

Abstract
Combined neutron and X‐ray powder diffraction techniques highlighted the sorption capacity of the acidic L zeolite towards the L‐lysine amino acid. The role of zeolite channels in the stabilization of the lysine absorbed and the effect of water on protein structure are elucidated at atomistic level. The stabilization of the L α‐helical conformation is related to strong H‐bonds between the tail aminogroups of lysine molecules and the Brønsted acid site as well as to complex intermolecular H‐bond system between water molecules, zeolite and amino acid. This finding is relevant in the catalytic synthesis of polypeptide, as well as in industrial biotechnology by qualitatively predicting binding behaviour
Keywords: L-lysine adsorption, L zeolite, neutron and synchrotron X-ray diffraction, α-helical conformation, Brønsted acid site
The adsorption capacity of the acidic L zeolite towards the L‐lysine was investigated by combining neutron and X‐ray powder diffraction techniques. The stabilization of the L α‐helical conformation is related to strong H−bonds between the tail aminogroups of lysine molecules and the Brønsted acid site as well as to complex intermolecular H‐bond system between water molecules, zeolite and amino acid.
1. Introduction
Amino acids, the building blocks of proteins, have recently attracted a lot of attention in many fields such as drug and gene delivery, solid‐phase peptide synthesis, and biocompatible materials synthesis.1, 2 They are widely spread in natural waters, and represent key indicators of the extent of organic matter alteration and microbial activity in both terrestrial and marine environments.3, 4 The knowledge of the interaction between amino acids and solid surfaces is of fundamental importance to increase the level of understanding of much more complex systems involving proteins and/or bacteria at the solid/liquid interface.5 Amino acids show various adsorption behaviours including electrostatic attraction, covalent bonding, hydrogen bonding, and hydrophobic interactions.6
A variety of materials have been reported to adsorb amino acids, such as mesoporous materials,7, 8 clays,9, 10, 11, 12 titanium dioxide,13, 14 hydroxyapatite,15, 16, 17 hematite;18 pyrite;19, 20 silica gel,21 and zeolites.22, 23 Results so far obtained on zeolites are really promising due to peculiar structural features, high porosity and large surface area, thermal/ mechanical/ biological stability and a chemical stability to increase both their selectivity and activity.24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 In order to gain a deeper understanding on the zeolites efficiency in amino acid adsorption, further knowledge of the interaction between microporous sorbents and amino acid molecules is essential.
Zeolite L micropore structure (K6Na3Al9Si27O72 ⋅ 21H2O, framework type LTL) consists of a one‐dimensional channel system delimited by 12‐membered rings (maximum diameter=10.5 Å).35 Its structural features make it a potential candidate in the adsorption of long‐chain molecules. L‐lysine is a basic amino acid which can be the ideal benchmark to check the possible intermolecular interaction with the zeolite L during adsorption in water solution.
The study herein presented clarify the role of zeolite channels in the stabilization of the lysine absorbed, the effect of water on protein structure and how water molecules arrange around protein molecules. New information on the nature of sorbent/sorbate interactions in an aqueous environment are provided on the basis of Rietveld analyses based on both X‐ray (from synchrotron facility) and neutron diffraction data.
This combined approach was chosen to directly investigate the precise location of light neighbouring elements (e. g., H, C, N) in the periodic table. In addition, the high incoherent scattering contribution to the background by H atoms was avoided via sample deuteration, which has a much lower incoherent neutron scattering cross‐section. Replacing all hydrogen atoms involved in hydrogen bonding by deuterium atoms yield the possible determination of their positions using neutron diffraction with resolutions comparable to those obtained with diffraction from X‐rays. To the best of our knowledge, this is the first time that neutron diffraction technique has been employed to elucidate at atomistic level the interactions between zeolite framework and acid site of amino acids.
2. Results and Discussion
The adsorbed quantity for unit mass of adsorbent was measured at different contact time, starting from L‐lysine (L−Lys) solution at three different initial concentrations (Table 1).
Table 1.
