

Abstract
Rationale: Endothelial injury may provoke emphysema, but molecular pathways of disease development require further discernment. Emphysematous lungs exhibit decreased expression of HIF-2α (hypoxia-inducible factor-2α)-regulated genes, and tobacco smoke decreases pulmonary HIF-2α concentrations. These findings suggest that decreased HIF-2α expression is important in the development of emphysema.
Objectives: The objective of this study was to evaluate the roles of endothelial-cell (EC) HIF-2α in the pathogenesis of emphysema in mice.
Methods: Mouse lungs were examined for emphysema after either the loss or the overexpression of EC Hif-2α. In addition, SU5416, a VEGFR2 inhibitor, was used to induce emphysema. Lungs were evaluated for HGF (hepatocyte growth factor), a protein involved in alveolar development and homeostasis. Lungs from patients with emphysema were measured for endothelial HIF-2α expression.
Measurements and Main Results: EC Hif-2α deletion resulted in emphysema in association with fewer ECs and pericytes. After SU5416 exposure, EC Hif-2α–knockout mice developed more severe emphysema, whereas EC Hif-2α–overexpressing mice were protected. EC Hif-2α–knockout mice demonstrated lower levels of HGF. Human emphysema lung samples exhibited reduced EC HIF-2α expression.
Conclusions: Here, we demonstrate a unique protective role for pulmonary endothelial HIF-2α and how decreased expression of this endogenous factor causes emphysema; its pivotal protective function is suggested by its ability to overcome VEGF antagonism. HIF-2α may maintain alveolar architecture by promoting vascular survival and associated HGF production. In summary, HIF-2α may be a key endogenous factor that prevents the development of emphysema, and its upregulation has the potential to foster lung health in at-risk patients.
Keywords: hypoxia-inducible factor-2α, hepatocyte growth factor, emphysema
At a Glance Commentary
Scientific Knowledge on the Subject
Emphysema is globally responsible for significant morbidity and mortality, but current therapies target symptom relief rather than root causes. A recent epigenetic study showed that genes regulated by HIF-2α (hypoxia-inducible factor-2α) inversely correlate with disease severity and that inhaled tobacco smoke drives its expression down. These findings cumulatively suggest that the acquired loss of pulmonary HIF-2α expression may contribute to emphysema pathogenesis.
What This Study Adds to the Field
Here, we describe a genetic model in which the induced loss of endothelial HIF-2α results in an emphysematous phenotype, including alveolar airspace enlargement, microvascular attrition, and inflammation. Similar in pathology, human emphysema lung samples exhibit reduced endothelial HIF-2α expression. Endothelial Hif-2α–knockout mice demonstrate lower levels of hepatocyte growth factor, a molecule that is important for tissue repair; reduction of this factor could contribute to alveolar destruction. By contrast, Hif-2α–overexpressing mice are protected from the development of disease. These findings suggest that the acquired loss of HIF-2α, possibly because of cigarette smoking, contributes to emphysema and that targeting this process could limit disease progression.
Chronic obstructive pulmonary disease (COPD) afflicts more than 300 million patients globally and is expected to become the leading cause of death in 15 years (1). However, current therapies target symptom relief rather than the prevention or reversal of the disease (2). Individuals with COPD generally display one of the following two phenotypes: 1) emphysema, which is characterized by enlarged alveoli and lung parenchymal loss, or 2) chronic bronchitis, which involves inflammation of the small airways and associated chronic cough. The pathogenesis for emphysema involves nonspecific inflammation propagated by cigarette smoke injury, increased oxidative stress, an imbalance of proteases/antiproteases, and subsequent alveolar cell apoptosis, resulting in the histologic hallmark of enlarged alveolar spaces (3). Although multifactorial in evolution, there is hope that dominant molecular pathways can be identified to better prevent, diagnose, and treat this pervasive condition. To this end, a recent epigenetic study demonstrates that, among all the screened factors, only genes regulated by HIF-2α (hypoxia-inducible factor-2α) correlated significantly with several COPD severity markers, including FEV1, BODE (Body Mass Index, Airflow Obstruction, Dyspnea, and Exercise) index, and DlCO (4). These investigators showed that tobacco smoke decreases lung HIF-2α expression in a mouse model, illustrating how the expression of this factor could become impaired in adult life. However, the mechanisms by which HIF-2α and its downstream signaling cascades could impact emphysema pathophysiology are unknown.
HIF-2α belongs to the HIF family of heterodimeric transcription factors, which are responsible for regulating cellular adaptive responses to hypoxia (5–10). In normoxia, the α subunit (HIF-2α or HIF-1α) is hydroxylated and targeted for ubiquitination and proteasomal degradation via interaction with the von Hippel-Lindau protein. Iron chelators, such as deferoxamine (DFX), prevent hydroxylation of the α subunits, resulting in stabilization and expression of HIFs under normoxic conditions (11). In hypoxic environments, the α subunits accumulate, translocate to the nucleus, dimerize with the constitutively expressed β subunit (HIF-1β), bind to hypoxia-response elements, and thereby result in the augmented transcription of hundreds of genes (12). Although the HIFα subunits share structural homology, they regulate both overlapping and unique downstream genes and play cell- and context-specific roles (13, 14).
Our group recently demonstrated that endothelial-cell (EC) HIF-2α is an angiocrine factor necessary to maintain tracheal microvasculature; silencing EC Hif-2α results in microvascular attrition in large airway capillaries because of the disruption of ANGPT1 (angiopoietin 1)/TIE2 (tunica interna endothelial cell kinase 2) signaling (15). The rationale for studying endothelial HIF-2α in emphysema was suggested by the recent epigenetic study (4) and strengthened by the following factors: 1) the negative impact of tobacco smoke on total lung HIF-2α (4), 2) the associated decreased pulmonary vascular blood flow with increased EC death in this disease (16, 17), and 3) the prominent expression of HIF-2α in healthy ECs (18). For these reasons, we hypothesized that EC HIF-2α plays a fundamental role in protecting the lungs from emphysema.
In this study, we have used transgenic mouse models of disease to demonstrate that 1) the loss of EC Hif-2α, but not of Hif-1α, resulted in an emphysematous phenotype, 2) the loss of EC Hif-2α exacerbated alveolar enlargement after systemic exposure to the VEGFR2 inhibitor SU5416, and 3) the overexpression of EC Hif-2α prevented emphysema after SU5416 exposure. Emphysematous lungs from patients similarly exhibited reduced endothelial HIF-2α expression. Cumulatively, our data suggest that EC HIF-2α plays a unique role in maintaining the alveolar structure in health and after injury.
Methods
Animals
All animal procedures were approved by the administrative panel on laboratory and animal care of Stanford and the institutional animal care and utilization committee of the Veterans Association of Palo Alto. Five transgenic mouse lines were created to characterize the roles of HIF-1α and HIF-2α in emphysema. VE-Cadherin-CreERT2 (estrogen-controlled Cre under the regulation of VE-cadherin promoter) mice, which have been widely used to mediate gene recombination in endothelial lineage cells for vascular research (19), were crossed with Hif-1αflox/flox or Hif-2αflox/flox mice to generate EC-specific HIF loss-of-function colonies (EC HIF-1α knockout [Hif-1α ECKO], EC HIF-2α knockout [Hif-2α ECKO], and EC HIF-1α and EC HIF-2α knock out [Hif-1α,2α ECKO]). VE-Cadherin-CreERT2 mice were bred with Lox-stop-Lox-Hif-1α or Lox-stop-Lox-Hif-2α mice to generate EC-specific HIF gain-of-function colonies (EC HIF-1α overexpression [Hif-1α ECOE] and EC HIF-2α overexpression [Hif-2α ECOE]). Cre-negative, loxp-positive littermates were used as wild-type control animals. To activate Cre recombinase, tamoxifen (Sigma) was injected subcutaneously at a dose of 200 mg/kg for 3 consecutive days. Because these animals were generated in our lab and have been used for other studies, we have previously verified the knockout efficiency of this model to be 85–95% (15). To generate an emphysematous phenotype, SU5416 (Sigma) was injected subcutaneously three times/week at a dose of 20 mg/kg. To evaluate if HIF stabilization could prevent SU5416-induced emphysema, an iron chelator, DFX, was injected intraperitoneally three times/week at a dose of 100 mg/kg. All animals were 8–10 weeks old. Detailed timelines of experimental protocols are outlined in Figure E1 in the online supplement.
Tissue Processing
Mice were anesthetized with ketamine (50 mg/kg) and xylazine (10 mg/kg). The abdominal cavity was opened to expose the inferior vena cava (IVC). An incision was made in the IVC, creating a conduit for exsanguination. The chest cavity was exposed, and ice-cold phosphate-buffered saline (PBS) was injected into the right ventricle to remove circulating blood cells from the vasculature. The lungs were inflated with low–melting point agarose (for immunofluorescence staining) or 10% formalin (for trichrome and hematoxylin and eosin staining). Agarose-inflated samples were bathed in ice-cold saline to facilitate the setting of the agarose. The lung block was excised, and agarose-inflated samples were incubated in 4% paraformaldehyde for 24 hours, followed by 30% sucrose for 24 hours. Finally, they were embedded in optimal cutting temperature (OCT) solution (Sakura Finetek) and stored at −80°C until sectioned for staining. Formalin-inflated samples were stored in 10% formalin for 48 hours and embedded in paraffin before sectioning and trichome or hematoxylin and eosin staining.
