

In this study, Simond et al. developed a Cre-inducible murine model expressing a point-activated ESR1Y541S (ESR1Y537S in humans) driven by its endogenous promoter and provide critical insights into the tissue specific roles of ER during development.
Keywords: ERα, ESR1, point mutation, GEMM, development
Abstract
Mutations in the estrogen receptor α (ERα) occur in endocrine-resistant metastatic breast cancer. However, a major gap persists with the lack of genetically tractable immune competent mouse models to study disease. Hence, we developed a Cre-inducible murine model expressing a point-activated ESR1Y541S (ESR1Y537S in humans) driven by its endogenous promoter. Germline expression of mutant ESR1Y541S reveals dramatic developmental defects in the reproductive organs, mammary glands, and bones of the mice. These observations provide critical insights into the tissue-specific roles of ERα during development and highlights the potential use of our model in further developmental and cancer studies.
ER-positive breast cancer was the first histological subtype to be identified and accounts for 70% of diagnoses (Toy et al. 2013; Jeselsohn et al. 2015). ERα is a member of the family of nuclear receptors. When estrogen binds to the ligand-binding domain (LBD) of the ER, the receptor dimerizes and translocates to the nucleus (Beekman et al. 1993). In the nucleus it binds directly to estrogen response elements (EREs) in the DNA (classical model) or binds to other proteins that interact with separate areas of DNA (nonclassical model) leading to the transcriptional activation or repression of associated genes (O'Lone et al. 2004). In the clinic, ER-positive breast cancer is generally treated with the use of a selective estrogen receptor modulator (SERM): tamoxifen (Jaiyesimi et al. 1995; Chang 2012). However, 30% of patients display de novo resistance and the majority of initial responders eventually develop acquired resistance (Ariazi et al. 2006; Riggins et al. 2007; Jeselsohn et al. 2015). In up to 20% of endocrine-resistant metastatic tumors, this is caused by mutations in the LBD (i.e., ESR1Y537S in humans, ESR1Y541S in mice) of ERα, which leads to constitutive activation of the receptor (Weis et al. 1996; Jeselsohn et al. 2018).
Although a number of cell lines expressing activated forms of ERα exist, a genetically engineered mouse model (GEMM) was lacking (Martin et al. 2017; Toy et al. 2017; Jeselsohn et al. 2018). To functionally address the role of the mutant ESR1Y541S, we generated mice expressing a Cre-inducible ESR1Y541S allele in a germline fashion using the ubiquitous βActin promoter to drive Cre expression.
Results and Discussion
Germline expression of a single allele of ESR1Y541S results in runting of female and male carriers. The experimental male mice display prominent nipples and a closer anal–genital region, making them phenotypically indistinguishable from their female counterparts (Fig. 1; Supplemental Fig. S1). Experimental mice are significantly smaller in length and weight compared with control animals (Fig. 1C). Interestingly, only 15% of female experimental mice survive past 150 d of age compared with 85% of experimental male and 100% of wild-type mice (Fig. 1D). Pathological examination of female carriers reveals ovarian, uterine, and fallopian tube cysts, which upon rupturing could lead to their premature death (Figs. 1D, 2A). Even though rare, male mice are also found to die more often than their control counterparts. Notably, one male mouse (309 d old) was found to have developed a large cyst in its abdomen that seemed to be filled with seminal fluid. As such, it is possible that cysts also develop in male mice (data not shown).
Figure 1.
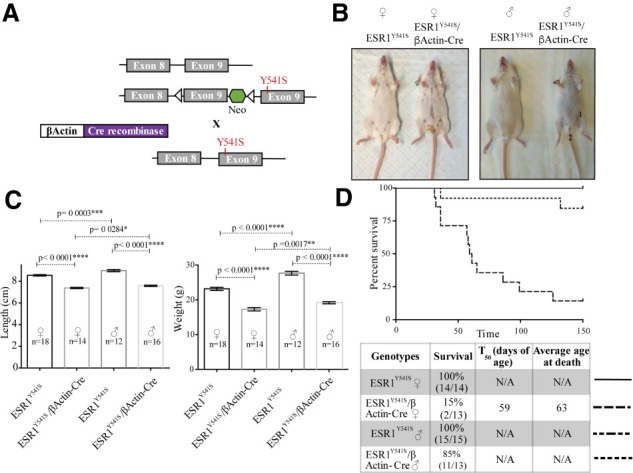
Germline expression of an ESR1Y541S knock-in feminizes male mice and mice of both sexes are runted. (A) Simplified breeding strategy. Animals expressing Cre-recombinase driven by the βActin promoter are crossed with mice expressing a conditional ESR1Y541S knock-in. (B) Images of wild-type and experimental female and male mice showing prominent nipples (1) and closer anal–genital region (2). Mice were sacrificed at 10 wk of age. (C) Length and weight of female and male wild-type and experimental mice. (D) Survival curve and summary table of female and male mice kept up to 150 d.