Parameters obtained by non‐linear fitting of the uptake data on L zeolite using a PSO model. The error is given as confidence interval at 95 % of probability.
|
Ci [mg L−1] |
k 2 [g/mg⋅min] |
qe [mg L−1] |
R 2 |
|---|---|---|---|
|
50 |
0.0340±0.0064 |
41.0±1.1 |
0.9962 |
|
75 |
0.0200±0.0022 |
62.0±1.3 |
0.9945 |
|
100 |
0.0120±0.0068 |
67.0±1.9 |
0.9877 |
The adsorption process reaches equilibrium in roughly 1 h (Figure 1). To quantify the kinetic constant, a pseudo second order (PSO) model was employed to model the data. The saturation capacity is about 0.62 mmol g−1 (Table 2), corresponding to ∼9.7 % in weight, higher than that reported for Lys adsorption on mesoporous siliceous materials (MCM‐41).36
Figure 1.
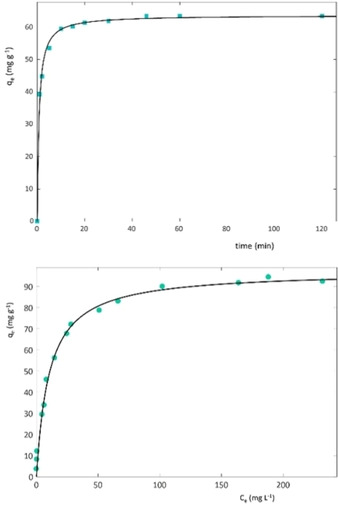
Uptake of Lys vs. time (a) and adsorption isotherm (b) on L zeolite.
Table 2.
Binding constants and saturation capacity for the adsorption of Lys. The error is given as confidence interval at 95 % of probability.
|
Material |
qs (mg g−1) |
B (L mg−1) |
R2 |
|---|---|---|---|
|
Zeolite L |
96.9±4.1 |
0.097±0.018 |
0.9913 |
Samples of saturated zeolite with both L–Lys and deuterated lysine (dL‐Lys) were used for structural X‐ray and neutron diffraction investigations, respectively.
After amino acid adsorption, the hexagonal symmetry has been retained, with all the observed reflections correctly indexed i]n the holohedral P6/mmm space group. Refined lattice parameters were: a=b=18.4287(1) Å, c=7.5383(1) Å, and V=2217.13(3) Å3. At variance with the previous data, unit‐cell parameters from data collected on the dL‐Lys sample investigated by neutron diffraction were a=b=18.4117(2) Å, c=7.5278(2) Å, and V=2209.98(8) Å3, respectively. Details of synchrotron X‐ray and neutron data collection and refinements agreement indices (R‐values) are reported in Table 1SI). Atomic coordinates, atomic fraction and ADPs of framework atoms obtained through structural refinement of data from synchrotron X‐ ray diffraction are supplied in supporting information (Tables 2–4 SI). The resulting slight difference can be readily explained by considering the different data collection conditions, i. e., neutron data have been collected at 4 K, whereas synchrotron data at room conditions. The refined tetrahedral occupancies from neutron diffraction data as well as the <T1−O> and <T2−O> mean bond distances (Table SI4) indicated an enrichment of Al at T2 site. With the exception of slight variations in the thermal motions of the organic molecules, a general convergence between the two diffraction techniques has been observed, thus providing a good picture of the real location of extraframework ions and molecules. Iterating the Rietveld refinement for X‐ray and neutron data by using only a silica framework model (i. e., no adsorbed guest molecule has been included yet) yields to a fit to the diffraction patterns with residual values: Rwp=0.2189, Rp=0.1729 for X‐ray and Rwp=0.2829, Rp=0.2138 for neutron, respectively.
At this stage, an investigation of the residual electron density by means of Difference Fourier maps was carried out for both X‐ray and neutron data in order to detect and locate the adsorbed extraframework species. As a matter of fact, refined structural models provide a potassium cation distribution similar to that reported by Gigli et al.37 K+ ions (∼8 atoms per unit cell, a.u.c.) are hosted at the KB, KC and KD sites, located at the centre of the 6MR, 8MR and close to the edge of the 12MR, respectively (Figure 2). In details, KB is six‐fold coordinated by the O3 framework oxygen atoms (KB−O3=2.87(1) Å [x6]), KC four‐fold coordinated by the O5 (KC−O5=2.69(1) Å [x4]), and KD seven‐fold coordinated by the O4 and O6 oxygen atoms and one coadsorbed water (W1) molecule (KD−O4=3.19(1) Å [x4]), the O6 (KD−O6=3.02(1) Å [x2], KD−W1=2.53(1) Å), the bond length are reported in bracket, (Figure 2)
Figure 2.