Microvascular Perfusion and Permeability Analysis
After general anesthesia was achieved, 100 μl of fluorescein isothiocyanate (FITC)–conjugated lectin (Vector Laboratories) were slowly injected into the IVC. After a period of 3 minutes (to allow for adequate circulation), 100 μl of Cy3-conjugated microspheres (#R50; Thermo Fisher Scientific) were injected. After another 3 minutes, the vasculature was flushed with PBS, and the lungs were inflated with a 1:1 solution of OCT and 30% sucrose and harvested as described above. Samples were embedded in OCT and frozen at −80°C, and 10-μm sections were prepared before imaging.
Morphologic Assessment
Average alveolar length was calculated for each sample using ImageJ software. After calibration to a known length, a 400-μm line was drawn, and alveolar intercepts were counted. A total of five lines were drawn randomly on each image, and alveolar intersections were counted. For each sample, five images were analyzed. The mean linear intercept (MLI) was calculated by dividing the line length by the total number of intercepts. For the quantification of FITC-lectin and microsphere fluorescence, total fluorescence intensity from multiple high-power fields was calculated and averaged. Intensity was then normalized to control. For quantification of HIF-2α immunofluorescence staining of CD31+ cells in human samples, HIF-2α intensity was calculated as area density (total intensity/area); intensity was then normalized to control. The total number of infiltrated immune cells in the lung was quantified based on at least six fields per sample. ImageJ was used for collagen density assessment of trichrome-stained images. Briefly, images were converted to grayscale, and thresholds were set to the blue channel. After the measurement of thresholded areas, the relative intensity of blue-stained collagen in the alveolar interstitium was calculated.
Immunohistochemistry
Frozen sections of murine samples were used for immunohistochemistry. Ten-μm sections were used for immunofluorescence staining. Antibodies used included anti-CD31 (1:400; BD Pharmigen), anti-NG2 (1:200; Sigma), anti-HGF (anti–hepatocyte growth factor) (1:50; Abcam), anti–cleaved caspase (1:50; Cell Signaling), and anti–HIF-2α (1:100; Novus). Secondary antibodies were conjugated with Alexa Fluor 488 or Cy3 (1:200; Invitrogen).
Lung samples from human control subjects and patients with COPD were used. The control tissue samples were provided by the Pulmonary Hypertension Breakthrough Initiative. The lung samples from patients with COPD were obtained from the Lung Transplant Research Consortium. Samples were deparaffinized, rehydrated with serial incubations in graded ethanol, and subjected to antigen retrieval. After fixation, sampled were treated with peroxidase blocking, permeabilized, and stained with anti-CD31 (1:50; Dako) and anti–HIF-2α (1:100; Novus). Secondary antibodies were conjugated with Alexa Fluor 488 or Cy3 (1:200; Invitrogen). DAPI (Vector Laboratories) was used for nuclear staining. Photomicrographs were captured using a Zeiss LSM 710 laser scanning confocal microscope with Zeiss LSM Image Browser software.
Real-Time qRT-PCR
Noninflated lung samples were incubated in RNAlater solution (Invitrogen) overnight at 4°C. RNA was purified and isolated using the Qiagen Shredder and RNeasy Mini Kit according to the manufacturer’s protocol. Total RNA (1 μg) was reverse-transcribed with Multiscribe Reverse Transcriptase (Thermo Fisher Scientific) per the manufacturer’s instructions on a T100 ThermalCycler (BioRad). SYBR Green Master Mix (Applied Biosystems) was combined with diluted reverse transcription reactions and 100 nM of the forward and reverse primers specific for each gene of interest. Detection was completed using the ABI Prism 7700 sequence detector (Applied Biosystems). Data were analyzed with SDS analysis software (Applied Biosystems), gene expression was normalized to 18S expression, and the 2−ΔΔCt method was used to calculate fold-changes. Primer sequences are shown in Table E1.
Pulmonary Function Testing
Evaluation of pulmonary function was conducted using a Buxco pulmonary function analysis system. Measurements were obtained as per the manufacturer’s instructions. Animals were first anesthetized with an intraperitoneal injection of ketamine (50 mg/kg) and xylazine (10 mg/kg). The trachea was exposed and cannulated with a 17-gauge cannula, and the animal was placed into the chamber. Fast flow volume measurements were conducted to evaluate the airflow obstruction, and pressure volume measurements were performed to obtain the lung volumes. Parameters for analysis included FEV (in microliters) at 20, 50, and 100 ms and FVC. Measurements were obtained in control animals and animals 28 days after HIF-2α–gene knockout.
Tobacco Treatment and Single-Cell RNA-Sequencing Analysis
C57BL/6 mice aged 8–10 weeks were exposed to cigarette smoke for 2 months using the inExpose robot (SCIREQ) and 3R4F reference cigarettes (University of Kentucky). The corresponding nonsmoker control animals underwent the same treatment but with smoke-free air. Lungs were dissected, and ECs were isolated for single-cell RNA sequencing as previously described (20).
Statistical Analysis
GraphPad Prism version 8.0 was used for statistical analysis. The differences between two groups at a single time point were compared using the Mann-Whitney test. For comparisons between multiple experimental groups at a single time point, the Kruskal-Wallis test followed by Dunn’s multiple comparisons test for post hoc analyses or one-way ANOVA followed by Dunnett’s test were used. All differences between groups were considered statistically significant at P < 0.05.
Results
Induced Loss of Endothelial Hif-2α Results in Emphysema
VE-CadherinCreERT2, Hif-2αflox/flox (Hif-2α ECKO) mice received tamoxifen injections on 3 consecutive days to induce endothelial-specific deletion of Hif-2α (Figure E1). Lungs were harvested at 14 and 28 days after tamoxifen injections and assessed for structural alterations. Trichrome staining of inflated lungs demonstrated that EC Hif-2α–knockout mice develop progressive alveolar space enlargement (Figure 1A). MLI analysis (21) confirmed a statistically significant increase in alveolar size (Figure 1B). Apoptosis of alveolar structural cells is an important upstream event in the pathogenesis of emphysema (22). We assessed alveolar cell expression of the apoptosis marker activated caspase 3 and found increased alveolar cell death in EC Hif-2α–knockout lungs (Figures 1C and 1D). Transcripts of common emphysema markers MMP-8 (matrix metalloproteinase 8), MMP-9, and elastase (23, 24) were increased, suggesting that the loss of EC Hif-2α activates proteases and breaks down alveolar structural proteins (Figure 1E). Pulmonary collagen was increased in EC Hif-2α–knockout animals at Day 28 (Figure 1F). In addition, pulmonary function testing measurements confirmed that EC Hif-2α–knockout mice have an obstructive ventilatory defect, as evidenced by a reduced FEV100/FVC ratio (Figure 1G). Collectively, these data suggest that the loss of endothelial Hif-2α causes an emphysematous phenotype at tissue, cellular, and molecular levels.
Figure 1.

Induced loss of endothelial Hif-2α (hypoxia-inducible factor-2α) results in emphysema. (A) Representative trichrome staining of lung sections from control mice and endothelial-cell HIF-2α–knockout (Hif-2α ECKO) mice 14 days or 28 days after Hif-2α deletion. (B) Mean linear intercept of the lungs of control mice and Day 14 (d14) and Day 28 (d28) Hif-2α ECKO mice (n = 5). (C) Representative immunofluorescence staining of lung sections from control mice and d14 and d28 Hif-2α ECKO mice for activated caspase 3. DAPI (blue) stains the nucleus. (D) Quantification of activated caspase 3 intensity comparing groups shown in C (n = 4–5). (E) Fold-change of the transcripts of MMP-8 (matrix metalloprotein 8), MMP-9, MMP-12, and elastase in control mice and d14 and d28 Hif-2α ECKO mice (n = 4). (F) Relative interstitial collagen intensity in control and d14 and d28 Hif-2α ECKO mice by quantifying images of trichrome-stained section (n = 5). (G) Pulmonary function test results of control and d28 Hif-2α ECKO mice (n = 5). Data are presented as mean ± SEM. *P < 0.05 and **P < 0.01 by the Mann-Whitney test. Scale bars, 40 μm. ns = not significant.
Induced Loss of Endothelial Hif-2α Causes Alveolar Microvasculature Damage
Given previous data suggesting that induced loss of EC Hif-2α results in microvascular attrition in the upper airway (15), we sought to evaluate the effect of EC Hif-2α knockout on the alveolar microvasculature. Immunofluorescence staining illustrated a progressive decrease in ECs, identified by CD31+ staining, and pericytes, identified by NG2+ staining, location, and morphology (Figures 2A–2C and E2A–E2C). To evaluate whether the loss of alveolar ECs and pericytes led to functional changes in the lung microvasculature, FITC-conjugated lectin and Cy3-tagged microspheres were used to assess the microvascular perfusion and leakage (15). FITC-lectin is a fluorescently labeled lectin that adheres to vascular endothelium, thus identifying perfused vessels in an area of interest. Cy3-conjugated microspheres are 0.05-μm polystyrene spheres that extravasate from the vasculature at porous sites; the presence of increased microspheres indicates a more permeable and damaged vasculature. EC Hif-2α deletion resulted in discontinuous lectin fluorescence and increased Cy3+ microsphere retention (Figures 2D–2F and E2D and E2E). Together, these data indicate that induced EC Hif-2α deletion leads to compromised alveolar microvascular function.