Figure 2.
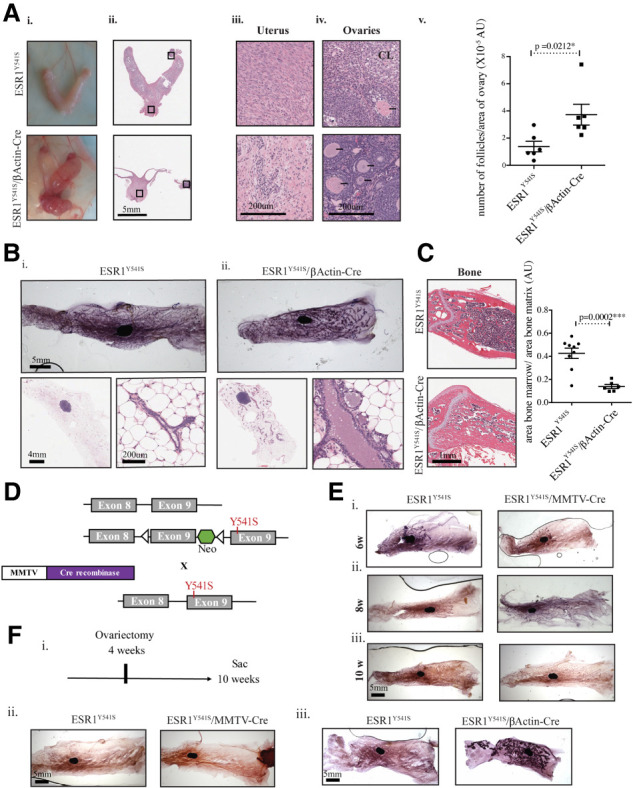
Female mice expressing ESR1Y541S develop uterine cysts, are infertile, and display abnormal mammary gland and bone development. (A, panels i–iv) Pictures and H&E staining of the female reproductive organs at different magnifications. (Panel v) Quantification of the number of follicles per surface area of the ovary in six samples per experimental condition. (B, panels i,ii) Mammary gland whole mounts of wild-type and experimental female mice as well as H&E sections at different magnifications. (C) H&E staining of bone tissue and quantification of the area of bone marrow/area of bone matrix in nine control samples and six experimental samples. (D) Simplified breeding strategy. Animals expressing Cre-recombinase driven by the MMTV promoter are crossed to mice expressing conditional ESR1Y541S. (E, panels i–iii) Mammary gland whole mounts of wild-type and experimental female mice at 6, 8, and 10 wk of age. (F, panel i) Schematic of ovariectomy timeline. (Panels ii,iii) Mammary gland whole mounts of ovariectomized mice.
A more exhaustive investigation of the experimental female phenotype reveals an abnormal distribution of epithelial cells in the uterus. The ovaries contain a greater number of follicles per surface area but lack the presence of the corpus luteum (CL), a structure that develops after ovulation (Niswender et al. 2000). Thus, their infertility may be due to their inability to ovulate (Fig. 2A). In addition, the mammary glands of experimental mice develop ductal ectasia and form ductal side buds, similar to mice overexpressing AIB-1, a known cofactor of ERα (Fig. 2B; Supplemental Fig S2A; Tikkanen et al. 2000). Besides sexual maturation, estrogen signaling is known to be essential in maintaining bone mineral density and proper distribution of osteoblasts (generates bone matrix) and osteoclasts (degrades bone matrix) (Gay et al. 2000; Khalid and Krum 2016). Consistent with the role of estrogens in stimulating osteoblast function, we found that experimental mice have a significant increase in bone area (lamellar and calcified collagen) (Fig. 2C; Waters et al. 2001; Khalid and Krum 2016). In contrast, no pathological difference was observed in the spleen, kidney, thymus, liver, and brain (Supplemental Fig. S2B–F). Additionally, there is no difference in cellular proliferation assayed by Ki67, or apoptosis assayed by cleaved caspase 3 (CC3) in the mammary glands, uterus, and ovaries. However, there is a significant decrease in the level of ERα in the uterus of experimental mice, likely due to the abundance of cystic cells without βActin expression (Supplemental Fig. S3). These observations indicate that expression of activated ERα has selective developmental effects on a number of tissues.
Given the profound effect of ESR1Y541S on ductal outgrowth, mammary epithelial expression of ESR1Y541S was evaluated. The ESR1Y541S mice were intercrossed to ones expressing Cre-recombinase driven by the mouse mammary tumor virus (MMTV) promoter (Fig. 2D). In contrast to what was observed in the germline strain, there is no difference in mammary ductal outgrowth in virgin female mice (6, 8, and 10 wk of age) or during pregnancy, lactation, and involution (days 2, 4, and 6) (Fig. 2E; Supplemental Fig. S4A). All male mammary fat pads remain duct-free (Supplemental Fig. S4B). To assess whether mammary ductal outgrowth is independent of ovarian estrogen in the context of mammary epithelial expression of ESR1Y541S, ovariectomy was performed on 4 wk old female mice, which resulted in attenuated ductal outgrowth. However, ovariectomy did not inhibit ductal outgrowth in the germline strain, demonstrating independence of exogenous estrogen (Fig. 2F; Supplemental Fig. S4C). The MMTV promoter is active in response to steroid hormones, thus after puberty, which argues that the developmental window of ESR1Y541S expression may dictate the severity of the phenotype (Otten et al. 1988; Wagner et al. 2001). Alternatively, expression of ESR1Y541S may be essential in both the epithelium and stroma for the development of mammary ducts (Mueller et al. 2002).
Pathologically, male mice with germline ESR1Y541S expression display a dramatic atrophy of the testes and seminal vesicles as well as an absence of the preputial glands, which are critical in sexual and dominance behavior (Fig. 3A; Bronson and Caroom 1971; Bronson and Marsden 1973). The reduction in size of the male sexual organs is consistent with previous results showing that the mass of seminal vesicles are reduced when ERα is overexpressed (Hruska et al. 2002). These mice are sterile, consistent with the absence of sperm, sertoli (support germ cells), and leydig cells (support cells that produce testosterone) (Fig. 3A; Lucas et al. 2011; Shima et al. 2013). In contrast to their control counterparts, 44% of experimental male mice develop an abnormal ductal tree (mostly hyperplasia) in at least one mammary gland, consistent with the feminization phenotype (Fig. 3B). Male carriers have similar hypertrophic bone cortices as the experimental female mice (Fig. 3C), but no other pathological differences were noted (Supplemental Fig. S5). The status of proliferation and apoptosis of the male reproductive tract was also evaluated. Elevated levels of ERα correlated with an increase in Ki67 in the testes and seminal vesicles, indicating a potential compensatory increase in proliferation of these atrophied organs (Supplemental Fig. S6). Although an overt pathological phenotype in the liver was absent, serum levels of liver enzymes were quantified. An increase in the levels of alkaline phosphatase (AP) in male experimental mice and a trend in female mice is observed. High AP levels without significant increase in other liver enzymes is likely due to the bone defect (Supplemental Fig. S7A; Stein and Lian 1993). In addition, there are no significant differences in the levels of steroid hormones assessed by ELISA (Supplemental Fig. S7B). Taken together, these data argue that germline expression of a single ESR1Y541S allele is sufficient to feminize male carriers.
Figure 3.
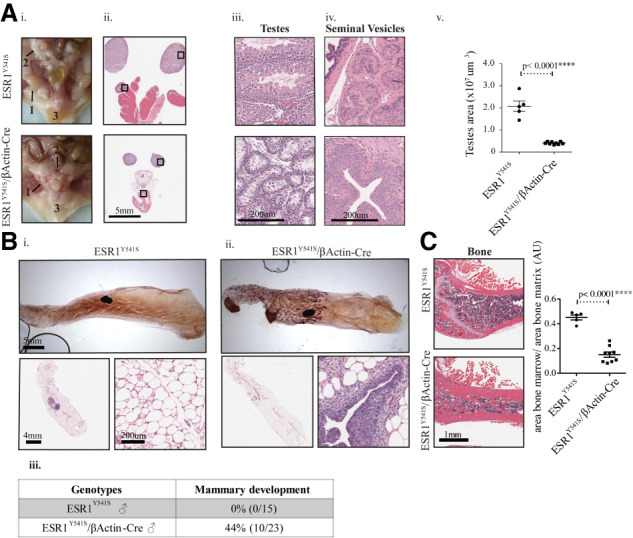
Male mice expressing ESR1Y541S develop atrophied reproductive organs and display abnormal ductal and bone development. (A, panels i–iv) Pictures and H&E staining of the male reproductive organs at different magnifications showing testes (1), seminal vesicles (2), and preputial glands (3). (Panel v) Quantification of the surface area of the testes in five samples for control mice and nine for experimental mice. (B, panels i,ii) Mammary gland whole mounts of wild-type and experimental male mice as well as H&E sections at different magnifications. (Panel iii) Table summary of the percentage and number of male mice that develop a ductal tree in at least one mammary gland. (C) H&E staining of bone tissue and quantification of the area of bone marrow/area of bone matrix in five control samples and nine experimental samples.