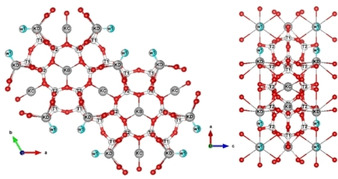
Projection of L framework along the c (left) and b axes (right), respectively. Light blue spere: water molecules; grey sphere: K ions; red sphere: oxygens atoms.
Along with W1 site, another Fourier residual peak was attributed to co‐adsorbed water molecules (W2 site), H‐bonded to O1 and O2 framework oxygens (W2–O1=3.25(1) [x2], and W2–O2=2.90(1) Å). These co‐adsorbed water molecules (∼8 molecules per unit cell) correspond to w2 and w8 sites of Barrer and Villiger, respectively.38. Four others residuals peaks, lying on the ab plane, were located within the 12MR channel (C1, C2, C3 and C4 sites, Table 4 in supporting information). These four independent sites are partially occupied (Table 4 SI) and generate a coronene‐like structure (see Figure 3). C2 site (multiplicity 12) hosts always carbon atoms, C1 site (multiplicity 12) can be occupied by carbon or nitrogen, C3 and C4 (multiplicity 24) sites by oxygen or nitrogen, respectively. For these reasons, all of these are refined with the scattering length of carbon. Their occupancies (Table 4 SI) give rise to 12 C, 4 N and 4 O atoms, corresponding to two lysine molecule per unit cell (Table 5 SI). Because of the crystal symmetry, twelve different lysine orientations are possible (six of these are symmetrically equivalent) but they don't occur simultaneously (due to the partial occupancy of the C1, C2, C3 and C4 sites) (see Figure 3 and Figure 1SI in supporting information).
Figure 3.
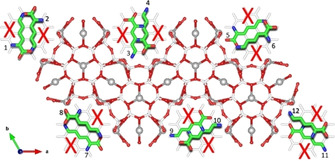
Projection along the c axis of the lysine coronene‐like structure. Green line: C atoms; red line: oxygens of lysine; blue line: nitrogen atoms; grey sphere: K ions; red sphere: framework oxygens atoms;
The difference Fourier map generated from neutron data exhibited more complex features than the X‐ray one because deuterium has a large scattering cross section for neutrons, while hydrogen or deuterium scattering is insignificant for X‐rays. The inclusion of the detected guest species within a new structural model refined into further Rietveld refinement cycles yields to a tremendous decrease of the structural refinement factors to Rwp=0.098, Rp=0.097 for X‐ray and Rwp=0.0296, Rp=0.0211 for neutron data (Table SI1). The final Rietveld fit of both synchrotron and neutron data are reported in Figure 4.
Figure 4.
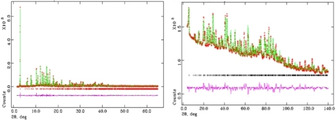
Rietveld refinement performed on X‐ray synchrotron (left) and neutron (right) powder diffraction data of sample L‐lysine. The experimental data are indicated by cross signs, the calculated pattern is the continuous line and the lower curve is the weighted difference between the calculated and observed patterns.
A geometry optimization tool as implemented in EXPO201439 was finally used to minimize the energy of the crystal structure as well calculate the position of the H/D atoms. The resulting optimized crystal structure was then used as input coordinates for GSAS40, 41 and refined again together with the framework until the geometry of both L–Lys and dL‐Lys was reasonable described. In order to avoid false minima and perform a physically meaningful structure refinement, L–Lys molecule coordinates were fixed in the final cycles for both X‐ray synchrotron and neutron Rietveld refinements thus limiting the scattering of refined ADPs. The geometry optimization procedure highlighted that the different relative orientations of L−Lys molecules proposed form a perfect α‐helical structure (Figure 5).
Figure 5.
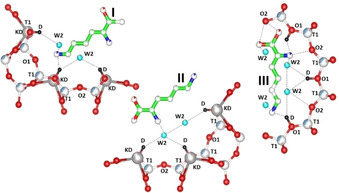
Optimized L framework projected along the c axis, showing the interactions between the guest molecules and the host zeolite framework. Green sphere: C atoms; light blue spere: water molecules; grey sphere: K ions; red sphere: oxygens atoms; black sphere: Brønsted acid site.