Figure 2.
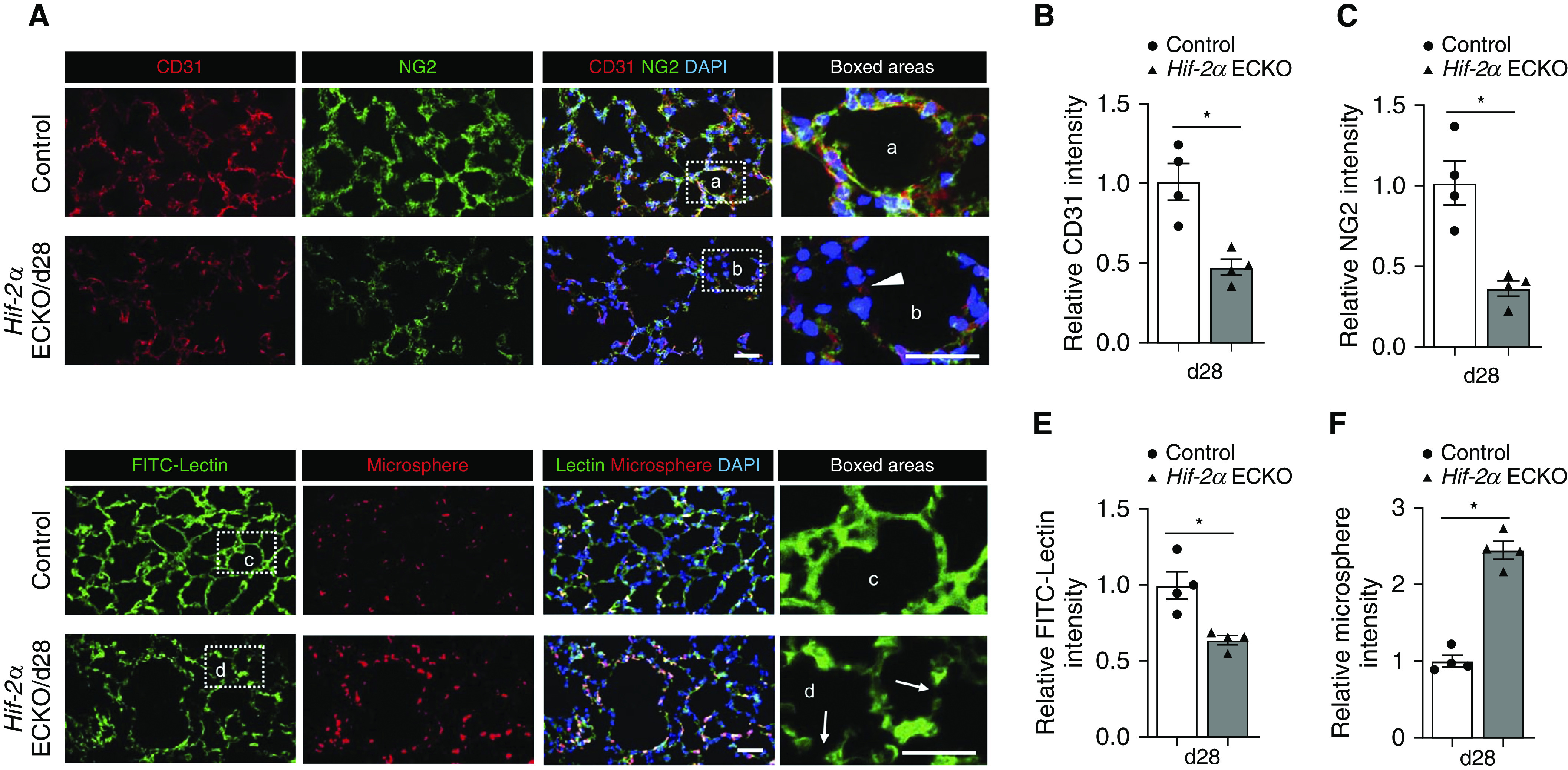
Induced loss of endothelial Hif-2α (hypoxia-inducible factor-2α) damages alveolar microvasculature. (A) Representative immunofluorescence staining of lung sections from control and Day 28 (d28) endothelial-cell HIF-2α–knockout (Hif-2α ECKO) mice. CD31 (red) identifies endothelial cells, and NG2 (green) identifies pericytes. The white arrowhead in the inset points to an area with low CD31 and NG2 staining in the alveoli of Hif-2α ECKO mice. DAPI (blue) stains the nucleus. (B and C) Quantification of CD31 (B) and NG2 (C) intensity comparing groups shown in A (n = 4). (D) Representative pulmonary microvascular fluorescein isothiocyanate (FITC)-lectin perfusion (green) and microsphere permeability (red) images in control and d28 Hif-2α ECKO mice. White arrows in the inset identify disrupted vasculature in the alveoli of Hif-2α ECKO mice. DAPI (blue) stains the nucleus. (E and F) Quantification of FITC intensity (E) and microspheres (F) comparing groups shown in D (n = 4). Data are presented as mean ± SEM. *P < 0.05 by Mann-Whitney test. Scale bars, 40 μm.
Induced Loss of Endothelial Hif-2α Alters Bronchial and Alveolar Airway Epithelium
COPD is characterized by a derangement of the bronchial epithelium, including basal cell hyperplasia, decreased ciliated cells, shortened ciliated cells, squamous cell metaplasia, and goblet-cell expansion (25). To further characterize the changes associated with the induced deletion of Hif-2α, we evaluated the bronchial airway epithelium. Decreased expression of EC Hif-2α decreased the height of E-cadherin+ epithelial cells (Figure 3A, insets a and d) and caused dystrophic changes to acetylated tubulin+ ciliated cells (Figure 3A, insets b and e). There was a notable decline in E-cadherin+ cell number in the alveolar compartment (Figure 3A, insets c and f), which supports the notion that EC Hif-2α deletion results in lung parenchymal loss. Quantification of data from Figure 3A is shown in Figures 3B–3D. In agreement with the immunofluorescence staining, high-resolution scanning electron microscopy images illustrated flattened cilia and an abnormal epithelial phenotype (Figure 3E). Consistent with the results of histological evaluation, real-time qRT-PCR analysis indicated a decline in markers for ciliated and goblet cells, including FOXJ1, DNAH5, and MUC5AC, but no obvious change in club-cell markers, such as SCGB1A1 and SCGB3A2 (Figures 3F and E3). Together, these findings suggest that downregulating EC Hif-2α in adult mice causes unique bronchial airway epithelial alterations in addition to pathologic emphysematous alveolar remodeling.
Figure 3.