To further identify developmental pathways affected by germline expression of ESR1Y541S, spleen, bone, mammary gland (female), uterus/fallopian tubes, ovaries, testes, and seminal vesicles from control and experimental mice were subjected to RNA-seq analysis. Strikingly, we found that the transcriptional profile of experimental male testes and seminal vesicles cluster with that of the ovaries and uterus/fallopian tubes, respectively, indicating that the male reproductive organs have adopted transcriptional features from their female counterparts. The rest of the transcriptional programs cluster by organ and experimental condition (Fig. 4A). Volcano plots show that many genes are found differentially regulated in between experimental conditions (Supplemental Fig. S8). Gene ontology (GO) analysis reveals signatures up-regulated in the testes and seminal vesicles that relate to cell migration, differentiation, and the extracellular matrix. In contrast, down-regulated genes are involved with sperm differentiation and movement in the testes, and involve the endoplasmic reticulum, Golgi, and glycosylation in the seminal vesicles. The ovaries and uterus/fallopian tubes display an up-regulation of inflammatory signatures, likely associated with the presence of cysts. In addition, the ovaries exhibit down-regulation of steroid metabolism and the uterus/fallopian tubes show down-regulation of organ morphogenesis (Supplemental Figs. S9, S10). In the bone, there is an up-regulated muscle and contractility signature and a down-regulation of B-cell activation in both female and male experimental mice. In the mammary gland, there is an up-regulation of a ribosomal signature and down-regulation of fatty acid metabolism. When isolating the top 40 differentially regulated genes in the mammary gland we decided to validate three significantly up-regulated genes in the experimental mice: lipocalin 2 (Lcn2), whey acidic protein (Wap), and β-casein (Csn2). These genes were chosen for their high number of reads and low P-values. Interestingly, Lcn2 is associated with the innate immune response and cancer progression (Flo et al. 2004; Yang et al. 2009). Both Wap and Csn2 are genes associated with the lactating mammary gland, suggesting that the fluid filling the ducts of the experimental mice may be milk (Supplemental Table ST1; Supplemental Figs. S11, S12; Campbell et al. 1984; Kumar et al. 1994). To address the alteration in the differentiation status of female and male reproductive organs, we performed immunostaining with an anti-Sox9 antibody, a protein that plays a critical role in the differentiation of sertoli cells and the maintenance of stem cells (Jo et al. 2014). We show an increase in Sox9 protein levels in the uterus/fallopian tubes, testes, and seminal vesicles, which is consistent with their undifferentiated state (Fig. 4B). These observations support the contention that expression of a mutated ESR1 allele has a dramatic impact on the transcriptional profiles of organs sensitive to ER signaling during development.
Figure 4.
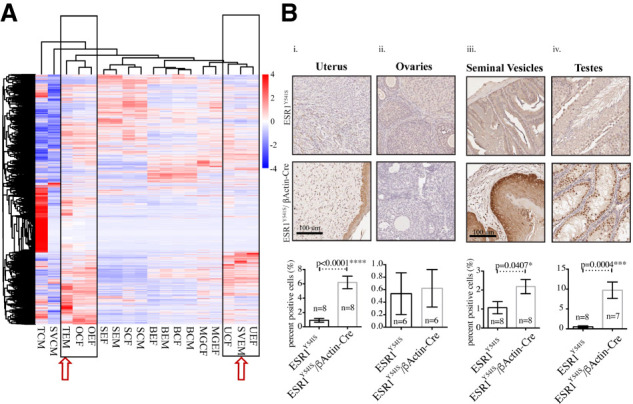
The reproductive organs of experimental male mice are more transcriptionally similar to female reproductive organs. (A) Heat map of differentially expressed genes. (T) testes, (SV) seminal vesicles, (O) ovaries, (S) spleen, (B) bone, (MG) mammary gland, (U) uterus/fallopian tubes, (E) experimental, (C) control, (F) female, (M) male. (B, panels i,ii) Immunohistochemistry for Sox9 in the uterus/fallopian tubes and the ovaries. (Panels iii,iv) Immunohistochemistry for Sox9 in the seminal vesicles and the testes. Analysis was done using Halo; moderate and strong staining levels were used to quantify the uterus, ovaries, and testes, and strong staining levels were used to quantify the seminal vesicles.
ESR1Y541S is rarely found in primary ER-positive breast tumors, but has been described in metastasis of endocrine resistant cancers (Takeshita et al. 2015). Notably, ER-positive cancer progression has been found to occur through the recruitment of myeloid-derived suppressor cells (MDSCs) to the tumor (Svoronos et al. 2017; Rothenberger et al. 2018). GO analysis of spleen samples identified up-regulation of signatures associated with chromosomal segregation and DNA replication in experimental mice and down-regulation of the lymphoid lineage (Supplemental Fig. S13A–D). Therefore, we investigated whether this mutation correlated with an up-regulation at the protein level of the myeloid lineage by assessing MDSCs in the spleen and blood identified with CD11b+, Ly6G+, and Ly6C+ by fluorescence-activated cell sorting (FACS) (Supplemental Fig. S13E). We found a significant up-regulation of MDSCs in the spleen of experimental mice and a similar trend in the blood (Fig. 5; Supplemental Fig. S14). Taken together, these data argue that in addition to altering the differentiation status of the reproductive tract, ESR1Y541S can have a profound effect on the immune system.
Figure 5.
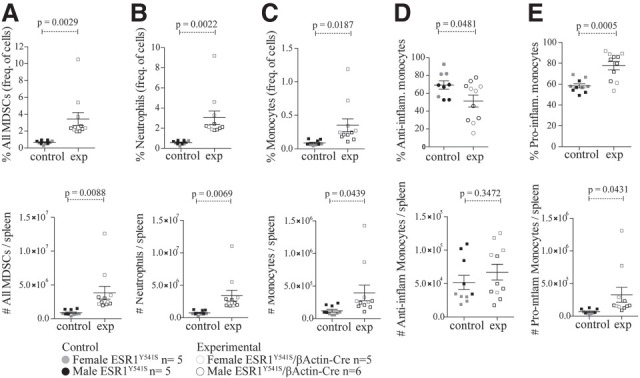
Animals with germline expression of ESR1Y541S have higher levels of MDSCs in the spleen. Percentage and number of MDSCs (A), neutrophils (B), monocytes (C), anti-inflammatory monocytes (D), and proinflammatory monocytes (E). Data from female and male mice were pooled.
It is important to note that the dramatic effects on tissue morphogenesis in this model is a consequence of expressing a single mutant allele at physiological levels, suggesting an integral role for the tight regulation of ESR1 activity during development. The observed feminization of the male ESR1Y541S GEMM is consistent with the longstanding view that estrogen signaling plays a critical role in the sexual differentiation of reproductive tracts in both sexes (Bondesson et al. 2015).
In addition, our observations that expression of ESR1Y541S can skew the MDSC population is in agreement with the concept that estrogen signaling can modulate the immune microenvironment (Ouyang et al. 2016). Further studies with myeloid-specific expression of ESR1Y541S should allow us to test this issue directly. Consistent with clinical observations that ESR1 mutations are rarely found in primary tumors, mammary epithelial expression of ESR1Y541S does not induce any pathological abnormality in the mammary gland. These data strongly argue that expression of ESR1Y541S is not sufficient to drive malignant transformation of the mammary epithelium but requires additional genetic alterations to facilitate transformation. In this regard, AIB-1 has been frequently reported as overexpressed and amplified in ER-positive breast cancer (Anzick et al. 1997). In addition, mammary epithelial loss of NF1, an ERα corepressor, has been reported to drive breast cancer development (Wallace et al. 2012). All these observations potentiate the use for our conditional ESR1Y541S mouse model in the study of development, behavior, and endocrine-resistant metastatic breast cancer in combination with other mouse models of mammary tumorigenesis.
Materials and methods
Generation of ESR1Y541S mice
This GEMM was generated using a targeting vector expressing wild-type ESR1 with loxP sites flanking wild-type exon 9 and a neomycin stop cassette. Two positive ES clones were sent to the Goodman Cancer Research Center (GCRC) Transgenic Facility for blastocyst injection to obtain chimeric ESR1Y541S mice. One clone had successful germline transmission. ESR1Y541S strain was backcrossed seven times on an FVB/N background. In the presence of Cre-recombinase, the loxP sites recombine excising wild-type exon 9 and the neomycin stop cassette, which proceeds a point mutated exon 9 (details are in the Supplemental Material; Andrechek et al. 2000).
Animal husbandry
Our mice were housed in the animal facility at the GCRC and our experiments followed our approved animal use protocol (AUP). The strains used in this study were ESR1, βActin-Cre, MMTV-Cre, and were all kept on an FVB/N background.
DNA extraction genotyping PCR
DNA was extracted from tail pieces at 2 wk of age and at sacrifice using salt precipitation as described previously (Simond et al. 2017). Primers used for genotyping were ESR1 F (GCCTTTGGAGTTGCTCATCC), ESR1 R (TTGTAGAGATGCTCCATGCC), gender F (CTGAAGCTTTTGGCTTTGAG), gender R (CCACTGCCAAATTCTTTGG), Cre F (TGCTCTCGTTTGCCG), and Cre R (ACTGTGTCCAGACCAGGC). All primers were used at a concentration of 10 µM. The PCR enzymes used were as follows: Qiagen Taq polymerase (Qiagen 20120X) was used for ESR1 and gender genotyping PCRs, and OneTaq polymerase (NEB M0480X) was used for Cre genotyping PCRs.
Tissue sample processing
Tissue samples were collected at necropsy and either flash-frozen in liquid nitrogen and stored at −80°C until further use or fixed immediately in 10% neutralized formalin for 24 h (brain, liver, mammary gland, kidney, spleen, and thymus) or in 4% paraformalyhyde for 24 h (bone). Fixed tissue was paraffin embedded and sectioned at a thickness of 4 µm by the histology core facility in the GCRC at McGill University. H&E staining was performed by the histology core facility.
Immunohistochemistry
Slides were deparaffinized and hydrated. The antigen retrieval step was done by submerging slides in sodium citrate (pH 6) (Vector Laboratories H-3300). Sections were blocked for 5 min with 1× power block (Biogenex) and then incubated with primary antibody. Next, a secondary antibody (SignalStain Boost IHC detection reagent HRP, rabbit 8114) was added. Once counterstain was deemed sufficient, samples were dehydrated and immediately mounted. Scanned slides were analyzed using the Halo software (details are in the Supplemental Material).
The primary antibodies used were Ki67 (1:50 rabbit mAb; Cell Signaling 12202), cleaved caspase 3 (1:200 rabbit mAb; Cell Signaling 9661), ERα (1:200 rabbit mAb; Santa Cruz Biotechnology 543 ), and Sox9 (1:400 rabbit mAb; Abcam 3697). All staining was done on formalin-fixed paraffin embedded tissue.
Quantification of ductal ectasia
H&E-stained images were used and a classifier was designed using the Halo software that could recognize the mammary epithelium and the lumen of the ducts. The surface area of the mammary epithelium or the lumen was divided by the total surface area of the mammary gland.
ELISA
For estradiol and progesterone, four samples were pooled and three independent pools were quantified. For testosterone, five independent serum samples were used for control and experimental conditions
The following kits were used: estradiol parameter assay kit KGE014 from R&D Systems, progesterone ELISA kit ADI-900-011from Enzo Life Sciences, and testosterone parameter assay kit KGE010 from R&D Systems.
Protocol from the companies were followed and plates were read on the Varioskan plate reader (Thermo Fisher Scientific)
RNA-seq analysis
Two independent samples per experimental condition were used and RNA was extracted using the RNeasy kit (Qiagen). RNA-seq analysis was performed by Novogene (details are in the Supplemental Material)
qRT-PCR
mRNA was reverse-transcribed into cDNA using TransScript (TransGen Biotech). Real-time quantitative PCR was performed using SYBR Green master mix (Roche), run on a LighCycler 480 in triplicates, and normalized to Gapdh (primers are listed in the Supplemental Material).
FACS analysis
Peripheral blood was collected by cardiac puncture after treating mice with 0.25 mg of ketamine and red blood cells were lysed using Vitalyse (Bio E, Inc.). Cells were stained and then fixed using IC fixation buffer (eBioscience). Full spleens were processed into single-cell suspensions and red blood cells were lysed with ACK lysis buffer (150 mM NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA, dissolved in H2O and adjusted to pH 7.2–7.4). Cells were then stained and fixed using IC fixation buffer (eBioscience). The following antibodies were used in an appropriate combination of fluorochromes: CD11b (clone M1/70; BioLegend), CD11c (clone N418; BioLegend), CD45 (clone 30-F11; BD), F4/80 (clone Bm8; BioLegend), Ly6C (clone HK1.4; BioLegend), and Ly6G (clone 1A8; BioLegend). Samples were analyzed with a BD LSRFortessa flow cytometer (BD Biosciences) and FlowJo software (Tree Star). Flow cytometry was performed on spleen and blood when mice reached 10 wk of age, collected in five separate sessions, and then pooled.
Statistical analysis
All experiments on animals were done nonrandomized and nonblinded. Data were analyzed using GraphPad Prism. The statistical analysis used throughout the study was an unpaired two-tailed student t-test. All error bars represent standard error of the mean (SEM). For all statistical tests, P-values <0.05 were considered statistically significant.
Data availability
RNA-seq data have been uploaded to the Sequence Read Archive (SRA) under accession number PRJNA624176.
Supplementary Material
Acknowledgments
We thank Cynthia Lavoie for performing the ovariectomies, Dr. Mitra Cowen and colleagues from the transgenic core at the Goodman Cancer Research Center (GCRC) for their assistance with the generation of the transgenic strain, staff at the Comparative Medicine and Animal Resources Center (CMARC) for assistance with studies involving mice, and the members of the Muller laboratory for their support and feedback. This work is supported by Canadian Institutes of Health Research (CIHR) Foundation Award FDN-148373 to W.J.M., Canada Research Chair in Molecular Oncology to W.J.M., and CIHR (PJT-152903) to M.J.R. M.J.R. received salary support from the Fonds de Recherche du Québec Santé-Chercheurs-Boursiers Junior 1 (32807) and CIHR New Investigator Award 360874. A.M.S. received a Dr. Victor K.S. Lui Studentship and the Michael D'Avirro Fellowship in Molecular Oncology Research.