Additionally, neutron refinement highlighted the occurrence of a Brønsted acid site (D∼1.3 a.u.c.) at the framework oxygen O1, pointing toward the 12‐MR channel running parallel to the c‐axis (Figure 5). The refinement evidenced the occurrence of interactions between the guest molecules and the host zeolite framework. The α‐carboxylic acid group strongly interacts (e. g., H‐bonds) with W2 water molecule (the O−O bond length 2.37(1) Å) and framework oxygen atoms O1 and O2 (bond distances 2.73(1) Å and 3.14 (1) Å, respectively).
The stabilization of I and III assemblies in Figure 5 is due to the presence of strong H‐bonds (2.66 Å) between the tail aminogroups of lysine molecules and the Brønsted acid site (D). The extra stabilization of lysine is emphasized by the simultaneous occurrence of H‐bonds between the nitrogen of the side chain lysyl ((CH2)4NH2) with the acid proton (2.66 (1) Å), the framework oxygen O2 (N−O=3.14 (1) Å), and the water molecule W2 (O−O=2.37 (1) Å).
The occurrence of sorption interactions between Lys and the zeolitic Brønsted‐acid sites (BASs) are recently observed in zeolite MFI framework by Chen et al.42, 43 The authors reported highlighted that difference in the local adsorption geometries of L‐ and D‐Lys on the zeolitic Brønsted acid sites (BASs) are dependent on both the site‐isolation effect and the confinement restraints exerted by the rigid frameworks, respectively.
In our work, the resulting adsorption scheme of stabilizations entails the formation of lysine‐water molecules that interact with neighbouring framework oxygen atoms and Brønsted acid sites. On the whole, the refinement of the extraframework site occupancies led to a total of 8 water molecules (Δw∼6.0 wt%) and 2.0 (Δw∼11.0 wt%) L‐lysine molecule per unit‐cell, respectively.
The total amount of the extraframework content determined through the Rietveld refinement (i. e., Δw∼17 wt%) is in good agreement with the weight loss registered by the TG analysis between 120 and 900 °C (Figure 6).
Figure 6.
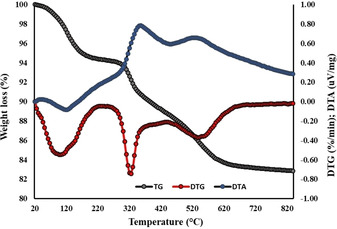
Thermal analysis: in grey and red circles the total weight loss curve (TG) and its first derivative (DTG), respectively; in blue the differential thermal analysis (DTA) curve.
The first weight loss (∼2 wt%), recorded at low temperature (i. e., 20–120 °C), corresponds to the release of H2O molecules bound at the zeolite surface, not detectable through Rietveld powder refinement.
The more relevant mass reduction (∼16 wt%), registered between 120 and 900 °C, is due to desorption of water molecules accompanied by the degradation of the amino acid molecule within the zeolite framework. The structural bonded water desorption reasonably starts at 120 °C, as testified by the presence of a shoulder at the right side of the DTG peak centred at 100 °C, whereas the L‐lysine stepped decomposition begins at higher temperature (∼200 °C) and continues up to the end of the thermal treatment.
The two exothermic peaks recorded by the DTA curve in the same thermal range are attributable to the oxidation and/or combustion of the compound that occurs upon heating.
3. Conclusions
In conclusion, adsorption experiments, neutron and X‐ray (collected at synchrotron facility) powder diffraction aided by thermal analysis, were applied for determining the sorption capacity of the acidic L zeolite towards the L‐lysine amino acid. Gathered results showed that the structural features, the channel system and the chemical properties of the host material favour the molecule adsorption process, although do not limit the diffusion of water molecules within the zeolite micropores. The role of zeolite channels in the stabilization of the lysine absorbed, the effect of water on protein structure and how water molecules arrange around protein molecules are elucidated at atomistic level. The stabilization of the L α‐helical conformation is due to the presence of strong H‐bonds between the tail aminogroups of lysine molecules and the Brønsted acid site as well as the occurrence of complex intermolecular H‐bond system between water molecules, zeolite and amino acid. This finding is relevant in the catalytic synthesis of polypeptide, as well as in the properties of zeolite‐amino acid hybrid materials. In addition, considering the total L‐lysine content encapsulated and the host‐guest interactions that stabilize the molecule within the pores, the L zeolite can be considered a proper sorbent material to operate in amino acids fractionation. These results can be transferred to investigation of stereochemistry in related microporous materials, providing new information on the adsorption of amino acids in zeolites from aqueous phases and could be used to support industrial biotechnology by qualitatively predicting binding behaviour.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was carried out in the framework of the project Protecting the Enclosed Parts of the Sea in Adriatic from pollution, PEPSEA (Project ID: 10047424) within the Interreg V A Italy Croatia Cross‐border Cooperation Programme 2014‐2020. FFABR–2017 funded by the University of Ferrara is also acknowledged for funding. Authors thank the staff of D2B (ILL) and MCX (Elettra) beamlines for the help in neutron and X‐ray diffraction data collection, respectively.The constructive reviews of referees were highly appreciated.
