

Abstract
Telomere dysfunction is associated with multiple fibrotic lung processes, including chronic lung allograft dysfunction (CLAD)—the major limitation to long-term survival following lung transplantation. Although shorter donor telomere lengths are associated with an increased risk of CLAD, it is unknown whether short telomeres are a cause or consequence of CLAD pathology. Our objective was to test whether telomere dysfunction contributes to the pathologic changes observed in CLAD. Histopathologic and molecular analysis of human CLAD lungs demonstrated shortened telomeres in lung epithelial cells quantified by teloFISH, increased numbers of surfactant protein C immunoreactive type II alveolar epithelial cells, and increased expression of senescence markers (β-galactosidase, p16, p53, and p21) in lung epithelial cells. TRF1F/F (telomere repeat binding factor 1 flox/flox) mice were crossed with tamoxifen-inducible SCGB1a1-cre mice to generate SCGB1a1-creTRF1F/F mice. Following 9 months of tamoxifen-induced deletion of TRF1 in club cells, mice developed mixed obstructive and restrictive lung physiology, small airway obliteration on microcomputed tomography, a fourfold decrease in telomere length in airway epithelial cells, collagen deposition around bronchioles and adjacent lung parenchyma, increased type II aveolar epithelial cell numbers, expression of senescence-associated β-galactosidase in epithelial cells, and decreased SCGB1a1 expression in airway epithelial cells. These findings demonstrate that telomere dysfunction isolated to airway epithelial cells leads to airway-centric lung remodeling and fibrosis similar to that observed in patients with CLAD and suggest that lung epithelial cell telomere dysfunction may be a molecular driver of CLAD.
Keywords: airway fibrosis, telomere, club cells, DNA damage, senescence
Clinical Relevance
Fibrosis associated with lung transplantation is attributed primarily to alloimmunity. We show in this article that the genetic deletion of telomere shelterin protein TRF1 in club cells caused pathophysiology similar to that found in patients with chronic lung allograft dysfunction (CLAD). Currently, no convincing model exists for studying the molecular mechanism of telomere dysfunction relevant to CLAD. We provide data suggesting that changes associated with CLAD pathology are due to short telomeres in the airway epithelium. Our novel mouse model will be an advancement in the field for exploring the relationship between telomere dysfunction and CLAD.
Lung transplantation is a lifesaving therapy for patients with end-stage lung disease. Chronic lung allograft dysfunction (CLAD) is a common complication that limits long-term survival after lung transplantation (1), with 50% of patients succumbing to CLAD within 5 years (2). CLAD is a syndrome clinically defined primarily by a decline in lung function. CLAD is subdivided into obstructive and restrictive subtypes, known as bronchiolitis obliterans syndrome and restrictive allograft syndrome, respectively (3). Although these subtypes may overlap, they have characteristic pathologies and imaging findings. Bronchiolitis obliterans syndrome is characterized pathologically by constrictive or obliterative bronchiolitis, whereas restrictive allograft syndrome is characterized by a variable combination of constrictive bronchiolitis, pulmonary fibrosis, and pleuroparenchymal fibroelastosis (4, 5).
Although immune responses are believed to play a role in CLAD pathogenesis, augmentation of immunosuppression is generally not effective in reversing CLAD progression (6). Thus, understanding the alloimmune-independent mechanisms underlying the pathologies of CLAD may be useful for understanding this syndrome. The airway epithelium consists of club cells, ciliated cells, basal cells, and neuroendocrine cells. Of these, club cells show evidence of damage in patients with CLAD (7). In addition to secreting protective factors, club cells replicate and differentiate to replenish airway epithelial cells and, thus, may be under stress in the context of lung transplantation. As a potential marker of replicative stress (8), short telomeres in lung airway epithelial cells have been associated with the development of CLAD (9). Furthermore, explanted lung tissue from patients with CLAD shows evidence of telomere shortening (10).
Telomere dysfunction manifests clinically as fibroproliferative lung pathologies, of which usual interstitial pneumonia is the most common, but other patterns of fibrosis are also observed (11). Conditional deletion of telomere shelterin proteins in specific cell types has been shown to model pathologies of human diseases mediated by telomere dysfunction (12–15). For example, modeling telomere dysfunction in surfactant protein C–expressing cells results in lung fibrosis. To test the hypothesis that telomere shortening in airway epithelial cells is sufficient to induce CLAD pathology, we developed a mouse model in which the shelterin protein family member TRF1 (telomere repeat binding factor 1) was selectively deleted in club cells.
Some of the results of these studies have been previously reported in the form of a preprint (bioRxiv, 28 August 2019 https://www.biorxiv.org/content/10.1101/746768v1).
Methods
Human Specimens
Fibrotic lung tissue was obtained at the time of lung transplantation from patients with a pathological diagnosis of CLAD. Control human lungs consisted of lungs not used by the Northern California Transplant Donor Network; our studies indicate that these lungs are physiologically and pathologically normal (16). Endobronchial and transbronchial biopsies were performed within 90 days post transplant, and sections were recut from formalin-fixed, paraffin-embedded tissue blocks, as previously described (9). Written informed consent was obtained from all subjects and was approved by the University of California, San Francisco Institutional Review Board (protocol number 13–10738).
Mice
TRF1F/F mice were purchased from The Jackson Laboratory, SCGB1a1-cre/ERT mice were provided by Jason Rock, at University of California, San Francisco. Mice were bred and housed in pathogen-free conditions in accordance with the guidelines of the Laboratory Animal Resource Center. All animal procedures were performed using protocols approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco.
Tamoxifen Administration
Tamoxifen (T0600; Toronto Research Chemicals) suspended in peanut oil was injected (250 mg/kg body weight once per week) intraperitoneally to SCGB-creTRF1F/F mice and TRF1F/F littermate controls beginning at 10 weeks of age.
Hydroxyproline Assay
Hydroxyproline levels were quantified as described previously (17).
Telomere Quantitative Fluorescence In Situ Hybridization Assay
Telomere lengths were measured on paraffin-embedded tissue sections by quantitative fluorescence in situ hybridization (Q-FISH) as described (15).
Senescence Associated-β-Galactosidase Assay
To test for senescence-associated-β galactosidase (SA-β-gal) (18), staining on tissues following protocol was used as described (19).
Lung Mechanics
Lung mechanics were measured as described by the manufacturer (flexiVent; SCIREQ Scientific Respiratory Equipment Inc.). To measure pulmonary physiology, mice were anesthetized with intraperitoneal ketamine/xylazine, and an adequate depth of anesthesia was assured by observing pedal withdrawal reflex. A tracheostomy was performed on the mouse in deep-state anesthesia. A tubing adaptor (20 gauge) was used to cannulate the trachea. The mouse was then attached to a rodent ventilator and pulmonary mechanics analyzer and ventilated at a tidal volume of 9 ml/kg, a frequency of 150 breaths/min, and a positive end-expiratory pressure of 3 cm H2O. The mouse was paralyzed with pancuronium bromide (0.1 mg/kg intraperitoneally) to facilitate static compliance measurement before tracheal cannulation and pancuronium administration. Lung mechanics were measured using forced oscillation maneuver algorithms using flexiVent software.
Statistics
Statistical analyses were performed with GraphPad Prism software (version 7.0).
P values were calculated using a two-tailed Student’s t test. Welch’s correction was applied for the analysis of unpaired groups. Differences in survival were analyzed by log-rank (Mantel-Cox) test. Data are represented as the mean ± SEM. P values represent the significance using the following symbols: ns = not significant, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Histopathology of CLAD Lungs
Masson’s trichrome staining on sections of explanted lung from human lung transplant recipients with CLAD revealed increased collagen deposition around the bronchioles (Figure 1A), parenchymal fibrosis (as quantified by the Ashcroft scoring method) (20) (Figure 1D), and concentric narrowing of airway lumens compared with normal lungs. Patchy bronchiolar obliteration was also observed in CLAD lungs (Figure 1A). Moreover, immunofluorescence staining identified α-smooth muscle actin (SMA)–expressing mesenchymal cells around the bronchioles in CLAD lungs, unlike normal lungs (Figure 1B). Immunostaining for surfactant protein C (SPC) revealed type II alveolar epithelial cell (AEC) hyperplasia (Figure 1C), with up to fivefold more type II AECs (Figure 1E) adjacent to the bronchioles.
Figure 1.
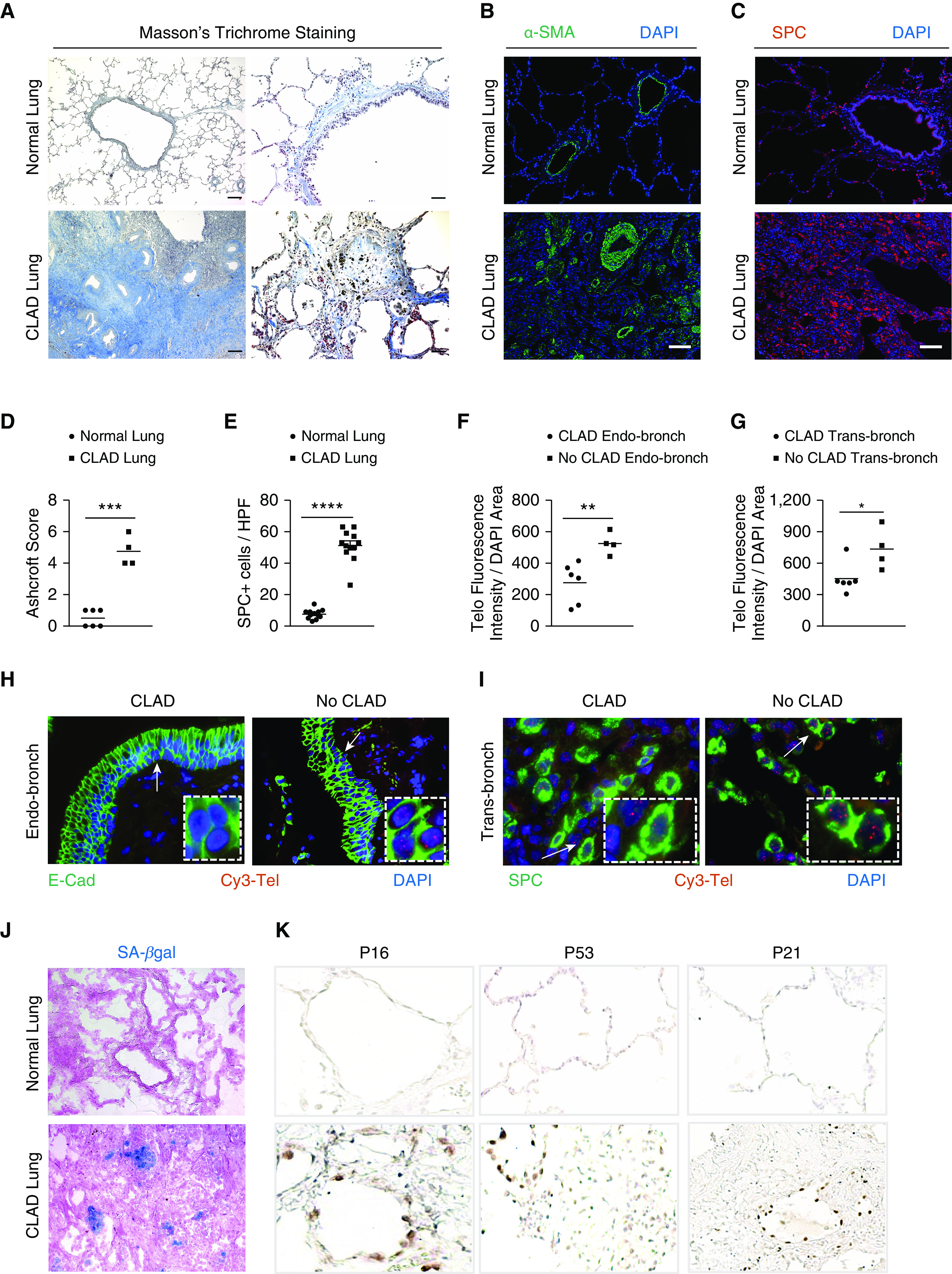
Chronic lung allograft dysfunction (CLAD) pathology in lung transplant recipients. (A) Masson’s trichrome staining in CLAD lungs and normal lungs. Collagen deposition (blue) is seen around the airways and in bronchiole, causing obliteration in CLAD lungs. Scale bars, 400 μm (left panels) and 100 μm (right panels). (B and C) Immunofluorescence staining on paraffin-embedded lung sections from normal and CLAD lungs with α-SMA antibody (B) and SPC antibody (C). Nuclei were stained with DAPI. N = 3. Scale bars, 100 μm. (D) Quantification of fibrosis using Ashcroft score. ***P < 0.001 (t test). (E) Quantification of the number of SPC immunoreactive cells per high-power field. Ten fields were used per group. ****P < 0.0001 (t test). (F and G) Quantification of telomere length by calculating telomere fluorescence intensity over the DAPI area in endobronchial and transbronchial biopsies diagnosed as CLAD and no CLAD. **P < 0.01 and *P < 0.05 (t test). (H and I) Representative images of telomere quantitative fluorescence in situ hybridization assay conducted on endobronchial and transbronchial biopsies diagnosed with CLAD and no CLAD. White arrows point to area depicted in the box. (J) Senescence-associated–β galactosidase staining (blue colored cells) on optimal cutting temperature–embedded cryosections counterstained with eosin. N = 6. (K) Immunohistochemistry on paraffin-embedded lung sections from normal and CLAD lungs with P16, P53, and P21 antibodies. N = 5. E-cad = E-cadherin; HPF = high-power field; SA-β-gal = senescence-associated–β galactosidase; SMA = smooth muscle actin; SPC = surfactant protein C.
Telomere Length and Markers of Senescence in CLAD Lungs
It has been shown that telomeres are short in the proximal airway epithelium of patients at risk for developing CLAD (9, 10). To determine whether telomeres are shortened in more than one epithelial cell subtype, endobronchial and transbronchial biopsies were used to measure telomere length by telomere Q-FISH assay. Telomere lengths of patients with CLAD were significantly shorter in the epithelial cells of endobronchial biopsies (Figure 1F), identified by immunostaining with E-cadherin (Figure 1H). In transbronchial biopsies, the quantification of telomere length in SPC immunoreactive cells also revealed significantly shorter telomeres in CLAD lungs compared with lungs with no CLAD (Figures 1G and 1I).
Telomere dysfunction mediates cellular senescence reprogramming (21, 22), which is implicated as a driver of lung remodeling and fibrosis (15, 23). To examine whether senescence programming is activated in CLAD lungs, they were stained with X-gal chromogen to identify SA-β-gal activity. Foci of SA-β-gal staining were found in CLAD lungs (Figure 1J), indicating the presence of senescent cells in the epithelium lining the airway that appeared to be obliterated. CLAD lungs were also immunostained for the senescence markers p16, p53, and p21 (Figure 1J), revealing immunoreactive epithelial cells in regions of lung fibrosis.
Telomere Uncapping in Club Cells Leads to Short Telomeres and Increases Mortality
Analysis of human samples demonstrated that telomere dysfunction and senescence programming are found in regions of lung remodeling and fibrosis observed in CLAD lungs. To test whether telomere dysfunction may mediate the remodeling, TRF1F/F mice were crossed with mice that inducibly express Cre recombinase in club cells upon treatment with tamoxifen (SCGB1a1-Cre ERT2 mice) to generate SCGB1a1-creTRF1F/F mice. These mice were treated with tamoxifen weekly for up to 9 months (Figure 2A). SCGB1a1-creTRF1F/F mice died between 7 and 9 months after beginning tamoxifen injections, in contrast with TRF1F/F mice, all of which survived (Figure 2B). Telomere length was found to be ∼fourfold shorter in club cells of SCGB1a1-creTRF1F/F mice compared with those of age-matched TRF1F/F controls (Figures 2C and 2D) when quantified by Q-FISH.
Figure 2.

The deletion of TRF1 (telomere repeat binding factor 1) in club cells leads to short telomeres and increased mortality. (A) Tamoxifen-inducible cre-mediated recombination approach to delete TRF1 in cre-expressed cells. Tamoxifen was administered intraperitoneally to mice weekly starting from 10 weeks of age at 250 mg/kg body weight. (B) Kaplan-Meier survival graph of TRF1F/F (telomere repeat binding factor 1 flox/flox) and SCGB1a1-creTRF1F/F mice treated with weekly injections of tamoxifen at 250 mg/kg body weight. ****P < 0.0001 (log-rank test). (C) Telomere quantitative fluorescence in situ hybridization assay conducted on paraffin-embedded lung sections collected after 8–9 months of tamoxifen administration. Cy3-Tel is a PNA probe to detect TTAGGG repeats. SCGB1a1 was used as an antibody to detect club cells. DAPI was used to detect nuclei. The boxed area of the upper panels is shown enlarged in the bottom panels. White arrows point to cells depicted in the boxed area. Scale bars, 20 μm. (D) Quantification of telomere length using telomere fluorescence intensity with reference to DAPI area. N = 3 mice/group. Each dot represents telomere fluorescence intensity in each club cell acquired from twenty different high-power fields. Data are pooled from three mice. ****P < 0.0001 (t test). E = endpoint; PNA = peptide nucleic acid.
Telomere Dysfunction in Club Cells Leads to Airway-Centric Lung Remodeling and Fibrosis
To evaluate the lung pathology of SCGB1a1-creTRF1F/F mice, lungs were stained with Masson’s trichrome stain. Increased collagen deposition was found predominantly around the small airways and extended into the adjacent lung parenchyma of SCGB1a1-creTRF1F/F lungs (Figures 3A and E1A in the data supplement). The obliteration of small airways (Figures E1B and E1C) and narrowing of the larger airway lumens were also observed (Figures 3A and E1A). Ashcroft scores for fibrosis were consistently elevated in SCGB1a1-creTRF1F/F mice relative to controls (Figure 3B). The quantification of collagen content in lungs by hydroxyproline assay revealed threefold more collagen in SCGB1a1-creTRF1F/F lungs (Figure 3C), and quantitative PCR of mouse lung mRNA demonstrated increased collagen 1 expression in SCGB1a1-creTRF1F/F lungs (Figure 3D).
Figure 3.

Airway-centric lung remodeling and fibrosis. (A) Masson’s trichrome staining was performed on lung sections from TRF1F/F and SCGB1a1-creTRF1F/F mouse lungs treated with weekly doses of tamoxifen at 250 mg/kg body weight. Deposition of collagen is indicated by blue stain. Arrows indicate areas of collagen deposition around bronchioles. Scale bars, 400 μm (left panels) and 100 μm (right panels). (B) Quantification of fibrosis using Ashcroft score. ****P < 0.0001 (t test). (C) Quantification of collagen deposition by hydroxyproline assay on lung tissues from TRF1F/F and SCGB1a1-creTRF1F/F mice. ***P < 0.001 (t test). (D) Quantitative PCR of collagen1 mRNA in lungs from TRF1F/F and SCGB1a1-creTRF1F/F mice 9 months after tamoxifen administration. **P < 0.01 (t test).
To assess overall lung morphometry, microcomputed tomography was performed on lungs extracted from SCGB1a1-creTRF1F/F and TRF1F/F control mice 9 months after tamoxifen administration. Coronal sections demonstrated increased density of tissue in the central regions of SCGB1a1-creTRF1F/F lungs, consistent with bronchiolar fibrosis (Figure 4A). Cross-sectional images show decreased airway frequency and diameter in SCGB1a1-creTRF1F/F lungs compared with control lungs (Figure 4A).
Figure 4.

Micro–computed tomography imaging and lung mechanics. (A) Lungs harvested from TRF1F/F and SCGB1a1-creTRF1F/F mice after treatment for 9 months with weekly injections of tamoxifen (250 mg/kg body weight) were subjected to micro–computed tomography imaging. Left lungs were used for imaging. Top: coronal section passing through the middle of the lung visualized in three dimensions. Air spaces are represented in black. Gray represents tissue density. Bottom: cross-sectional images showing air spaces, airways, and tissue density. Airways are marked with red border. Scale bars, 1 mm. (B–G) Lung mechanics measured by flexiVent. TRF1F/F and SCGB1a1-creTRF1F/F mice after treatment for 9 months with weekly injections of tamoxifen were subjected to measurement of total resistance (B), compliance (C), elastance (D), airway resistance (E), tissue damping (F), and pressure–volume loops (G). N = 4 mice/group. **P < 0.01 (t test). Crs = compliance; Ers = elastance; G = tissue damping; ns = not significant; Rn = airway resistance; Rrs = total resistance.
Lung Function of SCGB1a1-creTRF1F/F Mice
Because CLAD is defined in part by obstructive airway physiology, we evaluated the impact of club-cell telomere dysfunction on the lung function of SCGB1a1-creTRF1F/F mice compared with TRF1F/F control mice using the flexiVent instrument. The resistance of the overall respiratory system (Figure 4B) was higher in SCGB1a1-creTRF1F/F lungs than in control lungs. Lung compliance (Figure 4C) was significantly lower, with an analogous increase in elastance (Figure 4D) of the respiratory system, in SCGB1a1-creTRF1F/F lungs. To differentiate lung mechanics of central and peripheral lung compartments, broadband forced oscillations (constant-phase model) were used. In this model, newtonian resistance and tissue damping were increased in SCGB1a1-creTRF1F/F lungs relative to control lungs (Figures 4E and 4F). We did not observe a statistically significant difference in newtonian resistance, a parameter indicative of large conducting airway resistance (Figure 4E). Overall, this pattern of abnormality suggests increased resistance of small airways in the peripheral compartment and increased alveolar tissue stiffness. Quasistatic lung mechanics measured using pressure–volume curves (Figure 4G) showed that the static compliance, a parameter that assesses lung distensibility, was significantly reduced in SCGB1a1-creTRF1F/F lungs compared with TRF1F/F control lungs. In comparison, lung mechanics of SPC-cre TRF1F/F mice show similar trend as SCGB1a1-creTRF1F/F lungs, with the exception that tissue damping is not significantly different compared with TRF1F/F control lungs, confirming that distal airways are not remodeled and that the location of fibrosis is spatially different in SPC-cre TRF1F/F mice (Figure E2).
Immune Response Profile in SCGB1a1-Cre TRF1F/F Mice
BAL fluid collected from SCGB1a1-cre TRF1F/F mice showed elevated concentrations of the profibrotic marker TGF-β1 (Figure 5A), senescence-associated cytokine IL-6 (Figure 5B), and chemokine CCL2 (Figure 5C) compared with TRF1F/F mice. Protein levels of the club cell marker CC10 were significantly lower compared with those of TRF1F/F mice (Figure E5). Analysis of BAL fluid revealed greater than a sixfold increase in total cell count in SCGB1a1-cre TRF1F/F mice compared with TRF1F/F mice (Figure 5D). Differential cell count of BAL fluid indicated a significant increase in polymorphonuclear granulocytes (Figure 5E), macrophages (Figure 5F), and lymphocytes (Figure 5G). Immunophenotyping of whole lung homogenates revealed increased CD45+ leukocytes, including T cells, CD25+ cells, and natural killer (NK) cells (Figure 5H and Table E1) in SCGB1a1-cre TRF1F/F mice compared with TRF1F/F mice. Immunostaining of lung sections showed a modest increase in B220 immunoreactive cells, consistent with B-cell recruitment to SCGB1a1-cre TRF1F/F lungs, compared with TRF1F/F controls (Figure E3).
Figure 5.
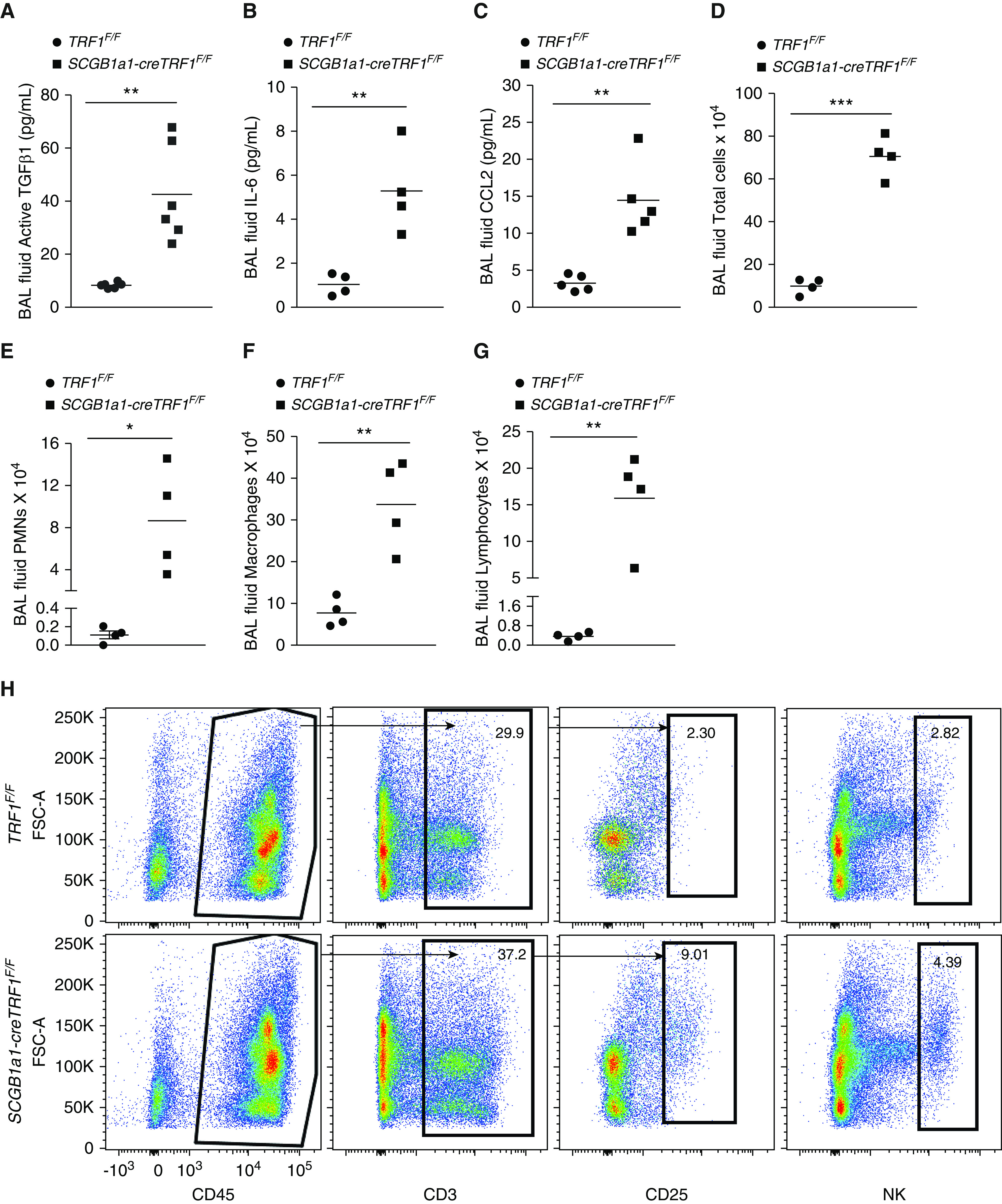
Mouse BAL fluid ELISA, cell count differential analysis, and lung immune cell analysis. (A–C) ELISA on BAL samples from TRF1F/F and SCGB1a1-creTRF1F/F mice to test for active TGF-B1 (A), IL-6 (B), and CCL2 (C). (D–G) Cell count and differentials of BAL fluid samples from lungs of TRF1F/F and SCGB1a1-creTRF1F/F mouse lungs treated with weekly doses of tamoxifen for 9 months; total cell count (D), PMNs (E), macrophages (F), and lymphocytes (G). N = 4 mice/group. *P < 0.05, **P < 0.01, and ***P < 0.001 (t test). (H) Immunophenotyping of lungs from TRF1F/F and SCGB1a1-creTRF1F/F at 9 months by flow cytometry. Percentage of CD3, CD25, and NK cell populations were derived from CD45-positive gate. Black arrows indicate the gating directionality. N = 3. Individual population numbers are mentioned in Table E1. FSC = forward scatter; NK cell = natural killer cell; PMNs = polymorphonuclear cells.
TRF1 Deletion in Club Cells Leads to DNA Damage and Activation of Cellular Senescence Programming
To determine the consequence of telomere dysfunction within club cells, immunostaining was performed for the DNA damage marker γ-H2AX. DNA damage foci localized to club cells (Figure 6A) of SCGB1a1-cre TRF1F/F lungs. Moreover, SA-β-gal staining to identify senescent cells showed more blue-stained cells in the airway epithelium in SCGB1a1-cre TRF1F/F lungs compared with TRF1F/F controls, consistent with activation of senescence programming (Figure 6B).
Figure 6.

Immunostaining of lung sections and detection of cellular senescence by SA-β-gal activity. (A) Immunostaining on sections of lungs harvested from TRF1F/F and SCGB1a1-creTRF1F/F mice treated with weekly tamoxifen injections for 9 months. SCGB1a1 immunostaining for club cells (red) and γH2AX (green). γH2AX foci (green) in the nuclei of club cells. Nuclei were stained with DAPI (blue). Scale bars, 20 μm. Boxed area is enlarged in bottom panel along with inset showing individual club cell. (B) SA-β-gal staining of sections of lungs harvested from TRF1F/F and SCGB1a1-creTRF1F/F mice after treatment for 9 months with weekly injections of tamoxifen (250 mg/kg body weight). Note the blue SA-β-gal+ cells (arrows) in lungs of SCGB1a1-creTRF1F/F mice.
Club Cell Telomere Dysfunction Leads to Heterogeneous Loss of Club Cells and Proliferation of SPC Immunoreactive Cells
Immunofluorescent costaining for SCGB1a1, SPC, and the cell proliferation marker Ki-67 showed fewer SCGB1a1 immunoreactive cells and increased expression of SPC and Ki67 in SCGB1a1-cre TRF1F/F lungs compared with TRF1F/F control lungs (Figure 7A). The quantification of cell numbers revealed a fivefold increase in total Ki67+ cells (Figure 7B), a twofold increase in SPC+ cells (Figure 7C), and an eightfold increase in dual SPC+ Ki67+ cells (Figure 7D) in SCGB1a1-cre TRF1F/F lungs compared with TRF1F/F control lungs. Within SPC cells, there was a fourfold increase in Ki67 positivity in SCGB1a1-cre TRF1F/F lungs compared with control lungs (Figure 7E). These results demonstrate a loss of SCGB1a1 immunoreactivity, hyperplasia of SPC immunoreactive cells, and increased proliferation of SPC immunoreactive cells. Interestingly, occasional SPC immunoreactive cells that also expressed the club cell marker SCGB1a1 were more localized in the airways in SCGB1a1-cre TRF1F/F lungs compared with controls (Figure E4). Moreover, quantitative PCR of lung lysates from SCGB1a1-cre TRF1F/F mice showed more than a fivefold increase in SMA mRNA expression amounts, an ∼1.5-fold increase in SPC, and a nearly threefold decrease in SCGB1a1 expression compared with those of TRF1F/F mice (Figure 7F). There was heterogeneous loss of SCGB1a1 immunoreactivity in the airway epithelium of SCGB1a1-cre TRF1F/F mice displaying select airways with near complete to partial loss of club cells and few airways with no loss (Figure E6). To address the heterogeneity, we quantified club-cell numbers, referencing the perimeter of basement membrane. Quantification of multiple random images from five different mice per group revealed ∼10-fold fewer club cells in SCGB1a1-cre TRF1F/F lungs than control lungs (Figure 7G). Immunofluorescent staining demonstrated increased SMA expression around airways (Figure 7H).
Figure 7.

Loss of club cells and type II alveolar epithelial cell hyperplasia around bronchioles. (A) Immunostaining on lung sections from TRF1F/F and SCGB1a1-creTRF1F/F mice treated with weekly doses of tamoxifen. Cell proliferation marker Ki67 (red), SCGB1a1 (green) for club cells, and SPC (far red changed to white for visibility). Inset: Ki67 marker in an SPC+ cell. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. (B–D) Quantification of total Ki67-positive cells (B), total SPC+ cells (C), and SPC and Ki67 double-positive cells (D). (E) Percentage Ki67+ cells over total SPC-positive cells. ***P < 0.001 (t test). (F) mRNA expression levels in SCGB1a1-creTRF1F/F lungs represented as log2 fold change over TRF1F/F control animals. (G) Quantification of club cells with reference to perimeter of basement membrane, based on multiple random images from a total of five mice per group. N = 5. ****P < 0.0001 (t test). (H) Immunofluorescence staining on paraffin-embedded lung sections costained with α-SMA and SCGB1a1. Scale bar, 50 μm.
Discussion
CLAD lungs have relatively short telomeres and express senescence markers in airway epithelial cells. Modeling this telomere dysfunction in club cells resulted in physiologic and pathologic changes that are similar to those found in patients with CLAD (Table E2). These changes included airway obstruction, with airway-centric fibrosis and obliteration of bronchioles; parenchymal fibrosis; expansion of SPC immunoreactive cells adjacent to the obliterated bronchioles; and regional lung fibrosis (24). Consistent with obliterated airways, the lung physiology of SCGB1a1-cre TRF1F/F lungs demonstrated increased tissue damping, reflecting increased peripheral airway resistance (25). Elements of CLAD-like pathology were recapitulated by modeling a single molecular defect in the absence of alloimmunity. These data suggest that the airway epithelial cell telomere dysfunction is a molecular driver of CLAD pathology.
Short telomeres are evident in diseases of tissue remodeling and fibrosis, including skin dyskeratosis (26), hypertrophic cardiomyopathy (27–29), liver cirrhosis (30), Duchenne muscular dystrophy (31), idiopathic pulmonary fibrosis (32), and CLAD (9, 10). Telomere dysfunction secondary to the deletion of shelterin protein TRF1 in select cell subtypes has modeled organ-specific pathologic changes attributed to human syndromes of telomere dysfunction. For example, the deletion of TRF1 in keratinocytes or hematopoietic cells caused hyperpigmentation and bone marrow failure, respectively (12, 13). In the lungs, the deletion of TRF1 in type II AECs leads to lung fibrosis in a subpleural distribution, with features similar to idiopathic pulmonary fibrosis (15). Distinct from the mouse model described here, lungs of animals with type II AEC telomere dysfunction have normal airway resistance values. Overall, these reports suggest that telomere dysfunction may be a molecular driver of many forms of tissue remodeling, with the specific pathologic appearance of the remodeling depending on the tissue and cellular subtype within the tissue where the telomere dysfunction is found. The findings reported in this article suggest that telomere dysfunction in airway epithelial cells may be a molecular driver of elements of lung remodeling that occurs following lung transplantation (7, 33–35).
The long-term deletion of TRF1 in vivo disrupts the shelterin complex, leading to telomere attrition (13). Similarly, the telomeres are short in airway epithelial cells of SCGB1a1-cre TRF1F/F transgenic mice. Why the telomeres are shorter in SCGB1a1-cre TRF1F/F transgenic mice is uncertain. One possibility is that telomere attrition is mediated by TZAP (telomeric-inc finger-Associated Protein), which preferentially binds to long telomeres with low concentration of shelterin complex, triggering a process known as “telomere trimming” and resulting in the rapid deletion of telomeric repeats (36). A second possibility is that increased cell turnover of epithelial cells causes telomere attrition. Future studies are necessary to differentiate these possibilities.
Telomere shortening and uncapping activates DNA damage machinery, cell-cycle checkpoints, and senescence reprogramming (21, 22). Consistent with these reports, we found γH2AX-positive DNA damage foci and positive staining for cellular senescence markers in tamoxifen-treated SCGB1a1-cre TRF1F/F transgenic mice, which could amplify local inflammatory responses (37). The accumulation of senescent cells correlated temporally with the development of fibrosis, suggesting their contribution to fibrosis. Short telomeres in club cells causing cellular senescence may lead to the release of senescence-associated secretory proteins such as IL-6 or CCL2 into the local lung mileu, leading to fibrotic remodeling around the airways. Whether the senescence-associated secretory proteins of airway epithelial cells contribute to lung remodeling requires further investigation.
Club cells are primarily located in the trachea and terminal bronchioles, where they function as stem cells by differentiating into other epithelial cell subtypes (e.g., type II AECs) in the context of various forms of lung injury (e.g., naphthalene injury) (38, 39). Similarly, bronchoalveolar stem cells expressing SPC and SCGB1a1 markers have been reported to repopulate injured airway epithelium (40). In our study, the generation of telomere dysfunction in club cells was associated with heterogeneous loss of cells expressing SCGB1a1 in bronchiolar epithelia and increased Ki67 staining in SPC immunoreactive cells adjacent to the airways. This raises the possibility that type II AECs or bronchoalveolar stem cells proliferate in response to the loss of club cells. An alternative explanation is that short telomeres and senescence reprogramming in club cells lead to their dedifferentiation, diminishing their ability to express SCGB1a1, and, hence, they could go undetected when probed. Differentiating these possibilities will require further investigation. Although it is not known to what extent the observed pathology is attributable to club-cell telomere dysfunction as opposed to loss of club cells, the observations of senescence, proliferation of SPC immunoreactive cells, and subsequent leukocyte recruitment all suggest a specific role for telomere dysfunction as the central driver.
Although multiple immune mechanisms may contribute to CLAD, this syndrome is generally linked to alloimmunity (41). Interestingly, the increased numbers of parenchymal and BAL T cells, B cells, CD25+ cells, and NK cells observed in SCGB1a1-cre TRF1F/F transgenic mice revealed an immune response in the lung consistent with that found in CLAD lungs, however in the absence of alloimmunity. We speculate that senescence reprogramming due to telomere dysfunction in airway epithelial cells leads to the release of an array of cytokines and chemokines that attract immune cells to the lung. Elevated numbers of T cells and NK cells could reflect a direct response to cell stress (42) or be due to T cell–attracting chemokines released by senescent club cells, such as CXCL-9, CXCL-10, and CXCL-11, that are commonly elevated in airway inflammation (43) and CLAD (44, 45). These findings have important implications for immunosuppression strategies in lung transplantation, because targeting alloreactive T cells might not alter the trajectory of a disease driven by club-cell telomere dysfunction.
Similar to all animal models, currently available mouse models of lung transplantation (46) have limitations. Animal models of tracheal transplantation (47–50) reproduce airway luminal narrowing but may not reflect the small airway disease, parenchymal changes, and vascularity of human allografts. Major histocompatibility complex (MHC)-mismatched orthotopic transplantation models reproduce features of acute rejection but not necessarily CLAD (51–54). Transplantation of B6D2F1/J lungs to DBA recipients has shown promise in development of CLAD-like pathology, but the DBA background may be a limitation because of the paucity of available genetic knockout models. Telomere dysfunction is a molecular defect identified in CLAD. Remarkably, the mouse model presented in this manuscript recapitulates changes found in CLAD by the genetic modification of telomere protein. Modeling the elements of CLAD pathology mediated by telomere dysfunction in airway epithelial cells has the advantage that surgery is not required. At the same time, because SCGB1a1-cre TRF1F/F transgenic mice model a single molecular pathway resulting in CLAD-like remodeling, this model will not inform about other important mechanisms relevant to lung transplantation, including ischemia-reperfusion injury, revascularization, alloimmunity, and immunosuppression.
In summary, telomere dysfunction in club cells of SCGB1a1-cre TRF1F/F transgenic mice causes progressive lung remodeling and airway-centric fibrosis similar to the lung remodeling and fibrosis characteristic of CLAD. The findings reported in this manuscript support the hypothesis that telomere dysfunction is a molecular driver of elements of lung remodeling and fibrosis found in patients with CLAD. In the context of lung transplantation, the lung epithelium undergoes high rates of epithelial cell turnover; this may lead to accelerated telomere attrition, senescence reprogramming of airway progenitor cells, and, ultimately, airway remodeling and fibrosis. The findings also suggest that CLAD and other forms of age-related lung remodeling, such as idiopathic pulmonary fibrosis, may share common molecular drivers. Although our study utilized club-cell telomere dysfunction to model CLAD, we do not exclude the possibility that telomere dysfunction or senescence reprogramming in other airway cellular subtypes may contribute to similar lung-remodeling features. The SCGB1a1-cre TRF1F/F spontaneous mouse model of lung remodeling may be useful for studying the contribution of molecular mechanisms of lung remodeling, driven by short telomeres in airway epithelial cells, to the airway-centric fibrosis observed in CLAD.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank all the patients and coordinators who participated in this study. They also thank Alfred Li at the Bone Imaging Research Core, UCSF, for assistance with microCT imaging.
Footnotes
Supported in part by U.S. National Institutes of Health grant HL139897 (P.J.W.), HL151552 (J.R.G.), the Nina Ireland Program for Lung Health, and Veterans Affairs Office of Research and Development grant CDA-2 IK2CX001034 (J.R.G.).
Author Contributions: R.P.N. and P.J.W. designed the study and drafted the manuscript. R.P.N. performed experiments, analyzed and interpreted data, and wrote the manuscript. G.G., N.A.-Z., J.E.M., and I.A. performed experiments and analyzed data. K.D.J., J.K., and J.R.G. contributed to study design, data interpretation, and manuscript revision. All authors reviewed and approved the final manuscript.
Data and material availability: All data related to this study are present in the paper or the data supplement.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0374OC on June 18, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Barker AF, Bergeron A, Rom WN, Hertz MI. Obliterative bronchiolitis. N Engl J Med. 2014;370:1820–1828. doi: 10.1056/NEJMra1204664. [DOI] [PubMed] [Google Scholar]
- 2.Van Herck A, Verleden SE, Sacreas A, Heigl T, Vanaudenaerde BM, Dupont LJ, et al. Validation of a post-transplant chronic lung allograft dysfunction classification system. J Heart Lung Transplant. 2019;38:166–173. doi: 10.1016/j.healun.2018.09.020. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, Waddell TK, Wagnetz U, Roberts HC, Hwang DM, Haroon A, et al. Restrictive allograft syndrome (RAS): a novel form of chronic lung allograft dysfunction. J Heart Lung Transplant. 2011;30:735–742. doi: 10.1016/j.healun.2011.01.712. [DOI] [PubMed] [Google Scholar]
- 4.Verleden GM, Raghu G, Meyer KC, Glanville AR, Corris P. A new classification system for chronic lung allograft dysfunction. J Heart Lung Transplant. 2014;33:127–133. doi: 10.1016/j.healun.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 5.Jonigk D, Rath B, Borchert P, Braubach P, Maegel L, Izykowski N, et al. Comparative analysis of morphological and molecular motifs in bronchiolitis obliterans and alveolar fibroelastosis after lung and stem cell transplantation. J Pathol Clin Res. 2016;3:17–28. doi: 10.1002/cjp2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verleden SE, Vos R, Vanaudenaerde BM, Verleden GM. Chronic lung allograft dysfunction phenotypes and treatment. J Thorac Dis. 2017;9:2650–2659. doi: 10.21037/jtd.2017.07.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly FL, Kennedy VE, Jain R, Sindhwani NS, Finlen Copeland CA, Snyder LD, et al. Epithelial clara cell injury occurs in bronchiolitis obliterans syndrome after human lung transplantation. Am J Transplant. 2012;12:3076–3084. doi: 10.1111/j.1600-6143.2012.04201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campisi J. From cells to organisms: can we learn about aging from cells in culture? Exp Gerontol. 2001;36:607–618. doi: 10.1016/s0531-5565(00)00230-8. [DOI] [PubMed] [Google Scholar]
- 9.Faust HE, Golden JA, Rajalingam R, Wang AS, Green G, Hays SR, et al. Short lung transplant donor telomere length is associated with decreased CLAD-free survival. Thorax. 2017;72:1052–1054. doi: 10.1136/thoraxjnl-2016-209897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everaerts S, Lammertyn EJ, Martens DS, De Sadeleer LJ, Maes K, van Batenburg AA, et al. The aging lung: tissue telomere shortening in health and disease. Respir Res. 2018;19:95. doi: 10.1186/s12931-018-0794-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48:1710–1720. doi: 10.1183/13993003.00308-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martínez P, Thanasoula M, Muñoz P, Liao C, Tejera A, McNees C, et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beier F, Foronda M, Martinez P, Blasco MA. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood. 2012;120:2990–3000. doi: 10.1182/blood-2012-03-418038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci USA. 2015;112:5099–5104. doi: 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1:e86704. doi: 10.1172/jci.insight.86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ware LB, Wang Y, Fang X, Warnock M, Sakuma T, Hall TS, et al. Assessment of lungs rejected for transplantation and implications for donor selection. Lancet. 2002;360:619–620. doi: 10.1016/s0140-6736(02)09774-x. [DOI] [PubMed] [Google Scholar]
- 17.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin αvβ6 binds and activates latent TGF β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 18.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 20.Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 22.Smogorzewska A, de Lange T. Different telomere damage signaling pathways in human and mouse cells. EMBO J. 2002;21:4338–4348. doi: 10.1093/emboj/cdf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Disayabutr S, Kim EK, Cha SI, Green G, Naikawadi RP, Jones KD, et al. miR-34 miRNAs regulate cellular senescence in type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verleden SE, Vos R, Vandermeulen E, Ruttens D, Bellon H, Heigl T, et al. Parametric response mapping of bronchiolitis obliterans syndrome progression after lung transplantation. Am J Transplant. 2016;16:3262–3269. doi: 10.1111/ajt.13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darrah RJ, Mitchell AL, Campanaro CK, Barbato ES, Litman P, Sattar A, et al. Early pulmonary disease manifestations in cystic fibrosis mice. J Cyst Fibros. 2016;15:736–744. doi: 10.1016/j.jcf.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, et al. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science. 2003;299:259–262. doi: 10.1126/science.1079447. [DOI] [PubMed] [Google Scholar]
- 27.Sharifi-Sanjani M, Oyster NM, Tichy ED, Bedi KC, Jr, Harel O, Margulies KB, et al. Cardiomyocyte-specific telomere shortening is a distinct signature of heart failure in humans. J Am Heart Assoc. 2017;6:e005086. doi: 10.1161/JAHA.116.005086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chatterjee S, de Gonzalo-Calvo D, Derda AA, Schimmel K, Sonnenschein K, Bavendiek U, et al. Leukocyte telomere length correlates with hypertrophic cardiomyopathy severity. Sci Rep. 2018;8:11227. doi: 10.1038/s41598-018-29072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang ACY, Chang ACH, Kirillova A, Sasagawa K, Su W, Weber G, et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc Natl Acad Sci USA. 2018;115:9276–9281. doi: 10.1073/pnas.1714538115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laish I, Mannasse-Green B, Hadary R, Biron-Shental T, Konikoff FM, Amiel A, et al. Telomere dysfunction in nonalcoholic fatty liver disease and cryptogenic cirrhosis. Cytogenet Genome Res. 2016;150:93–99. doi: 10.1159/000454654. [DOI] [PubMed] [Google Scholar]
- 31.Tichy ED, Sidibe DK, Tierney MT, Stec MJ, Sharifi-Sanjani M, Hosalkar H, et al. Single stem cell imaging and analysis reveals telomere length differences in diseased human and mouse skeletal muscles. Stem Cell Reports. 2017;9:1328–1341. doi: 10.1016/j.stemcr.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Liao F, Scozzi D, Furuya Y, Pugh KN, Hachem R, et al. An obligatory role for club cells in preventing obliterative bronchiolitis in lung transplants. JCI Insight. 2019;5:e124732. doi: 10.1172/jci.insight.124732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wendt C, Tram K, Price A, England K, Stiehm A, Panoskaltsis-Mortari A. Club cell secretory protein improves survival in a murine obliterative bronchiolitis model. Am J Physiol Lung Cell Mol Physiol. 2013;305:L642–L650. doi: 10.1152/ajplung.00021.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah RJ, Wickersham N, Lederer DJ, Palmer SM, Cantu E, Diamond JM, et al. Lung Transplant Outcomes Group. Preoperative plasma club (clara) cell secretory protein levels are associated with primary graft dysfunction after lung transplantation. Am J Transplant. 2014;14:446–452. doi: 10.1111/ajt.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li JS, Miralles Fusté J, Simavorian T, Bartocci C, Tsai J, Karlseder J, et al. TZAP: a telomere-associated protein involved in telomere length control. Science. 2017;355:638–641. doi: 10.1126/science.aah6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, et al. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. 2009;4:525–534. doi: 10.1016/j.stem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng D, Limmon GV, Yin L, Leung NH, Yu H, Chow VT, et al. Regeneration of alveolar type I and II cells from Scgb1a1-expressing cells following severe pulmonary damage induced by bleomycin and influenza. PLoS One. 2012;7:e48451. doi: 10.1371/journal.pone.0048451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Q, Liu K, Cui G, Huang X, Yao S, Guo W, et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat Genet. 2019;51:728–738. doi: 10.1038/s41588-019-0346-6. [DOI] [PubMed] [Google Scholar]
- 41.Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, et al. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant. 2011;30:624–631. doi: 10.1016/j.healun.2011.01.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calabrese DR, Lanier LL, Greenland JR. Natural killer cells in lung transplantation. Thorax. 2019;74:397–404. doi: 10.1136/thoraxjnl-2018-212345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenland JR, Wang P, Brotman JJ, Ahuja R, Chong TA, Kleinhenz ME, et al. Gene signatures common to allograft rejection are associated with lymphocytic bronchitis. Clin Transplant. 2019;33:e13515. doi: 10.1111/ctr.13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shino MY, Weigt SS, Li N, Derhovanessian A, Sayah DM, Huynh RH, et al. Impact of allograft injury time of onset on the development of chronic lung allograft dysfunction after lung transplantation. Am J Transplant. 2017;17:1294–1303. doi: 10.1111/ajt.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shino MY, Weigt SS, Li N, Palchevskiy V, Derhovanessian A, Saggar R, et al. CXCR3 ligands are associated with the continuum of diffuse alveolar damage to chronic lung allograft dysfunction. Am J Respir Crit Care Med. 2013;188:1117–1125. doi: 10.1164/rccm.201305-0861OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lama VN, Belperio JA, Christie JD, El-Chemaly S, Fishbein MC, Gelman AE, et al. Models of lung transplant research: a consensus statement from the National Heart, Lung, and Blood Institute workshop. JCI Insight. 2017;2:e93121. doi: 10.1172/jci.insight.93121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hertz MI, Jessurun J, King MB, Savik SK, Murray JJ. Reproduction of the obliterative bronchiolitis lesion after heterotopic transplantation of mouse airways. Am J Pathol. 1993;142:1945–1951. [PMC free article] [PubMed] [Google Scholar]
- 48.Belperio JA, Keane MP, Burdick MD, Lynch JP, III, Xue YY, Berlin A, et al. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. J Clin Invest. 2001;108:547–556. doi: 10.1172/JCI12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murakawa T, Kerklo MM, Zamora MR, Wei Y, Gill RG, Henson PM, et al. Simultaneous LFA-1 and CD40 ligand antagonism prevents airway remodeling in orthotopic airway transplantation: implications for the role of respiratory epithelium as a modulator of fibrosis. J Immunol. 2005;174:3869–3879. doi: 10.4049/jimmunol.174.7.3869. [DOI] [PubMed] [Google Scholar]
- 50.Jiang X, Khan MA, Tian W, Beilke J, Natarajan R, Kosek J, et al. Adenovirus-mediated HIF-1α gene transfer promotes repair of mouse airway allograft microvasculature and attenuates chronic rejection. J Clin Invest. 2011;121:2336–2349. doi: 10.1172/JCI46192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sui H, Olivier AK, Klesney-Tait JA, Brooks L, Tyler SR, Sun X, et al. Ferret lung transplant: an orthotopic model of obliterative bronchiolitis. Am J Transplant. 2013;13:467–473. doi: 10.1111/j.1600-6143.2012.04337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mimura T, Walker N, Aoki Y, Manning CM, Murdock BJ, Myers JL, et al. Local origin of mesenchymal cells in a murine orthotopic lung transplantation model of bronchiolitis obliterans. Am J Pathol. 2015;185:1564–1574. doi: 10.1016/j.ajpath.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinu T, Oishi H, Juvet SC, Cypel M, Liu M, Berry GJ, et al. Spectrum of chronic lung allograft pathology in a mouse minor-mismatched orthotopic lung transplant model. Am J Transplant. 2019;19:247–258. doi: 10.1111/ajt.15167. [DOI] [PubMed] [Google Scholar]
- 54.Smirnova NF, Conlon TM, Morrone C, Dorfmuller P, Humbert M, Stathopoulos GT, et al. Inhibition of B cell-dependent lymphoid follicle formation prevents lymphocytic bronchiolitis after lung transplantation. JCI Insight. 2019;4:e123971. doi: 10.1172/jci.insight.123971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.