Author contributions: C.L. performed all cloning for the generation of the ESR1Y541S strain. M.J.M. generated the data relating to the ESR1Y541S/MMTV-Cre cross for mammary gland whole-mount analysis at 6, 8, and 10 wk of age as well as mice during pregnancy, lactation, and involution (days 2, 4, and 6). S.A.C. and M.J.R. performed the FACS experiment quantifying the levels of MDSCs in the spleen and blood. A.M.S. performed all experiments except for the ones specified above. A.M.S. and W.J.M. designed the experiments and wrote the manuscript. All authors commented on the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.339424.120.
References
- Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. 2000. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. PNAS 97: 3444–3449. 10.1073/pnas.97.7.3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X-Y, Sauter G, Kallioniemi O-P, Trent JM, Meltzer PS. 1997. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277: 965–968. 10.1126/science.277.5328.965 [DOI] [PubMed] [Google Scholar]
- Ariazi EA, Ariazi JL, Cordera F, Jordan VC. 2006. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem 6: 181–202. 10.2174/156802606776173483 [DOI] [PubMed] [Google Scholar]
- Beekman JM, Allan GF, Tsai SY, Tsai M-J, O'malley BW. 1993. Transcriptional activation by the estrogen receptor requires a conformational change in the ligand binding domain. Mol Endocrinol 7: 1266–1274. [DOI] [PubMed] [Google Scholar]
- Bondesson M, Hao R, Lin C-Y, Williams C, Gustafsson J-Å. 2015. Estrogen receptor signaling during vertebrate development. Biochim Biophys Acta 1849: 142–151. 10.1016/j.bbagrm.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson FH, Caroom D. 1971. Preputial gland of the male mouse: attractant function. Reproduction 25: 279–282. 10.1530/jrf.0.0250279 [DOI] [PubMed] [Google Scholar]
- Bronson FH, Marsden HM. 1973. The preputial gland as an indicator of social dominance in male mice. Behav Biol 9: 625–628. 10.1016/S0091-6773(73)80056-2 [DOI] [PubMed] [Google Scholar]
- Campbell SM, Rosen JM, Hennighausen LG, Strech-Jurk U, Sippel AE. 1984. Comparison of the whey acidic protein genes of the rat and mouse. Nucleic Acids Res 12: 8685–8697. 10.1093/nar/12.22.8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M-S. 2012. Tamoxifen resistance in breast cancer. Biomol Ther 20: 256–267. 10.4062/biomolther.2012.20.3.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. 2004. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917–921. 10.1038/nature03104 [DOI] [PubMed] [Google Scholar]
- Gay CV, Gilman VR, Sugiyama T. 2000. Perspectives on osteoblast and osteoclast function. Poult Sci 79: 1005–1008. 10.1093/ps/79.7.1005 [DOI] [PubMed] [Google Scholar]
- Hruska KS, Tilli MT, Ren S, Cotarla I, Kwong T, Li M, Fondell JD, Hewitt JA, Koos RD, Furth PA et al. 2002. Conditional over-expression of estrogen receptor α in a transgenic mouse model. Transgenic Res 11: 361–372. 10.1023/A:1016376100186 [DOI] [PubMed] [Google Scholar]
- Jaiyesimi IA, Buzdar AU, Decker DA, Hortobagyi GN. 1995. Use of tamoxifen for breast cancer: twenty-eight years later. JCO 13: 513–529. 10.1200/JCO.1995.13.2.513 [DOI] [PubMed] [Google Scholar]
- Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. 2015. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 12: 573–583. 10.1038/nrclinonc.2015.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, Xiao T, Li W, Qiu X, Buchwalter G, et al. 2018. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 33: 173–186.e5. 10.1016/j.ccell.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo A, Denduluri S, Zhang B, Wang Z, Yin L, Yan Z, Kang R, Shi LL, Mok J, Lee MJ, et al. 2014. The versatile functions of Sox9 in development, stem cells, and human diseases. Genes Dis 1: 149–161. 10.1016/j.gendis.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid AB, Krum SA. 2016. Estrogen receptors α and β in bone. Bone 87: 130–135. 10.1016/j.bone.2016.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Clarke AR, Hooper ML, Horne DS, Law AJ, Leaver J, Springbett A, Stevenson E, Simons JP. 1994. Milk composition and lactation of β-casein-deficient mice. Proc Natl Acad Sci 91: 6138–6142. 10.1073/pnas.91.13.6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas TFG, Pimenta MT, Pisolato R, Lazari MFM, Porto CS. 2011. 17β-estradiol signaling and regulation of Sertoli cell function. Spermatogenesis 1: 318–324. 10.4161/spmg.1.4.18903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L-A, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, Gellert P, Buluwela L, Harrod A, Thornhill A, et al. 2017. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun 8: 1865 10.1038/s41467-017-01864-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SO, Clark JA, Myers PH, Korach KS. 2002. Mammary gland development in adult mice requires epithelial and stromal estrogen receptor α. Endocrinology 143: 2357–2365. 10.1210/endo.143.6.8836 [DOI] [PubMed] [Google Scholar]