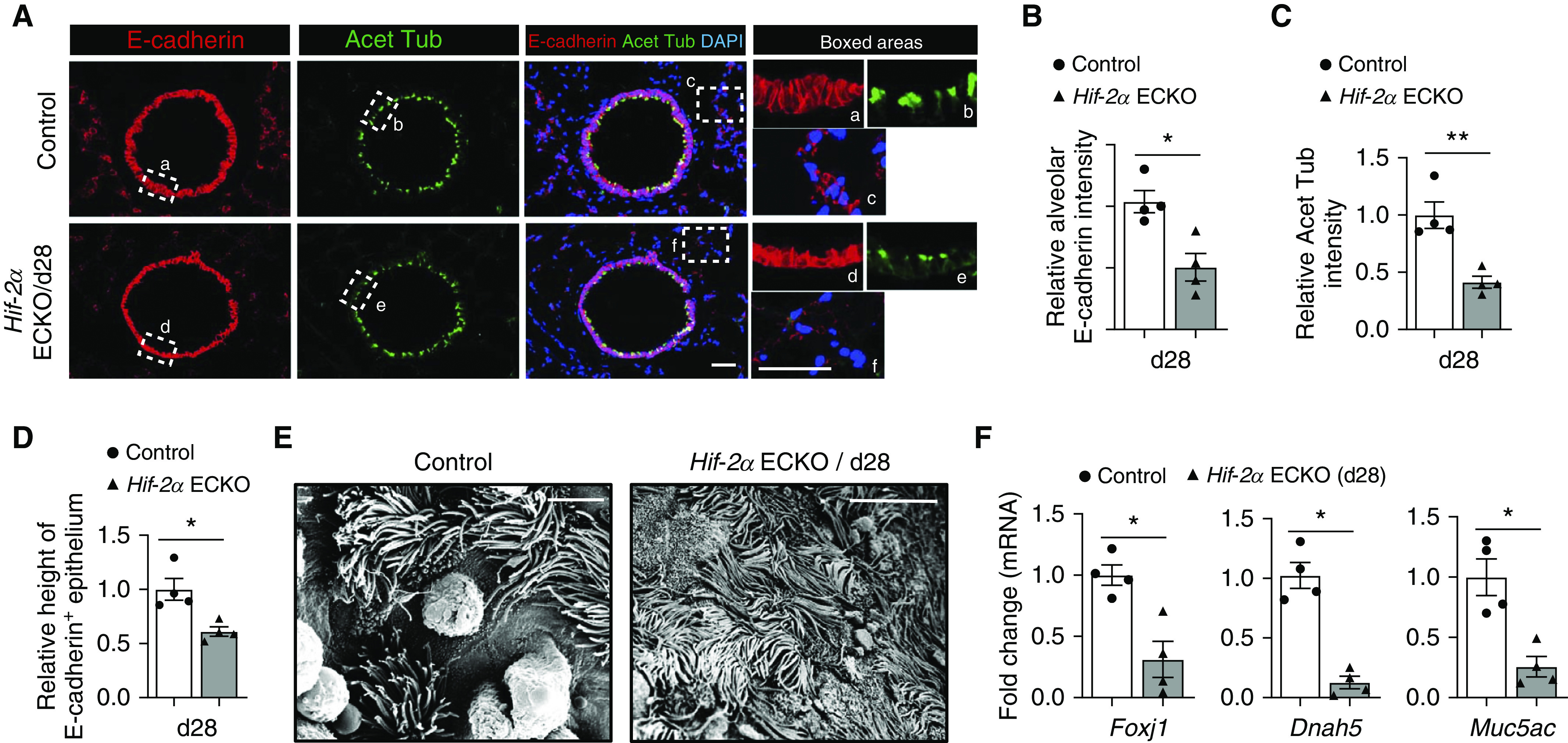
Induced loss of endothelial Hif-2α (hypoxia-inducible factor-2α) alters bronchial airway and alveolar epithelium. (A) Representative immunofluorescence staining of bronchial airway sections from control and Day 28 (d28) endothelial-cell HIF-2α–knockout (Hif-2α ECKO) mice. E-cadherin (red) identifies epithelial cells, and acetylated tubulin (green) identifies ciliated cells. Insets demonstrate the epithelial height and ciliated cell density of bronchial airways (a, b, d, and e) and alveolar epithelial cells (c and f). DAPI (blue) stains the nucleus. (B–D) Quantification of epithelial height (B), acetylated tubulin intensity (C), and E-cadherin intensity (D) comparing the groups shown in A (n = 4). (E) Scanning electron microscope images of control and d28 Hif-2α ECKO mice (F) mRNA fold-changes of markers for ciliated cells (Foxj1 and Dnah5) and goblet cells (Muc5ac), comparing lung tissues from control or d28 Hif-2α ECKO mice (n = 4). Data are presented as mean ± SEM. *P < 0.05 and **P < 0.01 by Mann-Whitney test. Scale bars, (A) 40 μm and (E) 6 μm. Acet Tub = acetylated tubulin.
Decreased Endothelial HIF-2α Expression in Patients with COPD
To explore the clinical relevance of the findings gleaned from the transgenic animal studies, we evaluated the expression of endothelial HIF-2α in lung tissue from healthy individuals relative to patients with early (Global Initiative for Chronic Obstructive Lung Disease [GOLD] 0–1) and advanced (GOLD 3–4) COPD. Demographic data for this study are shown in Table E2. Consistent with the animal results, COPD lungs demonstrated fewer CD31+ ECs and decreased expression of HIF-2α in ECs, which was especially pronounced in the samples from patients with advanced COPD (Figures 4A–4C and E4). These data indicate that the acquired loss of EC HIF-2α expression is relevant to clinical disease.
Figure 4.

Decreased endothelial HIF-2α (hypoxia-inducible factor-2α) expression in human lungs with chronic obstructive pulmonary disease. (A) Representative immunofluorescence staining of HIF-2α (green) and CD31 (red) of human lung tissue from control subjects and patients with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage 0–1 and GOLD stage 3–4. DAPI (blue) stains the nucleus, and white arrows point to endothelial cells expressing HIF-2α. (B and C) Quantification of relative CD31 intensity (B) and HIF-2α intensity (C) comparing the groups shown in A (n = 5–7). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA followed by Dunnett’s multiple comparisons test. Scale bars, 20 μm. ns = not significant.
Decreased HGF Expression after Induced Loss of Endothelial Hif-2α
HGF, a mitogen expressed by pericytes and ECs (26), is critical for the development and maintenance of alveoli during embryogenesis and postnatal life in rodents (27, 28). In addition, its expression is decreased in lungs of patients with COPD (29). Therefore, we hypothesized that EC HIF-2α deletion may induce emphysema via decreased HGF expression because of the loss of alveolar ECs and pericytes. RT-PCR analysis demonstrated significantly decreased whole lung HGF mRNA in mice with EC Hif-2α deletion (Figure 5A). We also noted a profound reduction of EC- and pericyte-derived HGF in lungs with EC Hif-2α deficiency (Figures 5B–5D). Together, these findings support a model in which decreased Hif-2α expression causes both EC and pericyte loss, which in turn leads to a decline in HGF production with decreased paracrine activity, a loss of alveolar epithelial cells, and abnormal alveolar anatomy (Figure 5E).
Figure 5.
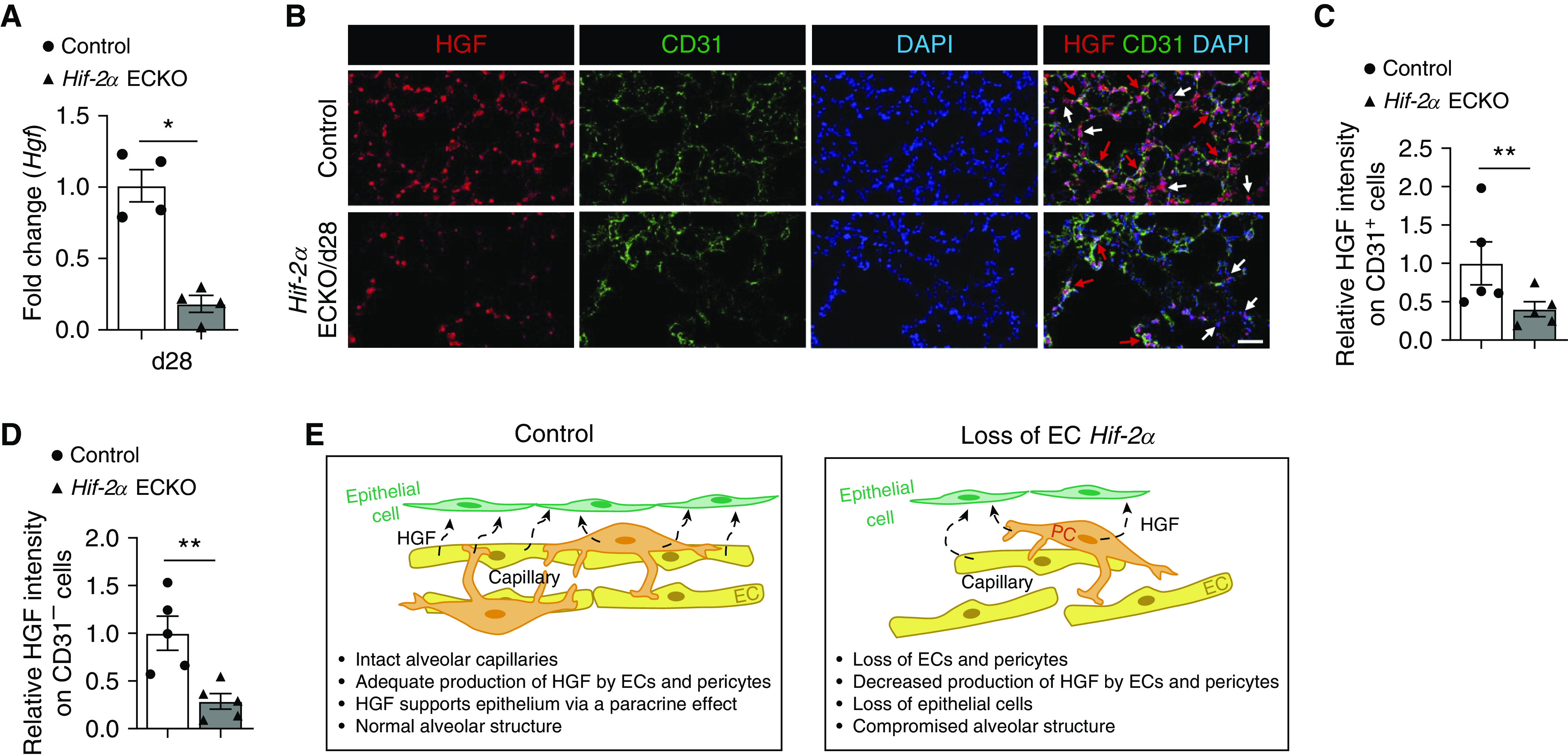
Decreased HGF (hepatocyte growth factor) expression in the lung after endothelial Hif-2α loss. (A) mRNA fold-change of Hgf in whole lungs of control animals and Day 28 (d28) endothelial-cell HIF-2α–knockout (Hif-2α ECKO) mice (n = 4). (B) Representative immunofluorescence staining of lung sections from control mice and d28 Hif-2α ECKO mice. HGF (red) identifies HGF expression, and CD31 (green) identifies endothelial cells (ECs). DAPI (blue) stains the nucleus. Red arrows point to HGF-expressing CD31+ cells. White arrows point to HGF-expressing CD31− cells. (C and D) Quantification of relative HGF expression in CD31+ cells (C) or CD31− cells (D) comparing groups shown in B (n = 5). (E) Schematic illustrating how the loss of EC Hif-2α results in alveolar disruption. In control animals, the alveolar capillaries are intact, and both ECs and pericytes produce HGF, which exerts a mitogenic effect on ECs via paracrine effects and maintains normal alveolar structure. The loss of EC Hif-2α leads to a loss of alveolar vascular cells, including both ECs and pericytes, which, in turn, causes a reduction in HGF production; decreased HGF-mediated mitogenic signaling induces EC death and compromised alveolar structure. Data are presented as mean ± SEM. *P < 0.05 and **P < 0.01 by Mann-Whitney test. Scale bars, 40 μm. PC = pericyte.