G. Beltrami, A. Martucci, L. Pasti, T. Chenet, M. Ardit, L. Gigli, M. Cescon, E. Suard, ChemistryOpen 2020, 9, 978.
Contributor Information
Prof. Annalisa Martucci, Email: mrs@unife.it.
Prof. Luisa Pasti, Email: psu@unife.it.
References
- 1. Casettari L., Vllasaliu D., Lam J. K., Soliman M., Illum L., Biomaterials 2012, 33, 7565–7583. [DOI] [PubMed] [Google Scholar]
- 2. Sun H., Meng F., Diam A. A., Hendriks M., Feijen J., Zhong Z., Biomacromolecules 2011, 12, 1937–1955. [DOI] [PubMed] [Google Scholar]
- 3. Vandewiele S., Cowie G., Soetaert K., Middelburg J. J., Deep-Sea Res. Pt II 2009, 56, 376–392. [Google Scholar]
- 4. de Haas H., van Weering T. C., de Stigter H., Cont. Shelf Res. 2002, 22, 691–717. [Google Scholar]
- 5. Humblot V., Méthivier C., Raval R., Pradier C. M., Surf. Sci. 2007, 601, 4189–4194. [Google Scholar]
- 6.PDR Network, LLC. PDR for Nonprescription Drugs, Dietary Supplements, and Herbs. 31st Ed. PDR Network, LLC, Montvale, NJ. 2010, p. 596.
- 7. Hartmann M., Chem. Mater. 2005, 17 4577–4593. [Google Scholar]
- 8. Krohn E., Tsapatsis M., Langmuir 2005, 21 8743–8750. [DOI] [PubMed] [Google Scholar]
- 9. de Santana H., Paesano A., da Costa A. C., Di Mauro E., De Souza E., Ivashita F. F., Zaia D. A., Amino Acids 2010, 38, 1089–1099. [DOI] [PubMed] [Google Scholar]
- 10. Benetoli L. O., de Souza C. M., da Silva K. L., de Souza Junior I. G., de Santana H., Jr Paesano A., Costa A. C., Zaia C. T. B. V., Zaia D. A. M., Origins Life Evol. Biospheres 2007, 37, 479–493. [DOI] [PubMed] [Google Scholar]
- 11. Parbhakar A., Cuadros J., Sephton M. A., Dubbin W., Coles B. J., Weiss D., Colloids Surf. A 2007, 307, 142–149. [Google Scholar]
- 12. Ding X., Henrichs S. M., Mar. Chem. 2002, 77, 225–237. [Google Scholar]
- 13. Jonsson C. M., Jonsson C. L., Estrada C., Sverjensky D. A., Cleaves H. J., Hazen R. M., Geochim. Cosmochim. Acta 2010, 74, 2356–2367. [Google Scholar]
- 14. Roddick-Lanzillotta A. D., McQuillan A. J., J. Collid. Interf. Sci. 2000, 227, 48–54. [DOI] [PubMed] [Google Scholar]
- 15. Lee W. H., Loo C. Y., Van K. L., Zavgorodniy A. V., Rohanizadeh R., J. Roy. Soc. Interface 2011, 9, 918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. El Shafei G. M., Moussa N. A., J. Colloid Interface Sci. 2001, 238, 160–166. [DOI] [PubMed] [Google Scholar]
- 17. Pan H., Tao J., Xu X., Tang R., Langmuir 2007, 23, 8972–8981. [DOI] [PubMed] [Google Scholar]
- 18. Kandori K., Sakai M., Inoue S., Ishikawa T., J. Colloid Interface Sci. 2006, 293, 108–115. [DOI] [PubMed] [Google Scholar]
- 19. Nair N. N., Schreiner E., Marx D., J. Am. Chem. Soc. 2006, 128, 13815–13826. [DOI] [PubMed] [Google Scholar]
- 20. Boehme C., Marx D., J. Am. Chem. Soc. 2003, 125, 13362–13363. [DOI] [PubMed] [Google Scholar]
- 21. Han D., Jia W., Liang H., J. Environ. Sci. 2010, 22, 237–241. [DOI] [PubMed] [Google Scholar]