- Niswender GD, Juengel JL, Silva PJ, Rollyson MK, McIntush EW. 2000. Mechanisms controlling the function and life span of the corpus luteum. Physiol Rev 80: 1–29. [DOI] [PubMed] [Google Scholar]
- Otten AD, Sanders MM, McKnight GS. 1988. The MMTV LTR Promoter is induced by progesterone and dihydrotestosterone but not by estrogen. Mol Endocrinol 2: 143–147. 10.1210/mend-2-2-143 [DOI] [PubMed] [Google Scholar]
- O'Lone R, Frith MC, Karlsson EK, Hansen U. 2004. Genomic targets of nuclear estrogen receptors. Mol Endocrinol 18: 1859–1875. 10.1210/me.2003-0044 [DOI] [PubMed] [Google Scholar]
- Ouyang L, Chang W, Fang B, Qin J, Qu X, Cheng F. 2016. Estrogen-induced SDF-1α production promotes the progression of ER-negative breast cancer via the accumulation of MDSCs in the tumor microenvironment. Sci Rep 6: 39541 10.1038/srep39541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH. 2007. Pathways to tamoxifen resistance. Cancer Lett 256: 1–24. 10.1016/j.canlet.2007.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberger NJ, Somasundaram A, Stabile LP. 2018. The role of the estrogen pathway in the tumor microenvironment. Int J Mol Sci 19: 611 10.3390/ijms19020611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima Y, Miyabayashi K, Haraguchi S, Arakawa T, Otake H, Baba T, Matsuzaki S, Shishido Y, Akiyama H, Tachibana T, et al. 2013. Contribution of Leydig and Sertoli cells to testosterone production in mouse fetal testes. Mol Endocrinol 27: 63–73. 10.1210/me.2012-1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simond AM, Rao T, Zuo D, Zhao JJ, Muller WJ. 2017. ErbB2-positive mammary tumors can escape PI3K-p110α loss through down-regulation of the Pten tumor suppressor. Oncogene 36: 6059–6066. 10.1038/onc.2017.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein GS, Lian JB. 1993. Molecular mechanisms mediating proliferation/differentiation interrelationships during progressive development of the osteoblast phenotype. Endocr Rev 14: 424–442. 10.1210/edrv-14-4-424 [DOI] [PubMed] [Google Scholar]
- Svoronos N, Perales-Puchalt A, Allegrezza MJ, Rutkowski MR, Payne KK, Tesone AJ, Nguyen JM, Curiel TJ, Cadungog MG, Singhal S, et al. 2017. Tumor cell–independent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov 7: 72–85. 10.1158/2159-8290.CD-16-0502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S, Omoto Y, Iwase H. 2015. Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Transl Res 166: 540–553.e2. 10.1016/j.trsl.2015.09.003 [DOI] [PubMed] [Google Scholar]
- Tikkanen MK, Carter DJ, Harris AM, Le HM, Azorsa DO, Meltzer PS, Murdoch FE. 2000. Endogenously expressed estrogen receptor and coactivator AIB1 interact in MCF-7 human breast cancer cells. PNAS 97: 12536–12540. 10.1073/pnas.220427297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, et al. 2013. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 45: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, Smith A, Wilson J, Morrow C, Wong WL, et al. 2017. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov 7: 277–287. 10.1158/2159-8290.CD-15-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K-U, Ward T, Davis B, Wiseman R, Hennighausen L. 2001. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res 10: 545–553. 10.1023/A:1013063514007 [DOI] [PubMed] [Google Scholar]
- Wallace MD, Pfefferle AD, Shen L, McNairn AJ, Cerami EG, Fallon BL, Rinaldi VD, Southard TL, Perou CM, Schimenti JC. 2012. Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics 192: 385–396. 10.1534/genetics.112.142802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters KM, Rickard DJ, Riggs BL, Khosla S, Katzenellenbogen JA, Katzenellenbogen BS, Moore J, Spelsberg TC. 2001. Estrogen regulation of human osteoblast function is determined by the stage of differentiation and the estrogen receptor isoform. J Cell Biochem 83: 448–462. 10.1002/jcb.1242 [DOI] [PubMed] [Google Scholar]
- Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. 1996. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol 10: 1388–1398. [DOI] [PubMed] [Google Scholar]
- Yang J, Bielenberg DR, Rodig SJ, Doiron R, Clifton MC, Kung AL, Strong RK, Zurakowski D, Moses MA. 2009. Lipocalin 2 promotes breast cancer progression. PNAS 106: 3913–3918. 10.1073/pnas.0810617106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been uploaded to the Sequence Read Archive (SRA) under accession number PRJNA624176.