Induced Loss of Endothelial Hif-2α Promotes Lung Inflammation
Inflammation is an important pathological alteration in COPD lungs and is characterized by increased numbers of both innate and adaptive immune cells and the activation of lung parenchymal and stromal cells, which produce proinflammatory cytokines and chemokines (30). Analysis of common proinflammatory cytokines TNF-α (tumor necrosis factor-α) and IL-1β indicated increased transcripts of both cytokines at Day 14 after EC Hif-2α gene deletion, with TNF-α remaining high at Day 28 (Figure E5A). Accordingly, there was an increase in CD45+ immune cell infiltration in the alveoli (Figures E5B and E5C), but not in the peribronchial regions (Figures E5D and E5E), at Day 28. These data indicate that EC HIF-2α is needed for quiescent alveolar immunity and that reduced HIF-2α expression has more overt damaging effects on alveoli than on bronchial airways.
Induced Loss of Endothelial Hif-1α Does Not Significantly Impact Alveolar Microvasculature or Size
Although the HIF-1α and HIF-2α isoforms share significant structural homology, they often play distinct cell-specific roles (12). There are a few studies indicating that HIF-1α is involved in COPD pathogenesis (31, 32). We asked whether EC Hif-1α loss-of-function results in a similar phenotype as EC Hif-2α deletion. Our results show that EC Hif-1α deletion did not cause a notable loss of either ECs or pericytes (Figures 6A–6C). Evaluation of alveolar structure by trichrome staining showed no significant change in alveolar size (Figures 6D and 6E). To determine whether the loss of EC Hif-1α may compound the changes induced by EC Hif-2α deletion, we created EC-specific Hif-1α and Hif-2α double-knockout animals. The assessment of alveolar microvasculature indicated that the deletion of both HIF isoforms in ECs did not exacerbate alveolar microvascular loss caused by EC Hif-2α deletion alone (Figure E6). Together, these data indicate that EC Hif-1α is not required for physiological alveolar microvascular homeostasis or maintenance of normal alveolar structure.
Figure 6.
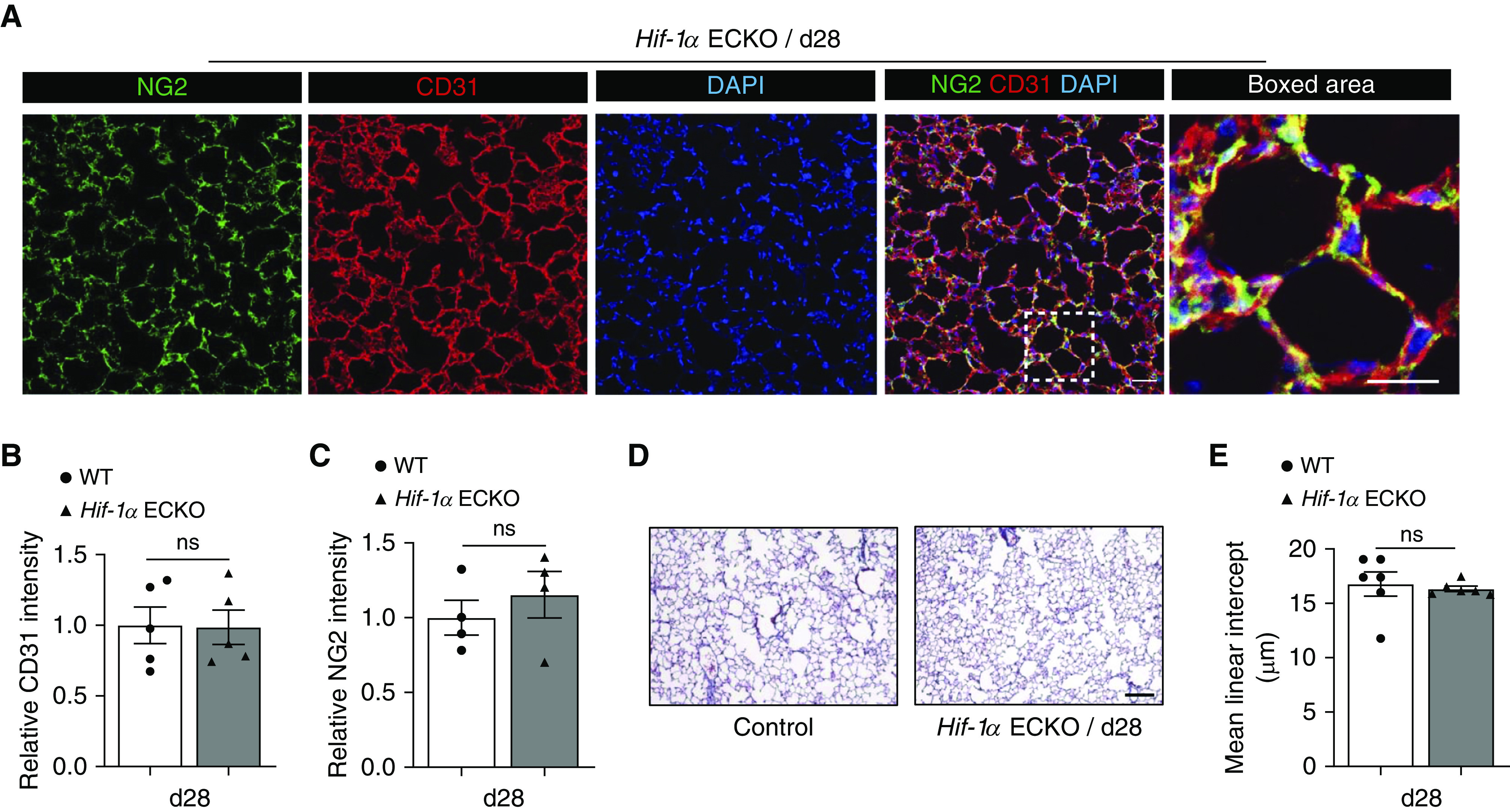
Induced loss of endothelial Hif-1α (hypoxia-inducible factor-1α) does not significantly impact alveolar microvasculature or size. (A) Representative immunofluorescence staining of lung sections from endothelial-cell HIF-1α–knockout (Hif-1α ECKO) mice 28 days after tamoxifen-mediated Cre recombination. CD31 (red) identifies endothelial cells, and NG2 (green) identifies pericytes. DAPI (blue) stains the nucleus. (B and C) Quantification of the intensity of CD31 (B) and NG2 (C) staining comparing the groups shown in A (n = 4). (D) Representative trichrome-stained lung sections from Day 28 (d28) Hif-1α ECKO mice. (E) Mean linear intercept measurements of control and d28 Hif-1α ECKO mice (n = 6). Data are presented as mean ± SEM. Mann-Whitney test was used. Scale bars, 40 μm. ns = not significant; WT = wild type.
Overexpression of Endothelial HIF-2α Prevents SU5416-induced Emphysema
Previous studies have shown that EC injury induced by the VEGFR2 inhibitor SU5416 leads to pulmonary cell apoptosis, alveolar enlargement, and an emphysematous phenotype (33). Given that microvascular loss may be germane to this model of emphysema, we questioned whether modulation of EC HIF-2α expression levels would impact the pathogenesis of SU5416-induced emphysema. A transgenic mouse strain with constitutive expression of EC Hif-2α (Hif-2α ECOE) (34) was created and used to determine the effect of EC HIF-2α augmentation on SU5416-induced emphysema. SU5416 resulted in an emphysematous phenotype with enlarged airspaces, as previously demonstrated (33). Although EC Hif-2α loss-of-function aggravated SU5416-induced air space enlargement, Hif-2α ECOE mice were protected from SU5416-induced pulmonary pathology (Figures 7A and 7B). Analyses of cellular apoptosis indicated that lungs from Hif-2α ECOE mice had fewer activated caspase 3+ cells after exposure to SU5416 (Figures 7C and 7D). The evaluation of EC and pericyte density demonstrated higher numbers of CD31+ cells and NG2+ cells in the lungs of Hif-2α ECOE mice (Figures 7E–7G). Assessment of EC HIF-2α expression indicated that SU5416 suppressed its expression in wild-type mice but not in Hif-2α ECOE mice (Figures E7A and E7B). Cigarette smoke exposure decreases expression of EC Hif-2α mRNA by ≈30% in the mouse lung, as measured by single-cell RNA-sequencing (Figure E7C). Together, these findings suggested that HIF-2α downregulation may represent a common molecular event that precedes cigarette- and SU5416-induced emphysema, and Hif-2α overexpression protected alveolar microvascular ECs from SU5416-induced apoptosis, which, in turn, prevented the development of emphysema. In addition, Hif-2α ECOE prevented SU5416-induced flattening of the bronchial airway epithelium (Figure E8), indicating that HIF-2α also protected the bronchial airway microvasculature and structural anatomy from SU5416-induced bronchial pathology. A similar observation was noted 14 days after SU5416 treatment (Figure E9), confirming the protective effect of EC HIF-2α against SU5416 in both bronchial airways and alveolar tissues at both early and late time points.