- 22. Boefka B., Piboon P., Limtrakl J., J. Mol. Struct. 2008, 889, 81–88. [Google Scholar]
- 23. Krohn J. E., Tsapatsis M., Langmuir 2006, 22 9350–9356. [DOI] [PubMed] [Google Scholar]
- 24. Pasti L., Rodeghero E., Sarti E., Bosi V., Cavazzini A., Bagatin R., Martucci A., RSC Adv. 2016, 6, 54544–54552. [Google Scholar]
- 25. Martucci A., Braschi I., Bisio C., Sarti E., Rodeghero E., Bagatin R., Pasti L., RSC Adv. 2015, 5, 86997–87006. [Google Scholar]
- 26. Martucci A., Braschi I., Marchese L., Quartieri S., Mineral. Mag. 2014, 78, 1115–1140. [Google Scholar]
- 27. Martucci A., Cremonini M. A., Blasioli S., Gigli L., Gatti G., Marchese L., Braschi I., Microporous Mesoporous Mater. 2013, 170 274–286. [Google Scholar]
- 28. Pasti L., Sarti E., Cavazzini A., Marchetti N., Dondi F., Martucci A., J. Sep. Sci. 2013, 36 1604–1611. [DOI] [PubMed] [Google Scholar]
- 29. Amorim R., Vilaca N., Martinho O., Reis R. M., Sardo M., Rocha J., Fonseca A. M., Baltazar F., Neves I. C., J. Phys. Chem. C 2012, 116 25642–25650. [Google Scholar]
- 30. Datt A., Fields D., Larsen S. C., J. Phys. Chem. C 2012, 116 21382–21390. [Google Scholar]
- 31. Martucci A., Pasti L., Marchetti N., Cavazzini A., Dondi F., Alberti A., Microporous Mesoporous Mater. 2012, 148, 174–183. [Google Scholar]
- 32. Braschi I., Blasioli S., Gigli L., Gessa C. E., Alberti A., Martucci A., J. Hazard. Mater. 2010. 178, 218–225. [DOI] [PubMed] [Google Scholar]
- 33. Damjanovic L., Rakic V., Rac V., Stosic D., Auroux A., J. Hazard. Mater. 2010, 184 477–484. [DOI] [PubMed] [Google Scholar]
- 34. Anderson M. A., Environ. Sci. Technol. 2000, 34, 725–727. [Google Scholar]
- 35. Baerlocher C., McCusker L. B., Olson D. H., Atlas of Zeolite Framework Types, 6th rev. ed., Elsevier, Amsterdam, 2007. [Google Scholar]
- 36. Gao Q., Xu W., Xu Y., Wu D., Sun Y., Deng F., Shen W., J. Phys. Chem. B 2008, 112, 2261–2267. [DOI] [PubMed] [Google Scholar]
- 37. Gigli L., Arletti R., Quartieri S., Di Renzo F., Vezzalini G., Microporous Mesoporous Mater. 2013, 177 8–16. [Google Scholar]
- 38. Barrer R. M., Villiger H., Zeitschrift für Krist. – Cryst. Mater. 1969, 128, 352–370. [Google Scholar]
- 39. Altomare A., Cuocci G., Giacovazzo C., Moliterni A., Rizzi R., Corriero N., Falcicchio A., J. Appl. Crystallogr. 2013, 46 1231–1235. [Google Scholar]
- 40. Toby B. H., J. Appl. Crystallogr. 2001, 34, 210–213. [Google Scholar]
- 41.A. C. Larson, R. B. Von Dreele, GSAS-Generalized structure analysis system (GSAS). Los Alamos National Laboratory Report LAUR, 2000, 86–748.
- 42. Chen T., Huang B., Day S., Tang C. C., Tsang S. C. E., Wong K. Y., Lo T. W. B., Angew. Chem. Int. Ed. Engl. 2020, 132 1109–1113. [DOI] [PubMed] [Google Scholar]
- 43. Chen T., Wun C. K. T., Day S. J., Tang C. C., & Lo T. W. B., Phys. Chem. Chem. Phys. 2020, Advance Article. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary