystic fibrosis. Pediatr Pulmonol 2004;37:400–6. [DOI] [PubMed] [Google Scholar]
- 38.Ferkol T, Rosenfeld M, Milla CE. Cystic fibrosis pulmonary exacerbations. Journal of Pediatrics 2006;148:259–64. [DOI] [PubMed] [Google Scholar]
- 39.Johnson C, Butler SM, Konstan MW, Morgan W, Wohl ME. Factors influencing outcomes in cystic fibrosis: a center-based analysis. Chest 2003;123:20–7. [DOI] [PubMed] [Google Scholar]
- 40.Ellaffi M, Vinsonneau C, Coste J, et al. One-year outcome after severe pulmonary exacerbation in adults with cystic fibrosis. Am J Respir Crit Care Med 2005;171:158–64. [DOI] [PubMed] [Google Scholar]
- 41.Texereau J, Jamal D, Choukroun G, et al. Determinants of mortality for adults with cystic fibrosis admitted in Intensive Care Unit: a multicenter study. Respir Res 2006;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sood N, Paradowski LJ, Yankaskas JR. Outcomes of intensive care unit care in adults with cystic fibrosis. Am J Respir Crit Care Med 2001;163:335–8. [DOI] [PubMed] [Google Scholar]
- 43.Vedam H, Moriarty C, Torzillo PJ, McWilliam D, Bye PT. Improved outcomes of patients with cystic fibrosis admitted to the intensive care unit. J Cyst Fibros 2004;3:8–14. [DOI] [PubMed] [Google Scholar]
- 44.Dobbin CJ, Milross MA, Piper AJ, Sullivan C, Grunstein RR, Bye PT. Sequential use of oxygen and bi-level ventilation for respiratory failure in cystic fibrosis. J Cyst Fibros 2004;3:237–42. [DOI] [PubMed] [Google Scholar]
- 45.Siuba M, Attaway A, Zein J, et al. Mortality in Adults with Cystic Fibrosis Requiring Mechanical Ventilation: Cross-Sectional Analysis of Nationwide Events. Ann Am Thorac Soc 2019. [DOI] [PubMed] [Google Scholar]
- 46.Liou TG, Adler FR, Cahill BC, et al. Survival effect of lung transplantation among patients with cystic fibrosis. JAMA 2001;286:2683–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayer-Hamblett N, Rosenfeld M, Emerson J, Goss CH, Aitken ML. Developing cystic fibrosis lung transplant referral criteria using predictors of 2-year mortality. Am J Respir Crit Care Med 2002;166:1550–5. [DOI] [PubMed] [Google Scholar]
- 48.Burgel PR, Lemonnier L, Dehillotte C, et al. Cluster and CART analyses identify large subgroups of adults with cystic fibrosis at low risk of 10-year death. Eur Respir J 2019;53. [DOI] [PubMed] [Google Scholar]
- 49.Coriati A, Sykes J, Nkam L, Hocine MN, Burgel PR, Stephenson AL. Validation of the French 3-year prognostic score using the Canadian Cystic Fibrosis registry. J Cyst Fibros 2019;18:396–8. [DOI] [PubMed] [Google Scholar]
- 50.Stanojevic S, Sykes J, Stephenson AL, Aaron SD, Whitmore GA. Development and External Validation of 1- and 2- year Mortality Prediction Models in Cystic Fibrosis. Eur Respir J 2019. [DOI] [PubMed] [Google Scholar]
- 51.Lehr CJ, Skeans M, Dasenbrook EC, et al. Effect of Including Important Clinical Variables on Accuracy of the Lung Allocation Score for Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 2019. [DOI] [PubMed] [Google Scholar]
- 52.Marshall BC, Butler SM, Stoddard M, Moran AM, Liou TG, Morgan WJ. Epidemiology of cystic fibrosis-related diabetes. J Pediatr 2005;146:681–7. [DOI] [PubMed] [Google Scholar]
- 53.Goss CH, Otto K, Aitken ML, Rubenfeld GD. Detecting Stenotrophomonas maltophilia does not reduce survival of patients with cystic fibrosis. Am J Respir Crit Care Med 2002;166:356–61. [DOI] [PubMed] [Google Scholar]
- 54.Goss CH, Rubenfeld GD, Otto K, Aitken ML. The effect of pregnancy on survival in women with cystic fibrosis. Chest 2003;124:1460–8. [DOI] [PubMed] [Google Scholar]
- 55.VanDevanter DR, Flume PA, Morris N, Konstan MW. Probability of IV antibiotic retreatment within thirty days is associated with duration and location of IV antibiotic treatment for pulmonary exacerbation in cystic fibrosis. J Cyst Fibros 2016;15:783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schechter MS, Shelton BJ, Margolis PA, Fitzsimmons SC. The association of socioeconomic status with outcomes in cystic fibrosis patients in the United States. Am J Respir Crit Care Med 2001;163:1331–7. [DOI] [PubMed] [Google Scholar]
- 57.Goss CH, Newsom SA, Schildcrout JS, Sheppard L, Kaufman JD. Effect of ambient air pollution on pulmonary exacerbations and lung function in cystic fibrosis. Am J Respir Crit Care Med 2004;169:816–21. [DOI] [PubMed] [Google Scholar]
- 58.Brody A, Sucharew H, Campbell J, et al. Computed tomography correlates with pulmonary exacerbations in children with cystic fibrosis. Am J Respir Crit Care Med 2005;172:1128–32. [DOI] [PubMed] [Google Scholar]
- 59.Smith JA, Owen EC, Jones AM, Dodd ME, Webb AK, Woodcock A. Objective measurement of cough during pulmonary exacerbations in adults with cystic fibrosis. Thorax 2006;61:425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 2015;373:1783–4. [DOI] [PubMed] [Google Scholar]
- 62.Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017;377:2013–23. [DOI] [PubMed] [Google Scholar]
- 63.Mayer-Hamblett N, Rosenfeld M, Treggiari MM, et al. Standard care versus protocol based therapy for new onset Pseudomonas aeruginosa in cystic fibrosis. Pediatr Pulmonol 2013;48:943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vandevanter DR, Yegin A, Morgan WJ, Millar SJ, Pasta DJ, Konstan MW. Design and powering of cystic fibrosis clinical trials using pulmonary exacerbation as an efficacy endpoint. J Cyst Fibros 2011;10:453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanders D, Bittner R, Rosenfeld M, Hoffman L, Redding G, Goss C. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med 2010;182:627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parkins MD, Rendall JC, Elborn JS. Incidence and risk factors for pulmonary exacerbation treatment failures in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa. Chest 2012;141:485–93. [DOI] [PubMed] [Google Scholar]
- 67.Schechter MS, Leonard A, Nash J, et al. Benchmarking: Signature Themes. Pediatr Pulmonol 2006;41:122–3.16369926 [Google Scholar]
- 68.Konstan M, Morgan W, Butler S, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr 2007;151:134–9, 9.e1. [DOI] [PubMed] [Google Scholar]
- 69.Byrnes CA, Vidmar S, Cheney JL, et al. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kusel MM, de Klerk N, Holt PG, Landau LI, Sly PD. Occurrence and management of acute respiratory illnesses in early childhood. J Paediatr Child Health 2007;43:139–46. [DOI] [PubMed] [Google Scholar]
- 71.van Ewijk BE, van der Zalm MM, Wolfs TF, et al. Prevalence and impact of respiratory viral infections in young children with cystic fibrosis: prospective cohort study. Pediatrics 2008;122:1171–6. [DOI] [PubMed] [Google Scholar]
- 72.Chattoraj SS, Ganesan S, Jones AM, et al. Rhinovirus infection liberates planktonic bacteria from biofilm and increases chemokine responses in cystic fibrosis airway epithelial cells. Thorax 2011;66:333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Ewijk BE, Wolfs TF, Aerts PC, et al. RSV mediates Pseudomonas aeruginosa binding to cystic fibrosis and normal epithelial cells. Pediatr Res 2007;61:398–403. [DOI] [PubMed] [Google Scholar]
- 74.Rosenfeld M, Ratjen F, Brumback L, et al. Inhaled hypertonic saline in infants and children younger than 6 years with cystic fibrosis: the ISIS randomized controlled trial. JAMA 2012;307:2269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mayer-Hamblett N, Retsch-Bogart G, Kloster M, et al. Azithromycin for Early Pseudomonas Infection in Cystic Fibrosis. The OPTIMIZE Randomized Trial. Am J Respir Crit Care Med 2018;198:1177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mott LS, Park J, Murray CP, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 2012;67:509–16. [DOI] [PubMed] [Google Scholar]
- 77.Stick S, Brennan S, Murray C, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr 2009;155:623–8.e1. [DOI] [PubMed] [Google Scholar]
- 78.Belessis Y, Dixon B, Hawkins G, et al. Early cystic fibrosis lung disease detected by bronchoalveolar lavage and lung clearance index. Am J Respir Crit Care Med 2012;185:862–73. [DOI] [PubMed] [Google Scholar]
- 79.Aurora P, Stanojevic S, Wade A, et al. Lung clearance index at 4 years predicts subsequent lung function in children with cystic fibrosis. Am J Respir Crit Care Med 2011;183:752–8. [DOI] [PubMed] [Google Scholar]
- 80.Kiska DL, Kerr A, Jones MC, et al. Accuracy of four commercial systems for identification of Burkholderia cepacia and other gram-negative nonfermenting bacilli recovered from patients with cystic fibrosis. J Clin Microbiol 1996;34:886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shelly DB, Spilker T, Gracely EJ, Coenye T, Vandamme P, LiPuma JJ. Utility of commercial systems for identification of Burkholderia cepacia complex from cystic fibrosis sputum culture. J Clin Microbiol 2000;38:3112–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saiman L, Burns JL, Larone D, Chen Y, Garber E, Whittier S. Evaluation of MicroScan Autoscan for identification of Pseudomonas aeruginosa isolates from cystic fibrosis patients. J Clin Microbiol 2003;41:492–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wong K, Roberts MC, Owens L, Fife M, Smith AL. Selective media for the quantitation of bacteria in cystic fibrosis sputum. J Med Microbiol 1984;17:113–9. [DOI] [PubMed] [Google Scholar]
- 84.Carson LA, Tablan OC, Cusick LB, Jarvis WR, Favero MS, Bland LA. Comparative evaluation of selective media for isolation of Pseudomonas cepacia from cystic fibrosis patients and environmental sources. J Clin Microbiol 1988;26:2096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Henry DA, Campbell ME, LiPuma JJ, Speert DP. Identification of Burkholderia cepacia isolates from patients with cystic fibrosis and use of a simple new selective medium. J Clin Microbiol 1997;35:614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Burns JL, Emerson J, Stapp JR, et al. Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin Infect Dis 1998;27:158–63. [DOI] [PubMed] [Google Scholar]
- 87.Davies JC, Moskowitz SM, Brown C, et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018;379:1599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Armstrong DS, Grimwood K, Carlin JB, Carzino R, Olinsky A, Phelan PD. Bronchoalveolar lavage or oropharyngeal cultures to identify lower respiratory pathogens in infants with cystic fibrosis. Pediatr Pulmonol 1996;21:267–75. [DOI] [PubMed] [Google Scholar]
- 89.Rosenfeld M, Emerson J, Accurso F, et al. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatr Pulmonol 1999;28:321–8. [DOI] [PubMed] [Google Scholar]
- 90.Kabra SK, Alok A, Kapil A, et al. Can throat swab after physiotherapy replace sputum for identification of microbial pathogens in children with cystic fibrosis? Indian J Pediatr 2004;71:21–3. [DOI] [PubMed] [Google Scholar]
- 91.De Boeck K, Alifier M, Vandeputte S. Sputum induction in young cystic fibrosis patients. Eur Respir J 2000;16:91–4. [DOI] [PubMed] [Google Scholar]
- 92.Henig NR, Tonelli MR, Pier MV, Burns JL, Aitken ML. Sputum induction as a research tool for sampling the airways of subjects with cystic fibrosis. Thorax 2001;56:306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suri R, Marshall LJ, Wallis C, Metcalfe C, Shute JK, Bush A. Safety and use of sputum induction in children with cystic fibrosis. Pediatr Pulmonol 2003;35:309–13. [DOI] [PubMed] [Google Scholar]
- 94.Ho SA, Ball R, Morrison LJ, Brownlee KG, Conway SP. Clinical value of obtaining sputum and cough swab samples following inhaled hypertonic saline in children with cystic fibrosis. Pediatr Pulmonol 2004;38:82–7. [DOI] [PubMed] [Google Scholar]
- 95.Burns JL, Saiman L, Whittier S, et al. Comparison of two commercial systems (Vitek and MicroScan-WalkAway) for antimicrobial susceptibility testing of Pseudomonas aeruginosa isolates from cystic fibrosis patients. Diagn Microbiol Infect Dis 2001;39:257–60. [DOI] [PubMed] [Google Scholar]
- 96.Burns JL, Saiman L, Whittier S, et al. Comparison of agar diffusion methodologies for antimicrobial susceptibility testing of Pseudomonas aeruginosa isolates from cystic fibrosis patients. J Clin Microbiol 2000;38:1818–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ciofu O, Giwercman B, Pedersen SS, Hoiby N. Development of antibiotic resistance in Pseudomonas aeruginosa during two decades of antipseudomonal treatment at the Danish CF Center. APMIS 1994;102:674–80. [PubMed] [Google Scholar]
- 98.Taccetti G, Campana S, Marianelli L. Multiresistant non-fermentative gram-negative bacteria in cystic fibrosis patients: the results of an Italian multicenter study. Italian Group for Cystic Fibrosis microbiology. Eur J Epidemiol 1999;15:85–8. [DOI] [PubMed] [Google Scholar]
- 99.Aaron SD, Vandemheen KL, Ferris W, et al. Combination antibiotic susceptibility testing to treat exacerbations of cystic fibrosis associated with multiresistant bacteria: a randomised, double-blind, controlled clinical trial. Lancet 2005;366:463–71. [DOI] [PubMed] [Google Scholar]
- 100.Aaron SD, Ferris W, Ramotar K, Vandemheen K, Chan F, Saginur R. Single and combination antibiotic susceptibilities of planktonic, adherent, and biofilm-grown Pseudomonas aeruginosa isolates cultured from sputa of adults with cystic fibrosis. J Clin Microbiol 2002;40:4172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smith AL, Fiel SB, Mayer-Hamblett N, Ramsey B, Burns JL. Susceptibility testing of Pseudomonas aeruginosa isolates and clinical response to parenteral antibiotic administration: lack of association in cystic fibrosis. Chest 2003;123:1495–502. [DOI] [PubMed] [Google Scholar]
- 102.Moskowitz SM, Foster JM, Emerson JC, Gibson RL, Burns JL. Use of Pseudomonas biofilm susceptibilities to assign simulated antibiotic regimens for cystic fibrosis airway infection. J Antimicrob Chemother 2005;56:879–86. [DOI] [PubMed] [Google Scholar]
- 103.Yau YC, Ratjen F, Tullis E, et al. Randomized controlled trial of biofilm antimicrobial susceptibility testing in cystic fibrosis patients. J Cyst Fibros 2015;14:262–6. [DOI] [PubMed] [Google Scholar]
- 104.Somayaji R, Parkins MD, Shah A, et al. Antimicrobial susceptibility testing (AST) and associated clinical outcomes in individuals with cystic fibrosis: A systematic review. J Cyst Fibros 2019;18:236–43. [DOI] [PubMed] [Google Scholar]
- 105.Moore RA, Hancock RE. Involvement of outer membrane of Pseudomonas cepacia in aminoglycoside and polymyxin resistance. Antimicrob Agents Chemother 1986;30:923–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lewin C, Doherty C, Govan J. In vitro activities of meropenem, PD 127391, PD 131628, ceftazidime, chloramphenicol, co-trimoxazole, and ciprofloxacin against Pseudomonas cepacia. Antimicrob Agents Chemother 1993;37:123–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aaron SD, Ferris W, Henry DA, Speert DP, Macdonald NE. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am J Respir Crit Care Med 2000;161:1206–12. [DOI] [PubMed] [Google Scholar]
- 108.Burns JL, Saiman L. Burkholderia cepacia infections in cystic fibrosis. Pediatr Infect Dis J 1999;18:155–6. [DOI] [PubMed] [Google Scholar]
- 109.Lang BJ, Aaron SD, Ferris W, Hebert PC, MacDonald NE. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with multiresistant strains of Pseudomonas aeruginosa. Am J Respir Crit Care Med 2000;162:2241–5. [DOI] [PubMed] [Google Scholar]
- 110.Krueger TS, Clark EA, Nix DE. In vitro susceptibility of Stenotrophomonas maltophilia to various antimicrobial combinations. Diagn Microbiol Infect Dis 2001;41:71–8. [DOI] [PubMed] [Google Scholar]
- 111.Saiman L, Chen Y, Tabibi S, et al. Identification and antimicrobial susceptibility of Alcaligenes xylosoxidans isolated from patients with cystic fibrosis. J Clin Microbiol 2001;39:3942–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Flume PA, Mogayzel PJ Jr., Robinson KA, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med 2009;180:802–8. [DOI] [PubMed] [Google Scholar]
- 113.VanDevanter DR, O’Riordan MA, Blumer JL, Konstan MW. Assessing time to pulmonary function benefit following antibiotic treatment of acute cystic fibrosis exacerbations. Respir Res 2010;11:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Horsley A, Jones AM, Lord R. Antibiotic treatment for Burkholderia cepacia complex in people with cystic fibrosis experiencing a pulmonary exacerbation. Cochrane Database Syst Rev 2016:CD009529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.West NE, Beckett VV, Jain R, et al. Standardized Treatment of Pulmonary Exacerbations (STOP) study: Physician treatment practices and outcomes for individuals with cystic fibrosis with pulmonary Exacerbations. J Cyst Fibros 2017;16:600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dovey M, Aitken ML, Emerson J, McNamara S, Waltz DA, Gibson RL. Oral corticosteroid therapy in cystic fibrosis patients hospitalized for pulmonary exacerbation: a pilot study. Chest 2007;132:1212–8. [DOI] [PubMed] [Google Scholar]
- 117.Smyth A, Tan KH, Hyman-Taylor P, et al. Once versus three-times daily regimens of tobramycin treatment for pulmonary exacerbations of cystic fibrosis--the TOPIC study: a randomised controlled trial. Lancet 2005;365:573–8. [DOI] [PubMed] [Google Scholar]
- 118.Smyth A, Lewis S, Bertenshaw C, Choonara I, McGaw J, Watson A. Case-control study of acute renal failure in patients with cystic fibrosis in the UK. Thorax 2008;63:532–5. [DOI] [PubMed] [Google Scholar]
- 119.Obrink-Hansen K, Jensen-Fangel S, Brock B, et al. Piperacillin/tazobactam continuous infusion at 12G/1.5G per day in CF patients results in target plasma-concentrations. J Cyst Fibros 2016;15:e13–4. [DOI] [PubMed] [Google Scholar]
- 120.Riethmueller J, Junge S, Schroeter TW, et al. Continuous vs thrice-daily ceftazidime for elective intravenous antipseudomonal therapy in cystic fibrosis. Infection 2009;37:418–23. [DOI] [PubMed] [Google Scholar]
- 121.Hubert D, Le Roux E, Lavrut T, et al. Continuous versus intermittent infusions of ceftazidime for treating exacerbation of cystic fibrosis. Antimicrob Agents Chemother 2009;53:3650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.VanDevanter DR, Heltshe SL, Spahr J, et al. Rationalizing endpoints for prospective studies of pulmonary exacerbation treatment response in cystic fibrosis. J Cyst Fibros 2017;16:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sanders DB, Solomon GM, Beckett VV, et al. Standardized Treatment of Pulmonary Exacerbations (STOP) study: Observations at the initiation of intravenous antibiotics for cystic fibrosis pulmonary exacerbations. J Cyst Fibros 2017;16:592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heltshe SL, West NE, VanDevanter DR, et al. Study design considerations for the Standardized Treatment of Pulmonary Exacerbations 2 (STOP2): A trial to compare intravenous antibiotic treatment durations in CF. Contemp Clin Trials 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bennett AV, Patrick DL, Lymp JF, Edwards TC, Goss CH. Comparison of 7-day and repeated 24-hour recall of symptoms of cystic fibrosis. J Cyst Fibros 2010;9:419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goss CH, Edwards TC, Ramsey BW, Aitken ML, Patrick DL. Patient-reported respiratory symptoms in cystic fibrosis. J Cyst Fibros 2009;8:245–52. [DOI] [PubMed] [Google Scholar]
- 127.Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the cystic fibrosis questionnaire-revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic pseudomonas aeruginosa airway infection.(Clinical report). Chest 2009;135:1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Flume PA, Mogayzel PJ, Robinson KA, et al. Cystic Fibrosis Pulmonary Guidelines: Treatment of Pulmonary Exacerbations. Am J Respir Crit Care Med 2009;180:802–8. [DOI] [PubMed] [Google Scholar]
- 129.VanDevanter DR, Pasta DJ, Konstan MW. Treatment and demographic factors affecting time to next pulmonary exacerbation in cystic fibrosis. J Cyst Fibros 2015;14:763–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Collaco JM, Green DM, Cutting GR, Naughton KM, Mogayzel PJ, Jr. Location and duration of treatment of cystic fibrosis respiratory exacerbations do not affect outcomes. Am J Respir Crit Care Med 2010;182:1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Waters V, Stanojevic S, Klingel M, et al. Prolongation of antibiotic treatment for cystic fibrosis pulmonary exacerbations. J Cyst Fibros 2015. [DOI] [PubMed] [Google Scholar]
- 132.Cystic Fibrosis Foundation Patient Registry 2011 Annual Data Report to the Center Directors. 2012.
- 133.Sanders DB, Bittner RC, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med 2010;182:627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Flume PA, Wainwright CE, Elizabeth Tullis D, et al. Recovery of lung function following a pulmonary exacerbation in patients with cystic fibrosis and the G551D-CFTR mutation treated with ivacaftor. J Cyst Fibros 2018;17:83–8. [DOI] [PubMed] [Google Scholar]
- 135.VanDevanter DR, Heltshe SL, Spahr J, et al. Rationalizing endpoints for prospective studies of pulmonary exacerbation treatment response in cystic fibrosis. J Cyst Fibros 2017;16:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wolter JM, Bowler SD, Nolan PJ, McCormack JG. Home intravenous therapy in cystic fibrosis: a prospective randomized trial examining clinical, quality of life and cost aspects. Eur Respir J 1997;10:896–900. [PubMed] [Google Scholar]
- 137.Schechter MS, VanDevanter DR, Pasta DJ, et al. Treatment Setting and Outcomes of Cystic Fibrosis Pulmonary Exacerbations. Ann Am Thorac Soc 2017. [DOI] [PubMed] [Google Scholar]
- 138.Foundation CF. Cystic Fibrosis Foundation Patient Registry 2015 Annual Data Report. 2016.
- 139.Roehrer E, Cummings E, Beggs S, et al. Pilot evaluation of web enabled symptom monitoring in cystic fibrosis. Inform Health Soc Care 2013;38:354–65. [DOI] [PubMed] [Google Scholar]
- 140.Roehrer E, Cummings E, Turner P, et al. Supporting cystic fibrosis with ICT. Stud Health Technol Inform 2013;183:137–41. [PubMed] [Google Scholar]
- 141.Cummings E, Hauser J, Cameron-Tucker H, et al. Enhancing self-efficacy for self-management in people with cystic fibrosis. Stud Health Technol Inform 2011;169:33–7. [PubMed] [Google Scholar]
- 142.Aitken ML, Caldwell E, E. W, Goss CH. Early Intervention in Pulmonary Exacerbation. Pediatr Pulmonol 2011;46:A331. [Google Scholar]
- 143.Keating D, Marigowda G, Burr L, et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018;379:1612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kirwan L, Fletcher G, Harrington M, et al. Longitudinal Trends in Real-World Outcomes after Initiation of Ivacaftor. A Cohort Study from the Cystic Fibrosis Registry of Ireland. Ann Am Thorac Soc 2019;16:209–16. [DOI] [PubMed] [Google Scholar]
- 145.Heltshe SL, Mayer-Hamblett N, Burns JL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis 2015;60:703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Frost FJ, Nazareth DS, Charman SC, Winstanley C, Walshaw MJ. Ivacaftor Is Associated with Reduced Lung Infection by Key Cystic Fibrosis Pathogens: A Cohort Study Using National Registry Data. Ann Am Thorac Soc 2019. [DOI] [PubMed] [Google Scholar]