Figure 7.
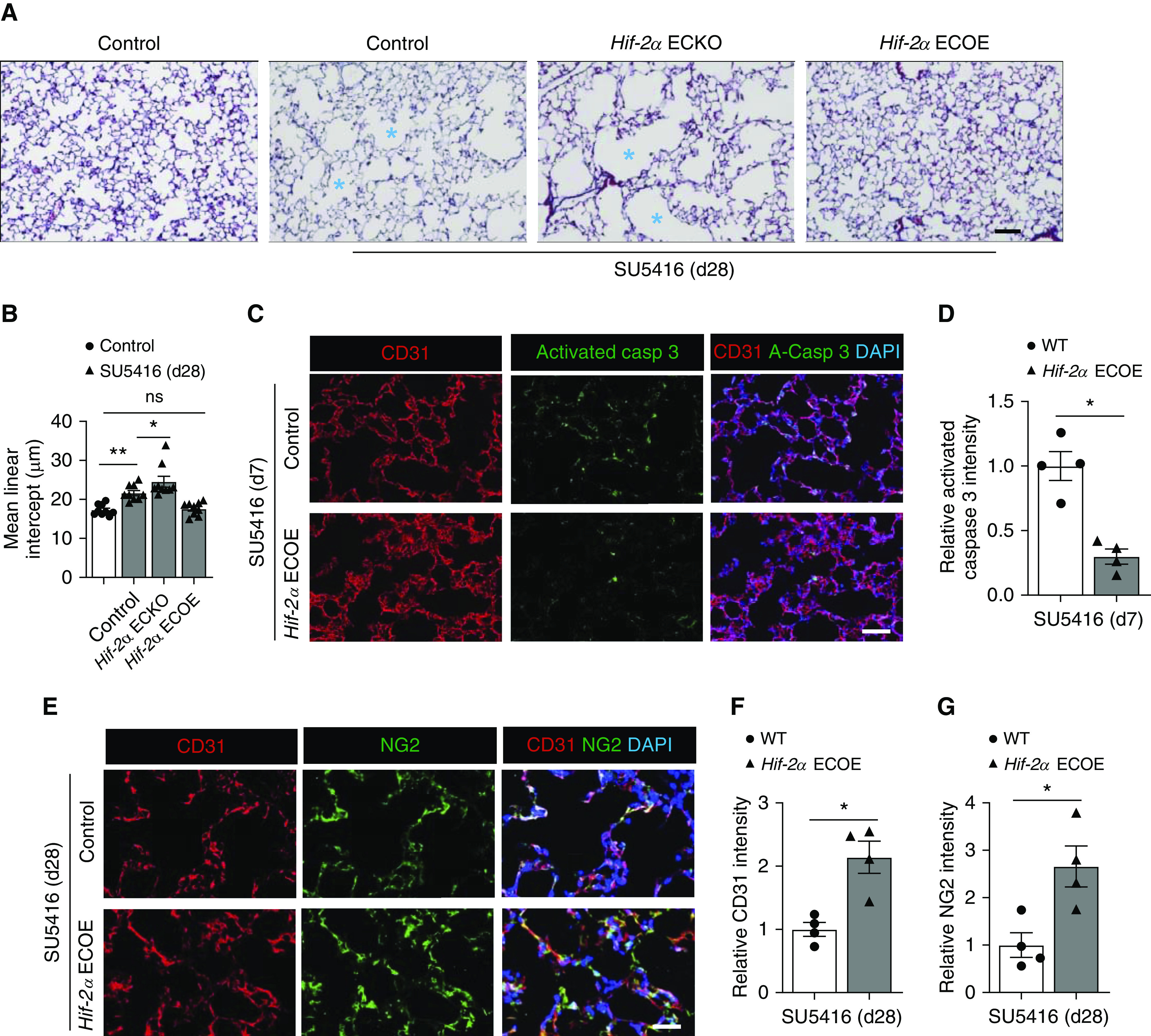
Endothelial Hif-2α (hypoxia-inducible factor-2α) overexpression prevents the development of emphysema after treatment with SU5416. (A) Representative trichrome staining of lung sections from control, endothelial-cell HIF-2α–knockout (Hif-2α ECKO), and endothelial-cell HIF-2α–overexpressing (Hif-2α ECOE) mice treated with SU5416. Lung sections were collected 28 d after SU5416 treatment. Control animals not exposed to SU5416 are shown for comparison. Blue asterisks denote enlarged alveolar spaces. (B) Mean linear intercept measurements of control, control + SU5416, Hif-2α ECKO + SU5416, and Hif-2α ECOE + SU5416 (n = 9). (C) Representative immunofluorescence staining of lung sections from control + SU5416 mice and Hif-2α ECOE + SU5416 mice. CD31 (red) identifies endothelial cells, and activated caspase 3 (green) identifies apoptotic cells. DAPI (blue) stains the nucleus. (D) Quantification of relative activated caspase 3 intensity comparing the groups shown in C (n = 4). (E) Representative immunofluorescence staining of lung sections from control + SU5416 mice and Hif-2α ECOE + SU5416 mice. CD31 (red) identifies vascular endothelial cells, and NG2 (green) identifies pericytes. DAPI (blue) stains the nucleus. (F and G) Quantification of relative intensity of CD31 (F) and NG2 (G) comparing the groups shown in E (n = 4). Data are presented as mean ± SEM. *P < 0.05 and **P < 0.01 by (B) the Kruskal-Wallis test or (D, F, and G) by Mann-Whitney test. Scale bars, 40 μm. casp = capsase; d28 = Day 28; ns = not significant; WT = wild type.
Iron Chelator Therapy with DFX Rescues SU5416-induced Emphysema in Control, but Not in Endothelial Hif-2α Knockout, Mice
Iron chelators are known to stabilize both HIF-1α and HIF-2α independent of tissue oxygenation status (35). Our prior study demonstrated that clinically used iron chelators promote microvascular repair after tracheal transplantation (36). We asked whether iron chelator–mediated HIF stabilization can similarly protect alveolar microvasculature against SU5416-induced damage and prevent emphysema. Administration of DFX to control mice with SU5416-mediated injury decreased pulmonary MLI measurements. However, the same DFX treatment regimen did not prevent air space enlargement in EC Hif-2α–knockout mice (Figure E10). Evaluation of lung HIF-2α expression indicated that DFX treatment failed to increase HIF-2α in Hif-2α ECKO mice (Figure E11), which likely accounted for the inability of DFX to prevent SU5416-induced alveolar space enlargement. Collectively, these data indicate that EC HIF-2α is required for the observed therapeutic effect of DFX in preventing SU5416-induced emphysema, suggesting that a HIF-2α–controlled signaling pathway is required to overcome the effect caused by the disruption of the VEGFR2 pathway in this model.
Discussion
Tobacco smoke is the leading risk factor for emphysema (37), but it remains a considerable challenge to discern how smoke-related toxins interact with homeostatic genetic programs to cause alveolar destruction (38). The identification of endogenous molecules responsible for the development of emphysema after tobacco smoke exposure would provide ideal interventional targets. In this report, we sought to determine how HIF-2α deficiency promotes emphysema and how HIF-2α may be therapeutically targeted.
Using a previously characterized transgenic mouse strain (15), we found that induced EC Hif-2α deletion causes air space enlargement with increased alveolar cell apoptosis, elevated immune cell infiltration in alveoli, heightened expression of proinflammatory cytokines in the lung, and augmented expression of genes implicated in emphysema pathogenesis. Pulmonary function tests further corroborated the findings from the tissue analysis. The relatively lower expression of MMP9, MMP12, and elastase and the lesser degree of cellular apoptosis at Day 28 compared with Day 14 indicate that high MMP and elastase expression precede alveolar cell injury. These findings also illustrate that induced Hif-2α deletion causes alveolar tissue damage, which is followed by pathologic tissue remodeling and increased collagen deposition. Although EC Hif-2α deletion clearly alters bronchial epithelium integrity, there is no obvious airway narrowing and no notable increase in peribronchial inflammatory cell infiltration. These data suggest that the loss of EC Hif-2α in adult mice is sufficient to induce an emphysema-like phenotype. This observed phenotype appears to resemble aging-associated COPD, in which there is a more pronounced alveolar pathology but less bronchial inflammation, thickening, and mucus production (39). A recent animal study indicated that tissue HIF-2α expression decreases in adults as they age (40). Therefore, loss of EC Hif-2α may accelerate alveolar destruction, similar to aging-triggered tissue degeneration, which further supports the idea that EC HIF-2α serves as an endogenous protective factor in the lungs.
Analyses of human lung tissue samples demonstrate a significant decrease in EC HIF-2α in GOLD 0–1 and GOLD 3–4 COPD lungs, with the latter exhibiting the lowest expression. These results from human samples are consistent with the single-cell RNA-sequencing analysis indicating decreased expression of EC Hif-2α mRNA in smoked mice. There have been several relevant studies indicating that HIF-2α can be downregulated in conditions with profound inflammation. For example, IFNγ and the endotoxin LPS suppress HIF-2α expression in macrophages (41). LPS also reduces HIF-2α expression in the lung (42). The airway parenchyma of a rejecting transplant is characterized by inflammation and decreased expression of HIF-2α (15). In addition, hypercapnia, a feature of advanced emphysema, suppresses HIF-2α levels (43). Tobacco toxin–induced tissue inflammation that provokes T-helper cell type 1 immunity and augmented tissue CO2 tension may reduce endothelial HIF-2α expression. Collectively, these data suggest that a decline in EC HIF-2α expression increases COPD severity and, therefore, that the induced EC Hif-2α-deletion mouse model has clinical relevance.
Our data suggest that reduced HIF-2α may be deleterious to lungs because of its downstream effect of decreased pulmonary HGF levels. HGF is a mitogen and survival factor with known relevance in tissue maintenance and regeneration (44). In the lungs, its expression is highest in the mesenchymal cells (i.e., pericytes) and ECs; through a paracrine effect, HGF acts on c-MET receptors expressed on type 2 pneumocytes and promotes alveolar morphogenesis (26). HGF replacement has been shown to induce angiogenesis and reverse emphysema in rodent models (45, 46). Consistent with other studies of emphysema (29), we noted significantly decreased HGF expression in Hif-2α ECKO animals, mainly because of the loss of alveolar pericytes and ECs. Our results suggest that the loss of EC Hif-2α causes a decrease in alveolar microvascular cells, including both ECs and pericytes, followed by decreased production of HGF and loss of epithelial cells, and culminates in alveolar space enlargement and compromised lung function (Figure 5E). The unique advantage of the Hif-2α ECKO model is that it recapitulates the effects of an environmental exposure (i.e., tobacco smoke), which is the most common cause of emphysema in the developed world. In the future, additional mechanistic studies delineating the pathways involved in the HIF-2α and HGF axis may lead to the development of novel therapies.
Although we established the importance of endothelial HIF-2α in health, we also sought to investigate its role in the lung after induced injury. The SU5416 emphysema model (17) posits that the promotion of EC apoptosis via blockade of VEGF/VEGFR2 signaling induces lung alveolar microvascular damage, followed by pneumocyte death. Importantly, VEGFR2 loss-of-function mutation was recently uncovered as a cause for emphysema and pulmonary hypertension in humans (47), strengthening SU5416-induced emphysema as a clinically relevant model (48). Our data show that loss or gain of endothelial Hif-2α expression respectively exacerbates or prevents SU5416-induced emphysema; these results indicate that HIF-2α protects ECs from VEGFR2 inhibition-induced cellular apoptosis. VEGF survival signals include the activation of the AKT pathway (49). Our prior study demonstrated that HIF-2α promotes EC survival by sustaining TIE2 signaling via pericyte-derived ANGPT1 (15). It has been established that AKT activation is the central pathway that mediates ANGPT1-induced TIE2 signaling (50). Therefore, it is possible that HIF-2α raises EC resistance to VEGFR2 inhibition by activating AKT through ANGPT1/TIE2 signaling. Although HGF primarily impacts epithelial cells, pericyte-produced HGF may also exert an effect on ECs and promote their survival (51) by activating the AKT pathway (52, 53). Collectively, our data suggest that augmented endothelial HIF-2α expression may allow sustained AKT activation and negate VEGFR2 inhibition-induced AKT suppression.
Although the effects of modulating both endothelial Hif-2α and Hif-1α expression were evaluated in this study, ultimately, only Hif-2α deletion resulted in emphysema, pericyte loss, and microvascular damage. Furthermore, the combined deletion of both endothelial Hif-1α and Hif-2α did not intensify the changes observed with Hif-2α loss alone. A reason for the observed functional differences could be that HIF-2α undergoes hydroxylation by prolyl hydroxylase domains less efficiently than HIF-1α, resulting in the stabilization of HIF-2α at higher oxygen concentrations (54). Together, these findings suggest that endothelial HIF-2α may play a more important role in emphysema, a condition with ongoing chronic hypoxia. However, our data do not exclude the possibility that HIF-1α derived from other cell types, such as macrophages or lymphocytes, may impact emphysema pathogenesis in ways that endothelial HIF-1α does not (55), given that HIF-1α polymorphisms have been linked to COPD (56). Upregulating both HIF-1α and HIF-2α with the iron chelator DFX prevented alveolar enlargement in control mice treated with VEGFR2 inhibitor but not in Hif-2α–knockout animals, further confirming that endothelial production of the HIF-2α isoform is important for alveolar maintenance. In this study, we evaluated the effect of a single dose of DFX to determine whether HIF-2α was the critical factor necessary for preventing emphysema, and future studies can benefit from the translational impact of this model by evaluating multiple doses to simulate more sustained HIF signaling.
Given the central role of the HIFs in regulating cellular responses to hypoxia, there has been significant interest in evaluating if these molecules could serve as therapeutic targets, either by increasing or inhibiting their expression. Currently, a phase III clinical trial of prolyl hydroxylase inhibitors (which result in constitutive expression of the HIF α subunits) as a treatment for anemia due to chronic kidney disease is underway (57). In the future, this small molecule inhibitor could potentially be reformulated for inhalational delivery to further validate our findings via a clinically feasible intervention. However, other studies have suggested that suppressing the HIFs may be beneficial. As multiple studies have suggested that inhibition of HIF-2α mitigates growth of clear-cell renal-cell carcinoma, there are currently two phase II trials (NCT03634540 and NCT02974738) evaluating the efficacy of a small molecule HIF-2α inhibitor as a chemotherapeutic agent. In addition, there has been a significant interest in using HIF-2α inhibitors as a potential therapy for pulmonary hypertension, given previous data suggesting that HIF-2α heterozygous-knockout mice are protected from the development of pulmonary hypertension in response to chronic hypoxic exposure (58). As such therapies evolve, given the findings described in our study, evaluation for pulmonary complications such as emphysema should also be considered.
Our study has several limitations. We focused on disease pathogenesis and prevention without exploring disease reversal, a process that is likely different than the sequential reversal of pathogenic steps causing alveolar destruction. Accordingly, future studies will evaluate the effects of HIF-2α modulation on established emphysema and can assess the effect of HIF modulation after chronic cigarette smoke exposure to more closely mimic human disease. In addition, given the proposed link between endothelial HIF-2α and pulmonary hypertension (59, 60) as well as the fact that chronic hypoxemia due to COPD can cause pulmonary hypertension, it would be insightful to characterize right ventricular pressures in the EC Hif-2α–overexpressing animals. Finally, the role of epithelial cell–derived HIF-2α warrants consideration, given its role in promoting differentiation of type II pneumocytes into type I pneumocytes (61).
Conclusions
Our findings fit into the compendium of recently reported results that a reduction of HIF-2α–regulated genes uniquely correlates with emphysema severity and that smoking is an environmental factor that reduces pulmonary levels of this HIF isoform (4). Our current effort can be joined with this prior study to conclude that endothelial HIF-2α serves a unique role as an endogenous factor that normally prevents the development of emphysema and that context-specific HIF-2α augmentation could benefit patients at-risk for alveolar loss.
Supplementary Material
Footnotes
Supported by NIH grants K12 HL120001–05 (to X.J.), HL095686 (to M.R.N.), and HL141105 (to M.R.N.), Stanford Chief Startup Funds, and Stanford Endowed Chair Funds (to M.R.N.). Funding for the Pulmonary Hypertension Breakthrough Initiative is provided under an NHLBI grant (R24HL123767) and by the Cardiovascular Medical Research and Education Fund.
