

Abstract
Rationale: Puerto Ricans have the highest childhood asthma prevalence in the United States (23.6%); however, the etiology is uncertain.
Objectives: In this study, we sought to uncover the genetic architecture of lung function in Puerto Rican youth with and without asthma who were recruited from the island (n = 836).
Methods: We used admixture-mapping and whole-genome sequencing data to discover genomic regions associated with lung function. Functional roles of the prioritized candidate SNPs were examined with chromatin immunoprecipitation sequencing, RNA sequencing, and expression quantitative trait loci data.
Measurements and Main Results: We discovered a genomic region at 1q32 that was significantly associated with a 0.12-L decrease in the lung volume of exhaled air (95% confidence interval, −0.17 to −0.07; P = 6.62 × 10−8) with each allele of African ancestry. Within this region, two SNPs were expression quantitative trait loci of TMEM9 in nasal airway epithelial cells and MROH3P in esophagus mucosa. The minor alleles of these SNPs were associated with significantly decreased lung function and decreased TMEM9 gene expression. Another admixture-mapping peak was observed on chromosome 5q35.1, indicating that each Native American ancestry allele was associated with a 0.15-L increase in lung function (95% confidence interval, 0.08–0.21; P = 5.03 × 10−6). The region-based association tests identified four suggestive windows that harbored candidate rare variants associated with lung function.
Conclusions: We identified common and rare genetic variants that may play a critical role in lung function among Puerto Rican youth. We independently validated an inflammatory pathway that could potentially be used to develop more targeted treatments and interventions for patients with asthma.
Keywords: admixed, FEV1, RNA sequencing, inflammatory, TMEM9 airway epithelial cells
At a Glance Commentary
Scientific Knowledge on the Subject
In the United States, asthma prevalence and severity vary significantly by race/ethnicity. Puerto Ricans have the highest childhood asthma prevalence (23.6%); however, it is not clear what causes Puerto Ricans to have such high occurrence. The structure of modern Hispanic/Latino populations is a mosaic of Native American, European, and African ancestral influences. A number of studies have reported the influence of genetic ancestry on the development of complex diseases. African genetic ancestry has been linked to decreased lung function, decreased drug response, and increased risk for the development of asthma in Latino populations.
What This Study Adds to the Field
We identified genomic regions that harbored candidate common and rare variants that could influence lung function among Puerto Rican youth with and without asthma using admixture-mapping, whole-genome sequencing, and functional genomics data. The findings of this study (i.e., the IL-6 and IL-1β inflammatory pathways mediated by TMEM9 gene expression) could potentially be used to treat Puerto Rican youth, a population that carries the majority of the asthma disease burden, and possibly benefit patients with asthma of other ethnicities, too. The findings of this manuscript therefore have important clinical and public health implications.
Asthma is an obstructive inflammatory lung disease that adversely influences the daily lives of 26 million Americans and costs $81 billion in annual health care (1). In the United States, asthma prevalence and severity vary significantly by race/ethnicity. Puerto Ricans have the highest childhood asthma prevalence (23.6%), followed by African Americans (18.1%), Mexican Americans (11.5%), and European Americans (9.5%) (2). These striking racial/ethnic disparities in asthma morbidity arise from a complex interplay of genetic and environmental risk factors (3–6). We previously demonstrated that the proportion of African genetic ancestry explained a substantial amount of the variance in baseline lung function in African Americans and Hispanics/Latinos, even after adjusting for environmental and social risk factors (7, 8).
Numerous studies have reported the influence of genetic ancestry on the development of complex diseases, including breast and prostate cancer, diabetes, cardiovascular disease, and asthma (9–15). Ancestry may reflect demographic history, unmeasured risk factors present in ancestral populations, and natural genetic variation, which can all be informative for understanding disease patterns within and across racial/ethnic groups. In addition, genetic risk alleles vary in frequency by race/ethnicity (16, 17). Therefore, among genetically admixed individuals, varying proportions of genetic ancestries may track with and therefore be a proxy of genetic risk alleles (7–15).
Modern Hispanic/Latino populations are a mosaic of Native American, European, and African ancestries. African genetic ancestry has been linked to decreased lung function, decreased drug response, and increased risk for the development of asthma in Latino populations (7, 8, 18). In contrast, increased Native American ancestry was associated with increased lung function in Latino children (19). It is possible that the large discrepancy in asthma prevalence and morbidity between Puerto Ricans and Mexican Americans may be partially explained by variation in genetic ancestry proportions. Previous studies have attempted to uncover genetic determinants of asthma in Puerto Ricans (20–24). However, it remains unclear why Puerto Ricans suffer such high asthma prevalence, morbidity, and mortality.
In this study, we used whole-genome sequencing (WGS) data to perform a genome-wide admixture-mapping analysis of lung function in asthma cases and controls from a Puerto Rican youth population from the island of Puerto Rico. Admixture mapping is a complementary method to traditional genome-wide allelic association tests. Typically, admixture linkage disequilibrium (LD) blocks are longer than haplotype blocks and therefore may have a higher chance of capturing disease-associated genes and reducing the multiple testing burden (25, 26). Admixture mapping identified multiple potential causal variants influencing lung function within two genomic regions. We used nasal epithelial cells from Puerto Rican youth and other available lung tissues to validate the functional roles of these variants and to explore possible biological pathways and mechanisms through which they could influence asthma prevalence and mortality. These results carry potentially important clinical implications for effective management of asthma in Puerto Rican youth.
Methods
Study Subjects
GALA II (Genes-Environment and Admixture in Latino Americans) is an ongoing case–control study of asthma in youth in the mainland United States and Puerto Rico (2006–present). Subjects were eligible to participate if they were 8–21 years of age and identified all four grandparents as Latino. In this study, only the subjects recruited from the island of Puerto Rico who had both lung function and genotype data were included in the main analysis (n = 836). Please see the online data supplement (Study Subjects) for study participants’ eligibility criteria.
Local institutional review boards approved the studies. All subjects and their legal guardians provided written informed assent/consent.
Pulmonary Function Testing
Pulmonary function testing was performed according to the American Thoracic Society standards with a KoKo PFT Spirometer (nSpire Health Inc.) to measure baseline lung function, particularly FEV1. All spirometric measures were evaluated by a pulmonologist in Puerto Rico and at the University of California, San Francisco. Lung function measurements were log-transformed to make the distribution less skewed (Figure E1 in the online supplement). Analyses were performed on log-transformed FEV1, which was treated as a continuous variable.
Local and Global Ancestry Estimation
More information about local and global ancestry estimations can be found in the supplement (Local and Global Ancestry Estimation).
Whole-Genome Sequencing
Genotypes were obtained from whole-genome sequences obtained as part of the TOPMed (Trans-Omics for Precision Medicine) sequencing program. Details regarding DNA extraction, WGS data generation, processing, and quality control steps can be found in the supplement (Whole Genome Sequencing Data Generation, Processing and Quality Control).
Admixture Mapping
Estimates of locus-specific ancestry were used to conduct admixture mapping. Each SNP was encoded with the number of alleles (0, 1, or 2 alleles) from a given ancestry. Additive linear regression models were used to test associations between local ancestry and lung function (measured FEV1, liters) adjusting for age (years), sex (0 = male; 1 = female), asthma status (0 = control; 1 = case), Native American and African genome-wide genetic ancestry proportions (%), and height (cm). We adopted a similar approach as Shriner and colleagues and our previous studies to determine the effective number of tests (n = 595) by fitting an autoregression model to the summary statistics generated from the admixture-mapping analysis using the coda package in R (8, 12, 27). A Bonferroni corrected threshold of genome-wide significance at alpha level was then defined as α = 0.05/595 = 8.4 × 10−5 and applied to correct for multiple testing and reduce false positive. More information can be found in the supplement (Admixture Mapping).
Fine Mapping: Conditional Analysis
Details of the fine-mapping conditional analysis can be found in the supplement (Fine Mapping: Conditional Analysis).
Region-based Association Analysis
Please see the supplement (Region-based association analysis) regarding region-based association analyses and drop-one variant analyses using SKAT-O (28).
H3K27ac ChIP-seq Assays in HBECs and Bronchial Smooth Muscle Cells
A detailed description of chromatin immunoprecipitation sequencing (ChIP-seq) analyses can be found in the supplement (Human bronchial epithelial cell culture and H3K27ac ChIP-seq assay).
Functional Annotation and Validation of Genetic Variants
Genomic annotation was conducted for the lead ancestry SNP, the SNPs identified from fine-mapping analyses, and all SNPs in linkage disequilibrium with them (r2 > 0.8). More information can be found in the supplement (Functional Annotation and Validation of Genetic Variants).
RNA Sequencing and eQTL Analysis
Generation of gene expression and expression quantitative trait loci (eQTL) data and expression analyses are summarized in the supplement [RNA sequencing and Expression Quantitative Trait Loci (eQTL) analysis].
All statistical analyses were conducted in R Studio (version 1.2.5019) and PLINK 1.9 (29, 30).
Results
Study Participants
Baseline characteristics of study participants, by asthma status, are displayed in Table 1. In the main analyses, there were a total of 836 study participants (720 cases with asthma and 116 controls without asthma). On average, patients with asthma were slightly younger than the controls (12.4 ± 3.3 and 13.1 ± 1.7 yr, respectively; P = 0.01). The mean lung function between asthma cases (2.3 ± 0.8 L) and controls (3.0 ± 0.7 L) were significantly different (P < 2.2 × 10−16). Subjects without asthma were significantly taller than the participants with asthma (157.5 ± 10.4 and 150.1 ± 13.6 cm, respectively; P = 3.2 × 10−8).
Table 1.
Descriptive Statistics for Puerto Rican Study Participants Recruited from the Island (n = 836)
| Asthma Cases | Asthma Controls | P Value | |
|---|---|---|---|
| No. of participants | 720 | 116 | |
| Age, mean ± SD, yr | 12.4 ± 3.3 | 13.1 ± 1.7 | 0.01* |
| Sex, M, n (%) | 408 (56.7) | 63 (54.3) | 0.9† |
| FEV1, mean ± SD, L | 2.3 ± 0.8 | 3.0 ± 0.7 | 2.2 × 10−16* |
| Height, mean ± SD, cm | 150.1 ± 13.6 | 157.5 ± 10.4 | 3.2 × 10−8* |
| Ancestry, mean ± SD, % | |||
| African | 22.9 ± 11.9 | 23.6 ± 8.1 | 0.5* |
| Native American | 10.5 ± 3.0 | 10.7 ± 2.8 | 0.4* |
| European | 66.5 ± 11.4 | 65.6 ± 8.0 | 0.4* |
Unpaired two-sample t test was conducted for continuous variables.
Two-proportion z-test was performed for categorical variables.
Admixture Mapping
A genome-wide single ancestry admixture mapping was conducted for each of the three reference ancestries (YRI, African; CEU, European; NAM, Native American). We discovered two genome-wide significant peaks for African and Native American ancestries after adjusting for covariates (Figure 1). The association test between local genetic ancestry and FEV1 identified a strong admixture signal on chromosomal region 1q32, indicating that each African ancestry allele was associated with a 0.12-L decrease in the volume of exhaled air (95% confidence interval, −0.17 to −0.07; P = 6.62 × 10−8) after transforming back to normal scale. Admixture mapping for Native American ancestry located a genomic region at 5q35.1 (Figure 1) that was significantly associated with a 0.15-L increase in lung function (95% confidence interval, 0.08–0.21; P = 5.03 × 10−6). The lead Native American ancestry SNP, rs12153426, is an intron variant within SLIT3. The lead African ancestry SNP (rs17696752) sits in a noncoding intergenic region (Figure 2).
Figure 1.
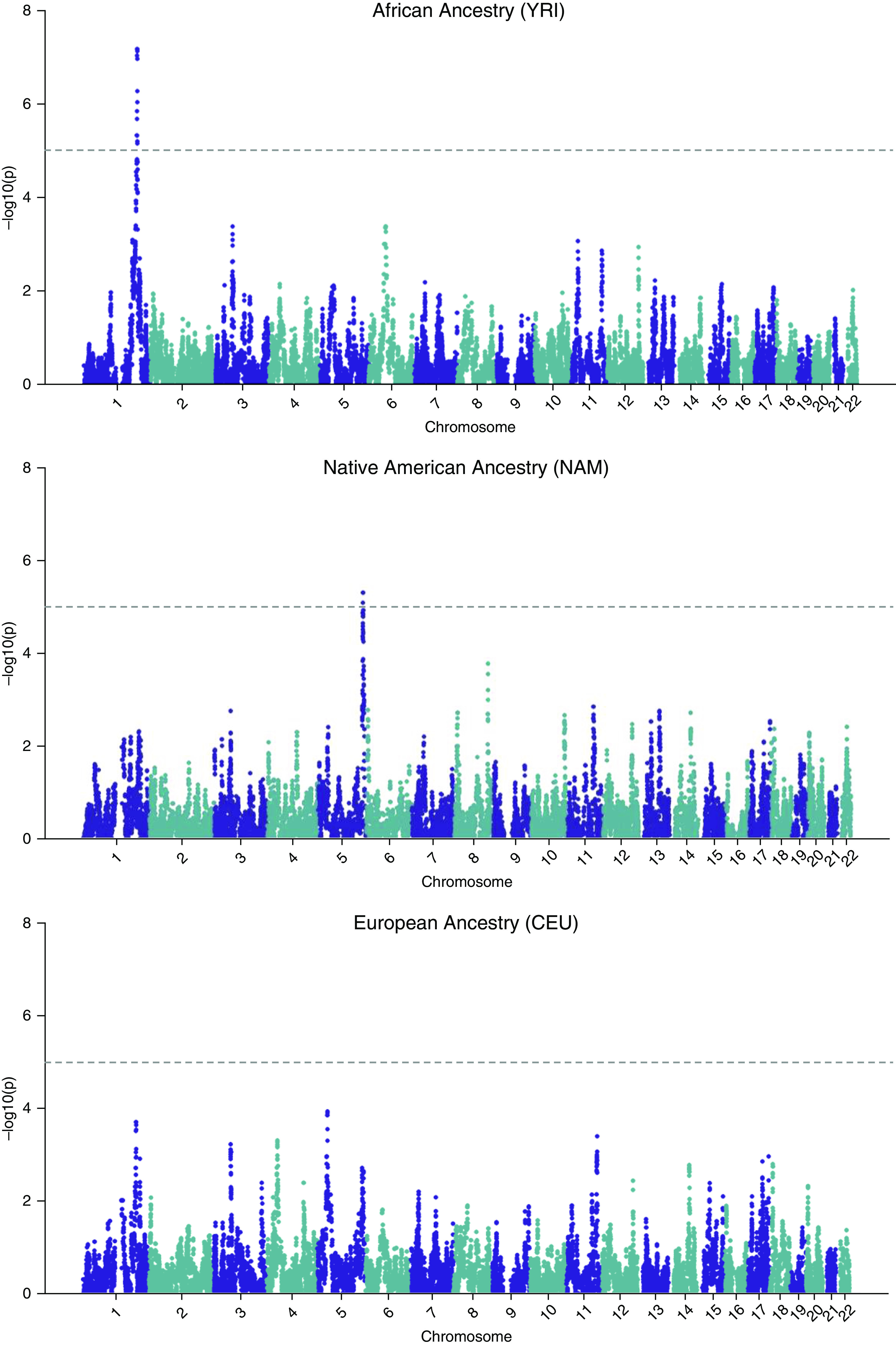
Admixture-mapping results. Genome-wide single ancestry admixture mapping for each reference ancestry (YRI, CEU, and NAM) with log-transformed FEV1 adjusting for covariates including age, sex, height, asthma status, and African and Native American ancestries is shown.
Figure 2.

Admixture-mapping peaks were discovered at 1q32 and 5q35.1. Locus zoom displaying the lead locus-specific ancestry signals (rs17696752 and rs12144397) and nearby genes is shown. chr = chromosome.
Fine Mapping: Conditional Analysis
Allelic association tests of lung function with WGS data were performed at the two genomic regions identified from admixture mapping. Conditional analyses on the association between lung function and locus-specific ancestry revealed that no single SNP was accountable for the observed admixture-mapping signals. Therefore, the joint effects of multiple SNPs on the locus-specific ancestry and lung function association were further investigated.
Stepwise linear regression models identified four SNPs mapped to African ancestry and seven SNPs mapped to Native American ancestry within the genomic regions harboring admixture signals (Table 2). The effect estimates shown in Table 2 represent the associations between the lead ancestry SNP and lung function, adjusting for additional SNPs and covariates. The individual allelic association tests between these SNPs and lung function are displayed in Table E1.
Table 2.
Fine Mapping of Genomic Regions Harboring Admixture Signal Using Sequenced Data
| β* | P Value | |
|---|---|---|
| African ancestry | ||
| rs17696752 | −0.02 | 6.62 × 10−8 |
| rs17696752 + rs16847664 | −0.02 | 5.29 × 10−6 |
| rs17696752 + rs16847664 + rs6679485 | −0.02 | 4.33 × 10−4 |
| rs17696752 + rs16847664 + rs6679485 + rs116270262 | −0.01 | 7.62 × 10−3 |
| rs17696752 + rs16847664 + rs6679485 + rs116270262 + rs10920079 | −0.01 | 0.09 |
| Native American ancestry | ||
| rs12153426 | 0.03 | 5.04 × 10−6 |
| rs12153426 + rs187841563 | 0.03 | 5.52 × 10−6 |
| rs12153426 + rs187841563 + rs11134502 | 0.03 | 5.12 × 10−6 |
| rs12153426 + rs187841563 + rs11134502 + rs73320187 | 0.02 | 2.96 × 10−3 |
| rs12153426 + rs187841563 + rs11134502 + rs73320187 + rs4323254 | 0.02 | 0.03 |
| rs12153426 + rs187841563 + rs11134502 + rs73320187 + rs4323254 + rs74677211 | 0.01 | 0.03 |
| rs12153426 + rs187841563 + rs11134502 + rs73320187 + rs4323254 + rs74677211 + rs62384405 | 0.01 | 0.03 |
| rs12153426 + rs187841563 + rs11134502 + rs73320187 + rs4323254 + rs74677211 + rs62384405 + rs12522512 | 0.01 | 0.21 |
Joint effects of multiple SNPs on lung function were evaluated using stepwise regression models. Conditional analyses on the locus-specific ancestry identified potential causal SNPs on chromosome 1 (1q32) and 5 (5q35.1).
β values represent the association between lead ancestry SNP (rs17696752 at 1q32 and rs12153426 at 5q35.1) and log-transformed FEV1 adjusting for covariates and additional SNPs in forward stepwise regression analyses. All regression models were adjusted for age, sex, height, asthma status, and African and Native American ancestries.
Fine Mapping: Region-based Association Analysis
The main results of the region-based association tests are presented in Table 3. On chromosome 1, we did not find any significant window after correcting for multiple testing (P < 2.28 × 10−5). However, two windows were suggestively associated with lung function (P < 4.60 × 10−4). The P values of these two suggestive windows represent lung function associations driven solely by rare variants. The region-based analysis on chromosome 5 identified two windows that were suggestively associated with lung function (P < 5.30 × 10−4).
Table 3.
Region-based Association Tests within Each Fine-Mapping Region
| Chr | Start Position | End Position | P Value | No. of Rare Markers | Nearest Gene(s) | Window ID |
|---|---|---|---|---|---|---|
| 1 | 196762063 | 196763063 | 2.97 × 10−5* | 1 | CFH, CHFR3 | 1 |
| 1 | 199359563 | 199360536 | 6.76 × 10−5* | 4 | LINC02789, LOC400600 | 2 |
| 5 | 169911224 | 169912224 | 1.01 × 10−4† | 6 | DOCK2, INSYN2B | 3 |
| 5 | 167726224 | 167727224 | 2.44 × 10−4† | 6 | TENM2 | 4 |
Definition of abbreviation: Chr = chromosome.
Genetic variants (minor allele frequency < 0.01) were aggregated in 1-kb sliding windows to examine the association with lung function adjusting for covariates using SKAT-O. Suggestive windows were reported within 1q32 and 5q35.1 regions.
Chromosome 1 (after multiple test correction): adjusted P value = 2.28 × 10−5, adjusted suggestive P value = 4.6 × 10−4.
Chromosome 5 (after multiple test correction): adjusted P value = 2.64 × 10−5, adjusted suggestive P value = 5.3 × 10−4.
For each suggestive window, we performed drop-one variant analysis to investigate the effects of each rare variant on lung function. When the rare variant rs1000558853 (minor allele frequency = 0.0006) was removed from window 2, the P value increased by nearly four orders of magnitude (P = 0.41), suggesting that most of the association with lung function for window 2 was contributed by rs1000558853 (Figure E2 and Table E2). The rare variants rs943656663 and rs539192870 also resulted in larger P values when they were excluded from windows 3 and 4, respectively. rs943656663 is an intron variant of TENM2, whereas rs539192870 falls within an intronic region of the genes DOCK2 and INSYN2B. The minor allele of the rare variant rs539192870 was found exclusively in Puerto Ricans (Figure E3).
Bioinformatics Functional Annotation for SNP Prioritization
The candidate SNPs identified from fine mapping, along with other SNPs in high LD with them (r2 > 0.80), were further investigated to understand their functional roles. Information from public data sources and H3K27ac ChIP-seq experiments indicated that four SNPs discovered by the African ancestry signal had regulatory functions in white blood cell and lung-related cell lines (Figure 3). We found that rs2400382 overlapped with a CEBPB (CCAAT/enhancer-binding protein β) transcription factor binding site in human lung fibroblast cells (IMR90) and H3K4me3 ChIP-seq and DNase I hypersensitivity signals (cCRE accession ID:EH38E1408465). The intron variants of MROH3P, rs10920079 and rs10920072, intersected with H3K27ac peaks in human bronchial epithelial cells (HBECs). The synonymous variant of CACNA1S, rs16847664, was also enriched with epigenetic markers in HBECs and may interact with TMEM9 based on high-throughput chromosome conformation capture data in IMR90 cells.
Figure 3.
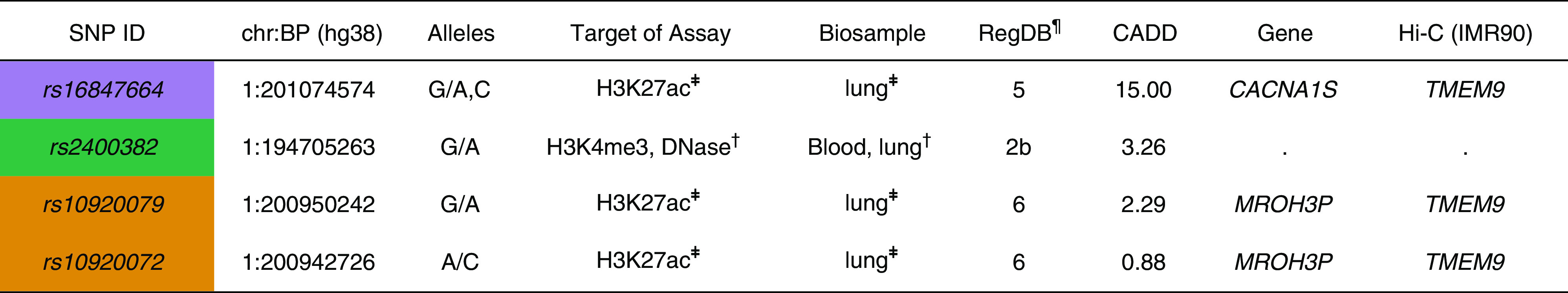
Functional annotation of four variants identified from fine-mapping analysis at 1q32. Each color represents SNPs identified from fine-mapping conditional analysis and their linkage disequilibrium SNPs (R2 > 0.8) with at least one or more than one regulatory element data. ¶RegulomeDB (RegDB) score represents the likelihood that a variant will be located within a functional region. The RegDB variant classification scheme is defined as follows (50): Likely to affect binding: 2b, TF binding + matched TF motif + matched DNase footprint + DNase peak. Minimal binding evidence: 4, TF binding + DNase peak; 5, TF binding or DNase peak; 6, motif hit. ‡Overlapping with H3K27ac chromatin immunoprecipitation sequencing peaks in human bronchial epithelial cells. †High signals of epigenetic markers in immune cells and lung-related tissues (Encyclopedia of DNA Elements data). BP = base pair position; CADD = Combined Annotation-Dependent Depletion; chr = chromosome; Hi-C = high-throughput chromosome conformation capture; TF = transcription factor.
Within the genomic region identified by admixture mapping with Native American ancestry, we identified 19 SNPs located in candidate regulatory regions (Figure 4). One of the candidate causal SNPs selected through fine-mapping analysis, rs6555875, was an intron variant of DOCK2 and fell within the H3K27ac peak area in bronchial smooth muscle cells. The high-throughput chromosome conformation capture data suggest that rs6555875 significantly interacts with FAM196B in IMR90 cells. The intron variant of RARS, rs11134516, overlapped with H3K4me3, H3K27ac, CTCF, and DNase peaks in white blood cells and lung-related tissues/cells according to Encyclopedia of DNA Elements data (cCRE accession ID:EH38E2429336). The ChIP-seq experiments in HBECs and bronchial smooth muscle cells further supported the regulatory potential of rs11134516. The RegulomeDB scores of rs185216867 and rs62384405 indicated that these SNPs were likely to affect transcription factor binding.
Figure 4.
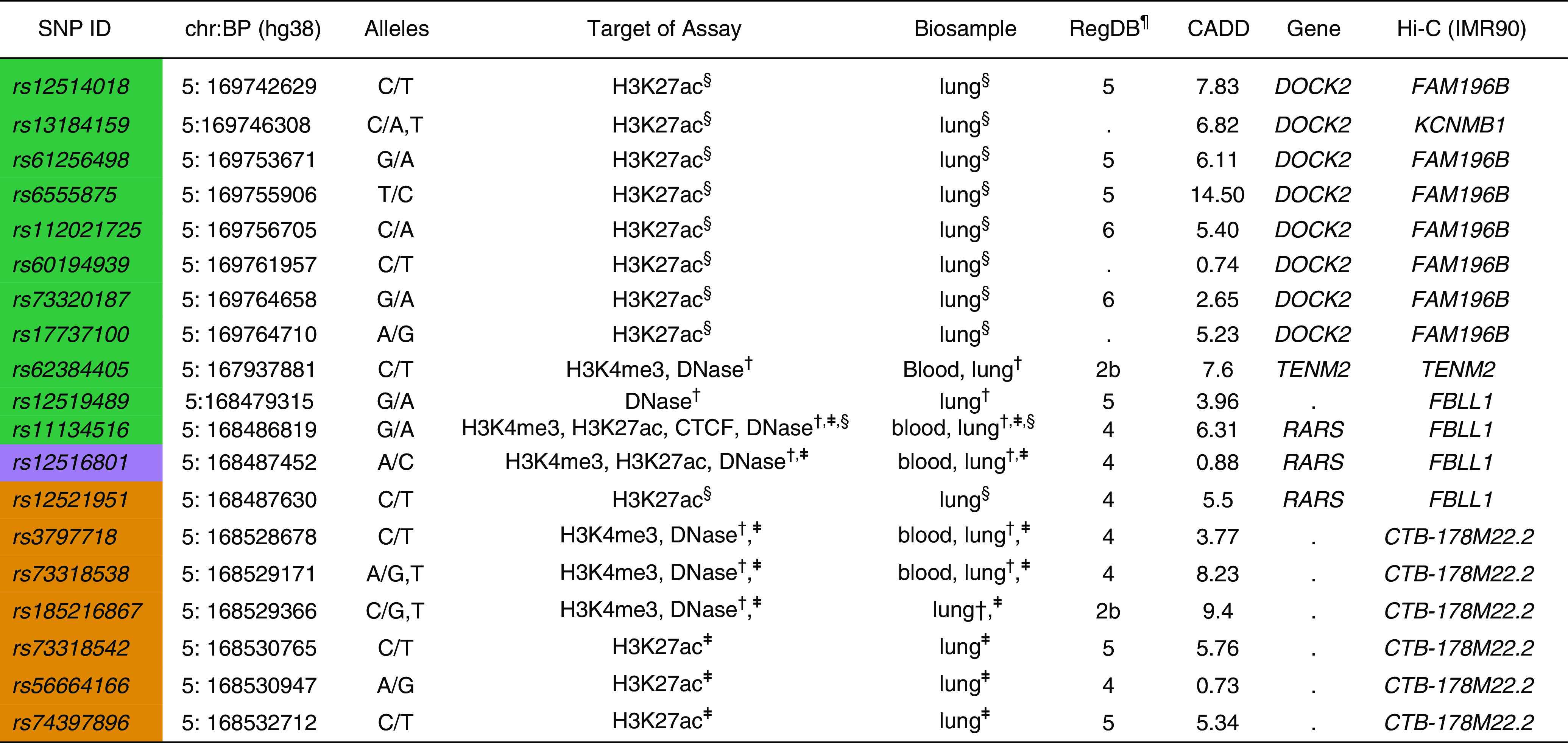
Functional annotation of 19 variants identified from fine-mapping analysis at 5q35.1. Each color represents SNPs identified from the fine-mapping conditional analysis and their linkage disequilibrium SNPs (R2 > 0.8) with at least one or more than one regulatory element data. ¶RegulomeDB (RegDB) score represents the likelihood that a variant will be located within a functional region. The RegDB variant classification scheme is defined as follows (50): Likely to affect binding: 2b, TF binding + matched TF motif + matched DNase footprint + DNase peak. Minimal binding evidence: 4, TF binding + DNase peak; 5, TF binding or DNase peak; 6, motif hit. §Overlapping with H3K27ac chromatin immunoprecipitation sequencing peaks in bronchial smooth muscle cells. †High signals of epigenetic markers in immune cells and lung (Encyclopedia of DNA Elements data). ‡Overlapping with H3K27ac chromatin immunoprecipitation sequencing peaks in human bronchial epithelial cells. For definition of abbreviations, see Figure 3.
Expression Analyses
We found two of the African ancestry–associated SNPs prioritized by admixture mapping (rs10920072 and rs10920079), which were in high LD with one another and were also cis-eQTL variants of the TMEM9 gene in nasal epithelial cells and MROH3P in esophagus mucosa (Figure E4 and Table 4). The minor alleles of these SNPs were found more frequently among the African population and associated with decreased expression of TMEM9 (Prs10920072 = 2.00 × 10−9 and Prs10920079 = 1.31 × 10−9) and decreased lung function after adjusting for covariates (Prs10920072 = 1.18 × 10−5 and Prs10920079 = 4.23 × 10−6). Using nasal epithelial gene expression data, we showed decreasing trends of TMEM9 expression and lung function with respect to one or two copies of the minor allele of rs10920072 and rs10920079 (Figure 5). TMEM9 expression was correlated with lung function (R2 = 0.18; P = 0.93 × 10−5) (Figure E5).
Table 4.
Expression Analysis Results: African Ancestry
| SNP ID | Chr | Position (hg38) | Minor Allele | Minor Allele Frequency |
Nasal Epithelial Cells |
Esophagus Mucosa (GTEx) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| YRI | CEU | PUR* | β | P Value | Gene | β | P Value | Gene | ||||
| rs10920079 | 1 | 200950242 | C | 0.47 | 0.05 | 0.33 | −0.19 | 1.31 × 10−9 | TMEM9 | −0.36 | 1.60 × 10−5 | MROH3P |
| rs10920072 | 1 | 200942726 | A | 0.66 | 0.06 | 0.42 | −0.21 | 2.00 × 10−9 | TMEM9 | −0.36 | 1.40 × 10−5 | MROH3P |
Definition of abbreviations: CEU = Utah Residents with Northern and Western European ancestry; Chr = chromosome; GTEx = Genotype-Tissue Expression Project; PUR = Puerto Ricans from Puerto Rico; YRI = Yoruba in Ibadan, Nigeria.
The expression quantitative trait locus mapping in nasal epithelial cells identified two significant cis-acting expression quantitative trait loci of TMEM9. These SNPs were also significantly associated with MROH3P expression in esophagus mucosa according to GTEx data. The minor allele of these SNPs tracked with African population, and these alleles were also found more frequently among our study participants than among people with European ancestry.
Minor allele frequency computed from the Puerto Rican study participants (n = 836).
Figure 5.

Expression analysis results. Expression of TMEM9 gene and lung function by minor allele of the candidate SNPs (rs10920072 and rs10920079) is shown.
Discussion
Findings from our previous studies have suggested that varying proportions of genetic ancestry may play a critical part in asthma prevalence and severity in African American and Latino populations (7, 8, 12, 19). Thus, we reasoned that the admixed population structure of Puerto Ricans could be leveraged to identify genetic variants associated with lung function. To our knowledge, our study is the first study to have delineated and validated potential biological pathways associated with lung function in Puerto Rican youth using admixture-mapping, whole-genome sequence, and functional genomics data. We discovered two genomic regions associated with lung function and candidate SNPs with important functional elements that may influence inflammatory pathways in lung-related tissues and peripheral blood cells.
We previously demonstrated that African genetic ancestry was associated with lower lung function in participants with and without asthma. Therefore, we performed an African-specific ancestry admixture-mapping analysis and discovered a genomic region at 1q32 that was associated with a risk of decreased lung function among the 836 Puerto Ricans with and without asthma. The lead locus-specific ancestry SNP, rs17696752, resides in a potential regulatory region in the intergenic region of the genes LINC02789 and NR5A2 based on overlap with a GATA3 ChIP-seq cluster in the human neuroblast cell line (SH-SY5Y); GATA3 is a master transcription factor involved with T-cell development and expression of T-helper cell type 2 cytokine genes (31). This SNP was also associated with decreased FEV1 in the U.K. Biobank data set (β = −0.01; P = 1.9 × 10−4) (32). The conditional analysis with sequenced data identified four candidate SNPs that could potentially explain the ancestry signal at 1q32. One of the SNPs was a synonymous variant of the CACNA1S gene, which plays a key role in skeletal muscle movement (contraction and relaxation) by generating calcium channels in muscle cells (33). It is not clear how CACNA1S is involved with lung function. However, rs3850625, a functionally deleterious missense variant of CACNA1S, was associated with FVC in the U.K. Biobank data set, further validating our study results (34). Previous genome-wide association studies identified genetic variants associated with asthma, lung function, and bronchodilator drug response at 1q32 (35–37). In addition, we previously identified this locus in a genome-wide association study of Puerto Rican asthma trios (father, mother, and affected child), independent of the current study, and found that the genomic region 1q32.2 was associated with asthma (21).
The potential biologic plausibility of the SNPs selected from the fine mapping above, as well as SNPs in high LD with them (r2 > 0.8), were further investigated by performing a bioinformatics search and functional experiments. We found four SNPs located in potential regulatory regions in lung-related tissues and peripheral blood cells. Two of our lung function–associated SNPs (rs10920079 and rs10920072) were eQTLs of TMEM9, and their minor alleles were significantly associated with decreased expression of TMEM9 in nasal epithelial cells from Puerto Rican youth. These two SNPs were also significantly associated with decreased MROH3P expression in esophagus mucosa according to GTEx data. The minor alleles of these SNPs were more prevalent in both African populations and our study participants compared with European populations. Another study demonstrated that expression of the TMEM9 gene was significantly associated with lung function (38). Although the direction of the association between TMEM9 and lung function depends on tissue and cell types, their findings indicate that TMEM9 gene expression could be used as an important biologic target for pulmonary diseases.
A recent study suggested that expression of TMEM9 regulates secretion of IL-6 and IL-1β, which are important cytokines involved with airway inflammation and asthma (39, 40). Our previous study has demonstrated that the interaction between genetic ancestry and variants in IL-6 and IL-6R modified the effects of bronchodilator drug response in patients with asthma (41). We further tested this hypothesis with our RNA-sequencing data and found that increased expression of TMEM9 was inversely correlated with both IL-6 and IL-1β expressions in nasal epithelial cells (Figures E6 and E7, respectively). Among our study subjects, having one or two copies of the minor allele of rs10920072 and rs10920079 appeared to reduce expression of TMEM9 and yield lower FEV1 values. Therefore, we speculate that the poor lung function of Puerto Rican youth could be mediated through the IL-6 and IL-1β inflammatory pathways and by TMEM9 gene expression.
We additionally discovered an admixture-mapping peak on chromosome 5q35.1, indicating that with each allele of Native American ancestral origin, lung function increased after adjusting for covariates. This is consistent with previous work demonstrating that Native American genetic ancestry was associated with improved lung function (19, 41). The Native American ancestry SNP with the smallest P value, rs12153426, fell within a potential regulatory region in intron 4 of SLIT3 based on overlap with a GATA3 ChIP-seq cluster in the human neuroblast cells (SH-SY5Y). SLIT3 is a protein-coding gene highly expressed in the lung (42). Previous studies demonstrated that expression of SLIT3 was involved with tumor suppression in lung cancer and possibly involved with organ developments including the diaphragm and kidney in mice (42–44). Fine-mapping analysis with sequenced data identified seven candidate SNPs to further explain the association between Native American genetic ancestry signal and lung function. Two SNPs (rs187841563 and rs73320187) were located within a potential regulatory region of intron 25 in DOCK2 based on overlap with ELF and P300 ChIP-seq clusters in B-lymphocyte cells. Expression of DOCK2 influences spatial rearrangement of the cytoskeleton, a necessary step prior to lymphocyte migration triggered by chemokine signaling upon antigen challenge (45).
The subsequent functional analysis of the seven candidate SNPs and their LD SNPs (r2 > 0.8) revealed that 19 SNPs had an evidence of regulatory function in lung-related tissues and peripheral blood cells. Based on relevant functional annotation, two SNPs (rs11134516 and rs12516801) appeared to have a strong enrichment of H3K4me3, H3K27ac, and DNase I hypersensitivity ChIP-seq signals, indicating a potential role in gene expression regulation. These two SNPs fell within an intronic region of RARS (arginyl-tRNA synthetase) and may interact with the FBLL1 gene. In addition, based on the RegulomeDB categories and Encyclopedia of DNA Elements epigenetics data, rs62384405 and rs185216867 may have regulatory functions, too. We do not know how rs62384405, an exon variant of a long noncoding RNA transcript overlapping with TENM2, influences lung function. However, a different variant residing within the intronic region of TENM2, rs73803919, was associated with a positive response to bronchodilator drug response (46). The exact mechanism(s) of how gene expression of RARS and FBLL1 influences lung function among our participants is an important direction for future study.
The region-based association tests did not find any window that was significantly associated with lung function after multiple testing correction. However, we identified four windows within the fine-mapping regions of chromosome 1 and 5 that passed the suggestive threshold levels, indicating that both common and rare variants in our admixture-mapping peaks can influence lung function.
Certain genetic variants may play important roles in development of childhood-onset asthma (47, 48). The mean (±SD) age for the asthma onset among our participants was 2.1 (±2.6) years. We found that the age of asthma onset significantly interacted with the lead Native American ancestry SNP (rs12153426) on lung function, suggesting that those who developed asthma later in life and had one or more than one copy of a Native American ancestry allele at the given locus resulted in positive lung function (βeta = 0.008; P = 0.001) (Table E3). Because our participants were composed of broad age groups (8–21 yr), we also examined the effects of age. After adjusting for covariates, age was significantly associated with lung function. However, the interactions between the lead ancestry SNPs and age were not statistically significant.
This study is among the few asthma genetic studies focusing exclusively on Puerto Rican youth recruited from the island of Puerto Rico. Applying the admixture-mapping technique over traditional genome-wide allelic association tests with genetically admixed Puerto Ricans enabled discovery of the disease-associated genetic variant(s) attributable to different ancestry haplotype blocks. In addition, our uniquely rich data set includes WGS, RNA sequencing, and eQTL in ciliated nasal epithelial cells from Puerto Rican youth and functional assays in lung-related tissues. We previously demonstrated that gene expression patterns between ciliated nasal epithelial cells and bronchial tissue biopsies overlap by 93% (49). Our WGS facilitated both common and rare variant analyses, thereby considering all potential causal genetic variants. Furthermore, we validated important biological mechanisms and pathways that could potentially be used to treat patients with asthma. Finally, the association between the 1q32.2 genomic region and lung function/asthma was also reported in two independent data sets, the U.K. Biobank and Puerto Rican families from the GALA I study (21, 32).
Although our study significantly contributes to improving our understanding of the genetic components of lung function among Puerto Rican youth, our findings should be interpreted with caution. First, our sample size was limited because we were studying a population of minority youth from a specific geographic location. Although our identified genomic regions reached genome-wide significance after correcting multiple testing, adequate power in future studies will invariably require larger sample sizes. Second, the findings from this study may not be generalizable or applicable to other populations. Although it is likely that the Native American ancestry SNPs that we identified in this study are population-specific, it is also possible that our candidate SNPs are relevant to other admixed Latino populations.
In summary, admixture mapping identified genomic regions that harbored candidate genetic variants that could influence lung function among the Puerto Rican youth with and without asthma. We validated multiple inflammatory pathways with our biological data, which has important clinical and public health implications.