Author Contributions: S.P. and X.J. planned experiments and conducted data analysis. S.P., A.B.T., P.D., M.X., and E.G. performed the experiments. A.G., E.C.B., G.L.S., and R.M.T. provided intellectual input and/or provided human tissue samples and contributed to the manuscript. S.P., W.T., X.J., and M.R.N. developed the idea for the study and wrote the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202001-0078OC on June 9, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Quaderi SA, Hurst JR. The unmet global burden of COPD. Glob Health Epidemiol Genom. 2018;3:e4. doi: 10.1017/gheg.2018.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Celli BR, MacNee W ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946. doi: 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- 3.Tuder RM, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc. 2006;3:503–510. doi: 10.1513/pats.200603-054MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoo S, Takikawa S, Geraghty P, Argmann C, Campbell J, Lin L, et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015;11:e1004898. doi: 10.1371/journal.pgen.1004898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol. 1998;275:L818–L826. doi: 10.1152/ajplung.1998.275.4.L818. [DOI] [PubMed] [Google Scholar]
- 8.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. 2020;21:268–283. doi: 10.1038/s41580-020-0227-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- 12.Semenza GL. Pharmacologic targeting of hypoxia-inducible factors. Annu Rev Pharmacol Toxicol. 2019;59:379–403. doi: 10.1146/annurev-pharmtox-010818-021637. [DOI] [PubMed] [Google Scholar]
- 13.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, et al. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- 15.Jiang X, Tian W, Tu AB, Pasupneti S, Shuffle E, Dahms P, et al. Endothelial hypoxia-inducible factor-2α is required for the maintenance of airway microvasculature. Circulation. 2019;139:502–517. doi: 10.1161/CIRCULATIONAHA.118.036157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hueper K, Vogel-Claussen J, Parikh MA, Austin JH, Bluemke DA, Carr J, et al. Pulmonary microvascular blood flow in mild chronic obstructive pulmonary disease and emphysema: the MESA COPD study. Am J Respir Crit Care Med. 2015;192:570–580. doi: 10.1164/rccm.201411-2120OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163:737–744. doi: 10.1164/ajrccm.163.3.2002117. [DOI] [PubMed] [Google Scholar]
- 18.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 19.Monvoisin A, Alva JA, Hofmann JJ, Zovein AC, Lane TF, Iruela-Arispe ML. VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn. 2006;235:3413–3422. doi: 10.1002/dvdy.20982. [DOI] [PubMed] [Google Scholar]
- 20.Xiang M, Grosso RA, Takeda A, Pan J, Bekkhus T, Brulois K, et al. A single-cell transcriptional roadmap of the mouse and human lymph node lymphatic vasculature. Front Cardiovasc Med. 2020;7:52. doi: 10.3389/fcvm.2020.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsia CC, Hyde DM, Ochs M, Weibel ER ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53. doi: 10.1186/1465-9921-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belvisi MG, Bottomley KM. The role of matrix metalloproteinases (MMPs) in the pathophysiology of chronic obstructive pulmonary disease (COPD): a therapeutic role for inhibitors of MMPs? Inflamm Res. 2003;52:95–100. doi: 10.1007/s000110300020. [DOI] [PubMed] [Google Scholar]
- 24.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 25.Jeffery PK. Structural and inflammatory changes in COPD: a comparison with asthma. Thorax. 1998;53:129–136. doi: 10.1136/thx.53.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato K, Diéguez-Hurtado R, Park DY, Hong SP, Kato-Azuma S, Adams S, et al. Pulmonary pericytes regulate lung morphogenesis. Nat Commun. 2018;9:2448. doi: 10.1038/s41467-018-04913-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calvi C, Podowski M, Lopez-Mercado A, Metzger S, Misono K, Malinina A, et al. Hepatocyte growth factor, a determinant of airspace homeostasis in the murine lung. PLoS Genet. 2013;9:e1003228. doi: 10.1371/journal.pgen.1003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Padela S, Cabacungan J, Shek S, Belcastro R, Yi M, Jankov RP, et al. Hepatocyte growth factor is required for alveologenesis in the neonatal rat. Am J Respir Crit Care Med. 2005;172:907–914. doi: 10.1164/rccm.200504-567OC. [DOI] [PubMed] [Google Scholar]
- 29.Kanazawa H, Tochino Y, Asai K, Hirata K. Simultaneous assessment of hepatocyte growth factor and vascular endothelial growth factor in epithelial lining fluid from patients with COPD. Chest. 2014;146:1159–1165. doi: 10.1378/chest.14-0373. [DOI] [PubMed] [Google Scholar]
- 30.Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138:16–27. doi: 10.1016/j.jaci.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 31.Yasuo M, Mizuno S, Kraskauskas D, Bogaard HJ, Natarajan R, Cool CD, et al. Hypoxia inducible factor-1α in human emphysema lung tissue. Eur Respir J. 2011;37:775–783. doi: 10.1183/09031936.00022910. [DOI] [PubMed] [Google Scholar]
- 32.Lee JW, Ko J, Ju C, Eltzschig HK. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med. 2019;51:1–13. doi: 10.1038/s12276-019-0235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106:1311–1319. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim WY, Safran M, Buckley MR, Ebert BL, Glickman J, Bosenberg M, et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 2006;25:4650–4662. doi: 10.1038/sj.emboj.7601300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haase VH. Hypoxia-inducible factor signaling in the development of kidney fibrosis. Fibrogenesis Tissue Repair. 2012;5:S16. doi: 10.1186/1755-1536-5-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang X, Malkovskiy AV, Tian W, Sung YK, Sun W, Hsu JL, et al. Promotion of airway anastomotic microvascular regeneration and alleviation of airway ischemia by deferoxamine nanoparticles. Biomaterials. 2014;35:803–813. doi: 10.1016/j.biomaterials.2013.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–773. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 38.Silverman EK. Genetics of COPD. Annu Rev Physiol. 2020;82:413–431. doi: 10.1146/annurev-physiol-021317-121224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brandsma CA, de Vries M, Costa R, Woldhuis RR, Königshoff M, Timens W. Lung ageing and COPD: is there a role for ageing in abnormal tissue repair? Eur Respir Rev. 2017;26:170073. doi: 10.1183/16000617.0073-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ebersole JL, Novak MJ, Orraca L, Martinez-Gonzalez J, Kirakodu S, Chen KC, et al. Hypoxia-inducible transcription factors, HIF1A and HIF2A, increase in aging mucosal tissues. Immunology. 2018;154:452–464. doi: 10.1111/imm.12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of HIF-alpha isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng H, He X, Tuo QH, Liao DF, Zhang GQ, Chen JX. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci Rep. 2016;6:20931. doi: 10.1038/srep20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selfridge AC, Cavadas MA, Scholz CC, Campbell EL, Welch LC, Lecuona E, et al. Hypercapnia suppresses the HIF-dependent adaptive response to hypoxia. J Biol Chem. 2016;291:11800–11808. doi: 10.1074/jbc.M116.713941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsumoto K, Funakoshi H, Takahashi H, Sakai K. HGF-met pathway in regeneration and drug discovery. Biomedicines. 2014;2:275–300. doi: 10.3390/biomedicines2040275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shigemura N, Sawa Y, Mizuno S, Ono M, Ohta M, Nakamura T, et al. Amelioration of pulmonary emphysema by in vivo gene transfection with hepatocyte growth factor in rats. Circulation. 2005;111:1407–1414. doi: 10.1161/01.CIR.0000158433.89103.85. [DOI] [PubMed] [Google Scholar]
- 46.Ono M, Sawa Y, Matsumoto K, Nakamura T, Kaneda Y, Matsuda H. In vivo gene transfection with hepatocyte growth factor via the pulmonary artery induces angiogenesis in the rat lung. Circulation. 2002;106(Suppl 1):I264–I269. [PubMed] [Google Scholar]
- 47.Eyries M, Montani D, Girerd B, Favrolt N, Riou M, Faivre L, et al. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur Respir J. 2020;55:1902165. doi: 10.1183/13993003.02165-2019. [DOI] [PubMed] [Google Scholar]
- 48.Botros L, Vonk Noordegraaf A, Aman J. Vanishing vessels aboding pulmonary disease: a role for VEGFR2. Eur Respir J. 2020;55:2000326. doi: 10.1183/13993003.00326-2020. [DOI] [PubMed] [Google Scholar]
- 49.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway: requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 50.DeBusk LM, Hallahan DE, Lin PC. Akt is a major angiogenic mediator downstream of the Ang1/Tie2 signaling pathway. Exp Cell Res. 2004;298:167–177. doi: 10.1016/j.yexcr.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 51.Barnes PJ. Hepatocyte growth factor deficiency in COPD: a mechanism of emphysema and small airway fibrosis? Chest. 2014;146:1135–1136. doi: 10.1378/chest.14-1194. [DOI] [PubMed] [Google Scholar]
- 52.Xiao GH, Jeffers M, Bellacosa A, Mitsuuchi Y, Vande Woude GF, Testa JR. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc Natl Acad Sci USA. 2001;98:247–252. doi: 10.1073/pnas.011532898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sulpice E, Ding S, Muscatelli-Groux B, Bergé M, Han ZC, Plouet J, et al. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biol Cell. 2009;101:525–539. doi: 10.1042/BC20080221. [DOI] [PubMed] [Google Scholar]
- 54.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41:518–528. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, Tang Y, Chen Y. HIF1A gene rs10873142 polymorphism is associated with risk of chronic obstructive pulmonary disease in a Chinese Han population: a case-control study. Biosci Rep. 2018;38 doi: 10.1042/BSR20171309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ariazi JL, Duffy KJ, Adams DF, Fitch DM, Luo L, Pappalardi M, et al. Discovery and preclinical characterization of GSK1278863 (daprodustat), a small molecule hypoxia inducible factor-prolyl hydroxylase inhibitor for anemia. J Pharmacol Exp Ther. 2017;363:336–347. doi: 10.1124/jpet.117.242503. [DOI] [PubMed] [Google Scholar]
- 58.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–1527. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. 2018;314:L256–L275. doi: 10.1152/ajplung.00096.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, et al. The endothelial prolyl-4-hydroxylase domain 2/hypoxia-inducible factor 2 axis regulates pulmonary artery pressure in mice. Mol Cell Biol. 2016;36:1584–1594. doi: 10.1128/MCB.01055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang Y, Kempen MB, Munck AB, Swagemakers S, Driegen S, Mahavadi P, et al. Hypoxia-inducible factor 2α plays a critical role in the formation of alveoli and surfactant. Am J Respir Cell Mol Biol. 2012;46:224–232. doi: 10.1165/rcmb.2011-0024OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.