Supplementary Material
Acknowledgments
Acknowledgment
The authors acknowledge the families and patients for their participation and thank the numerous healthcare providers and community clinics for their support and participation in GALA II. In particular, the authors thank study coordinator Sandra Salazar, as well as the recruiters who obtained the data: Duanny Alva, M.D., Gaby Ayala-Rodriguez, Lisa Caine, Elizabeth Castellanos, Jaime Colon, Denise DeJesus, Blanca Lopez, Brenda Lopez, M.D., Louis Martos, Vivian Medina, Juana Olivo, Mario Peralta, Esther Pomares, M.D., Jihan Quraishi, Johanna Rodriguez, Shahdad Saeedi, Dean Soto, and Ana Taveras. Whole-genome sequencing for the TOPMed program was supported by the NHLBI. WGS for “NHLBI TOPMed: Gene-Environment, Admixture and Latino Asthmatics Study” (phs000920) was performed at the New York Genome Center (3R01HL117004-02S3) and the University of Washington Northwest Genomics Center (HHSN268201600032I). Centralized read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I). The authors gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. WGS of part of GALA II was performed by New York Genome Center under The Centers for Common Disease Genomics of the Genome Sequencing Program (GSP) Grant (UM1 HG008901). The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. GSP is funded by the National Human Genome Research Institute, the NHLBI, and the National Eye Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Supported in part by the Sandler Family Foundation; the American Asthma Foundation; the Robert Wood Johnson Foundation Amos Medical Faculty Development Program; the Harry Wm. and Diana V. Hind Distinguished Professorship in Pharmaceutical Sciences II; National Institute of General Medical Sciences grant T32GM007546; NHLBI grants R01HL117004, R01HL128439, R01HL135156, 1X01HL134589, R35 HL145235, 1 U01HL138626, P01 HL132821, and P01 HL107202; National Institute of Allergy and Infectious Diseases grant U19 AI077439; National Institutes of Health and Environmental Health Sciences grants R01ES015794 and R21ES24844; National Institute on Minority Health and Health Disparities grants P60MD006902, R01MD010443, and R56MD013312; Department of Defense grant W81WH-16-2-0018; and National Human Genome Research Institute grant U01HG009080. M.J.W. was supported by NHLBI grant K01HL140218. K.L.K. was supported by NHLBI grant R01HL135156-S1, the University of California San Francisco Bakar Institute, the Gordon and Betty Moore Foundation (grant GBMF3834), the Alfred P. Sloan Foundation (2013-10-27) grant to University of California Berkeley through the Moore-Sloan Data Science Environment Initiative at the Berkeley Institute for Data Science, and the Tobacco-Related Disease Research Program (grants 24RT-0025 and 27IR-0030). M.J.W. was additionally supported by an Institutional Research and Academic Career Development Award (K12GM081266) and an NHLBI Research Career Development Award (K01HL140218).
Author Contributions: E.Y.L. conceived the work, performed data analyses, and prepared the manuscript. A.C.Y.M. contributed to data analysis and manuscript preparation. D.H. contributed data analyses and interpretation of results. S.S. and C.U. provided RNA sequencing data and interpretation of results. M.J.W. and K.L.K. contributed to manuscript preparation. W.E., L.B., and D.E. provided chromatin immunoprecipitation sequencing data. S.H. and C.E. contributed to DNA data processing. D.J., G.A., H.M.K., S.G., M.C.Z., and D.A.N. contributed to whole-genome sequencing data generation, processing, and annotations. E.Z. contributed to interpretation of results and provided methodological advice. J.R.-S. contributed to the collection of data from the recruitment regions in Puerto Rico. M.A.S. and E.G.B. supervised this work.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202002-0351OC on May 27, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.American Lung Association. The impact of asthma. 2020 [accessed 2020 May 21]. Available from: https://www.lung.org/lung-health-diseases/lung-disease-lookup/asthma/learn-about-asthma/impact-of-asthma.
- 2.Centers for Disease Control and Prevention. Lifetime asthma prevalence percents by age, United States: National Health Interview Survey, 2018. 2018 [accessed 2020 May 21]. Available from: https://www.cdc.gov/asthma/nhis/2018/table2-1.htm.
- 3.Lee JU, Kim JD, Park CS. Gene-environment interactions in asthma: genetic and epigenetic effects. Yonsei Med J. 2015;56:877–886. doi: 10.3349/ymj.2015.56.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neophytou AM, White MJ, Oh SS, Thakur N, Galanter JM, Nishimura KK, et al. Air pollution and lung function in minority youth with asthma in the GALA II (Genes-environments and admixture in Latino Americans) and SAGE II (study of African Americans, asthma, genes, and environments) studies. Am J Respir Crit Care Med. 2016;193:1271–1280. doi: 10.1164/rccm.201508-1706OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishimura KK, Galanter JM, Roth LA, Oh SS, Thakur N, Nguyen EA, et al. Early-life air pollution and asthma risk in minority children: the GALA II and SAGE II studies. Am J Respir Crit Care Med. 2013;188:309–318. doi: 10.1164/rccm.201302-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.von Mutius E. Gene-environment interactions in asthma. J Allergy Clin Immunol. 2009;123:3–11, quiz 12–13. doi: 10.1016/j.jaci.2008.10.046. [DOI] [PubMed] [Google Scholar]
- 7.Kumar R, Seibold MA, Aldrich MC, Williams LK, Reiner AP, Colangelo L, et al. Genetic ancestry in lung-function predictions. N Engl J Med. 2010;363:321–330. doi: 10.1056/NEJMoa0907897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pino-Yanes M, Thakur N, Gignoux CR, Galanter JM, Roth LA, Eng C, et al. Genetic ancestry influences asthma susceptibility and lung function among Latinos. J Allergy Clin Immunol. 2015;135:228–235. doi: 10.1016/j.jaci.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fejerman L, Chen GK, Eng C, Huntsman S, Hu D, Williams A, et al. Admixture mapping identifies a locus on 6q25 associated with breast cancer risk in US Latinas. Hum Mol Genet. 2012;21:1907–1917. doi: 10.1093/hmg/ddr617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freedman ML, Haiman CA, Patterson N, McDonald GJ, Tandon A, Waliszewska A, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci USA. 2006;103:14068–14073. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galanter JM, Gignoux CR, Torgerson DG, Roth LA, Eng C, Oh SS, et al. Genome-wide association study and admixture mapping identify different asthma-associated loci in Latinos: the genes-environments & admixture in Latino Americans study. J Allergy Clin Immunol. 2014;134:295–305. doi: 10.1016/j.jaci.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gignoux CR, Torgerson DG, Pino-Yanes M, Uricchio LH, Galanter J, Roth LA, et al. An admixture mapping meta-analysis implicates genetic variation at 18q21 with asthma susceptibility in Latinos. J Allergy Clin Immunol. 2019;143:957–969. doi: 10.1016/j.jaci.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torgerson DG, Capurso D, Ampleford EJ, Li X, Moore WC, Gignoux CR, et al. Genome-wide ancestry association testing identifies a common European variant on 6q14.1 as a risk factor for asthma in African American subjects. J Allergy Clin Immunol. 2012;130:622–629, e9. doi: 10.1016/j.jaci.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uribe-Salazar JM, Palmer JR, Haddad SA, Rosenberg L, Ruiz-Narváez EA. Admixture mapping and fine-mapping of type 2 diabetes susceptibility loci in African American women. J Hum Genet. 2018;63:1109–1117. doi: 10.1038/s10038-018-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wassel CL, Pankow JS, Peralta CA, Choudhry S, Seldin MF, Arnett DK. Genetic ancestry is associated with subclinical cardiovascular disease in African-Americans and Hispanics from the multi-ethnic study of atherosclerosis. Circ Cardiovasc Genet. 2009;2:629–636. doi: 10.1161/CIRCGENETICS.109.876243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown SA, Pereira N. Pharmacogenomic impact of CYP2C19 variation on clopidogrel therapy in precision cardiovascular medicine. J Pers Med. 2018;8:8. doi: 10.3390/jpm8010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dummer PD, Limou S, Rosenberg AZ, Heymann J, Nelson G, Winkler CA, et al. APOL1 kidney disease risk variants: an evolving landscape. Semin Nephrol. 2015;35:222–236. doi: 10.1016/j.semnephrol.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mak ACY, White MJ, Eckalbar WL, Szpiech ZA, Oh SS, Pino-Yanes M, et al. NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. Whole-genome sequencing of pharmacogenetic drug response in racially diverse children with asthma. Am J Respir Crit Care Med. 2018;197:1552–1564. doi: 10.1164/rccm.201712-2529OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno-Estrada A, Gignoux CR, Fernández-López JC, Zakharia F, Sikora M, Contreras AV, et al. Human genetics: the genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science. 2014;344:1280–1285. doi: 10.1126/science.1251688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brehm JM, Man Tse S, Croteau-Chonka DC, Forno E, Litonjua AA, Raby BA, et al. A genome-wide association study of post-bronchodilator lung function in children with asthma. Am J Respir Crit Care Med. 2015;192:634–637. doi: 10.1164/rccm.201501-0047LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choudhry S, Taub M, Mei R, Rodriguez-Santana J, Rodriguez-Cintron W, Shriver MD, et al. Genome-wide screen for asthma in Puerto Ricans: evidence for association with 5q23 region. Hum Genet. 2008;123:455–468. doi: 10.1007/s00439-008-0495-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forno E, Wang T, Yan Q, Brehm J, Acosta-Perez E, Colon-Semidey A, et al. A multiomics approach to identify genes associated with childhood asthma risk and morbidity. Am J Respir Cell Mol Biol. 2017;57:439–447. doi: 10.1165/rcmb.2017-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez-Pacheco N, Farzan N, Francis B, Karimi L, Repnik K, Vijverberg SJ, et al. Genome-wide association study of inhaled corticosteroid response in admixed children with asthma. Clin Exp Allergy. 2019;49:789–798. doi: 10.1111/cea.13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan Q, Brehm J, Pino-Yanes M, Forno E, Lin J, Oh SS, et al. A meta-analysis of genome-wide association studies of asthma in Puerto Ricans. Eur Respir J. 2017;49:1601505. doi: 10.1183/13993003.01505-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mani A. Local ancestry association, admixture mapping, and ongoing challenges. Circ Cardiovasc Genet. 2017;10:e001747. doi: 10.1161/CIRCGENETICS.117.001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shriner D. Overview of admixture mapping. Curr Protoc Hum Genet. 2017;94:1.23.1–1.23.8. doi: 10.1002/cphg.44. [DOI] [PubMed] [Google Scholar]
- 27.Shriner D. Curr Protoc Hum Genet. 2013. Overview of admixture mapping. Chapter 1:Unit 1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. NHLBI GO Exome Sequencing Project—ESP Lung Project Team. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.RStudio Team. RStudio: integrated development for R. Boston, MA: RStudio, Inc.; 2015. [Google Scholar]
- 31.Ray A, Cohn L. Th2 cells and GATA-3 in asthma: new insights into the regulation of airway inflammation. J Clin Invest. 1999;104:985–993. doi: 10.1172/JCI8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Open Target Genetics. UKB Neale v2. 2018 [accessed 2020 May 21]. Available from: https://genetics.opentargets.org/variant/1_199617282_G_A.
- 33.Genetics Home Reference. CACNA1S gene. 2020 [accessed 2020 May 21]. Available from: https://ghr.nlm.nih.gov/gene/CACNA1S.
- 34.Open Targets Genetics. UKB Neale v2. 2018 [accessed 2020 May 21]. Available from: https://genetics.opentargets.org/variant/1_201047168_G_A.
- 35.Ferreira MAR, Mathur R, Vonk JM, Szwajda A, Brumpton B, Granell R, et al. 23andMe Research Team; eQTLGen Consortium; BIOS Consortium. Genetic architectures of childhood- and adult-onset asthma are partly distinct. Am J Hum Genet. 2019;104:665–684. doi: 10.1016/j.ajhg.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, Melbourne CA, et al. Understanding Society Scientific Group. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet. 2019;51:481–493. doi: 10.1038/s41588-018-0321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyss AB, Sofer T, Lee MK, Terzikhan N, Nguyen JN, Lahousse L, et al. Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat Commun. 2018;9:2976. doi: 10.1038/s41467-018-05369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obeidat M, Nie Y, Fishbane N, Li X, Bossé Y, Joubert P, et al. Integrative genomics of emphysema-associated genes reveals potential disease biomarkers. Am J Respir Cell Mol Biol. 2017;57:411–418. doi: 10.1165/rcmb.2016-0284OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jevnikar Z, Östling J, Ax E, Calvén J, Thörn K, Israelsson E, et al. Unbiased Biomarkers in Prediction of Respiratory Disease Outcomes study group. Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J Allergy Clin Immunol. 2019;143:577–590. doi: 10.1016/j.jaci.2018.05.026. [DOI] [PubMed] [Google Scholar]
- 40.Wei W, Jiang F, Liu XC, Su Q. TMEM9 mediates IL-6 and IL-1β secretion and is modulated by the Wnt pathway. Int Immunopharmacol. 2018;63:253–260. doi: 10.1016/j.intimp.2018.07.036. [DOI] [PubMed] [Google Scholar]
- 41.Corvol H, De Giacomo A, Eng C, Seibold M, Ziv E, Chapela R, et al. Genetics of Asthma in Latino Americans (GALA) Study; Study of African-Americans, Asthma, Genes and Environments (SAGE) Investigators. Genetic ancestry modifies pharmacogenetic gene-gene interaction for asthma. Pharmacogenet Genomics. 2009;19:489–496. doi: 10.1097/FPC.0b013e32832c440e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C, Guo H, Li B, Sui C, Zhang Y, Xia X, et al. Effects of Slit3 silencing on the invasive ability of lung carcinoma A549 cells. Oncol Rep. 2015;34:952–960. doi: 10.3892/or.2015.4031. [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Zhang L, Wang D, Shen H, Jiang M, Mei P, et al. Congenital diaphragmatic hernia, kidney agenesis and cardiac defects associated with Slit3-deficiency in mice. Mech Dev. 2003;120:1059–1070. doi: 10.1016/s0925-4773(03)00161-8. [DOI] [PubMed] [Google Scholar]
- 44.Yuan W, Rao Y, Babiuk RP, Greer JJ, Wu JY, Ornitz DM. A genetic model for a central (septum transversum) congenital diaphragmatic hernia in mice lacking Slit3. Proc Natl Acad Sci USA. 2003;100:5217–5222. doi: 10.1073/pnas.0730709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genetics Home Reference. DOCK2 gene. 2020 [accessed 2020 May 21]. Available from: https://ghr.nlm.nih.gov/gene/DOCK2.
- 46.Catalog GWAS. Variant: rs73803919. 2015 [accessed 2020 May 21]. Available from: https://www.ebi.ac.uk/gwas/variants/rs73803919.
- 47.Calışkan M, Bochkov YA, Kreiner-Møller E, Bønnelykke K, Stein MM, Du G, et al. Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. N Engl J Med. 2013;368:1398–1407. doi: 10.1056/NEJMoa1211592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 49.Poole A, Urbanek C, Eng C, Schagema J, Jacobson S, O’Connor BP, et al. Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol. 2014;133:670–678, e12. doi: 10.1016/j.jaci.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.