

Abstract
Caveolae are prominent plasmalemmal invaginations in endothelial cells, especially in the lung vasculature, which comprises a vast surface area. PV1 (plasmalemmal vesicle–associated protein-1), a 60-kD glycoprotein expressed in endothelial cells, is essential for generating spoke-like diaphragmatic structures that span the neck region of endothelial caveolae. However, their role in caveolae-mediated uptake and endothelial-barrier function is unknown. Here, we generated mice with endothelial cell–specific deletion of PV1 through tamoxifen-induced Cdh5.Cre.ERT2 (endothelial-specific vascular cadherin.Cre.estrogen receptor 2)-mediated excision of the floxed PV1 allele. We observed that loss of PV1 specifically in endothelial cells increased lung vascular permeability of fluid and protein, indicating that PV1 is required for maintenance of lung vascular-barrier integrity. Endothelial-specific PV1 deletion also increased caveolae-mediated uptake of tracer albumin compared with controls, promoted Au-albumin accumulation in the bulb of caveolae, and induced caveolar swelling. In addition, we observed the progressive loss of plasma proteins from the circulation and reduced arterial pressure resulting from transudation of water and protein as well as edema formation in multiple tissues, including lungs. These changes seen after endothelial-specific PV1 deletion occurred in the absence of disruption of endothelial junctions. We demonstrated that exposure of wild-type mice to endotoxin, which is known to cause acute lung injury and increase protein permeability, also significantly reduced PV1 protein expression. We conclude that the key function of PV1 is to regulate lung endothelial permeability through its ability to restrict the entry of plasma proteins such as albumin into caveolae and their transport through the endothelial barrier.
Keywords: albumin, transcytosis, caveolin-1, endothelial-barrier function
Endothelial cells forming the intimal layer of blood vessels make contact with circulating blood cells and plasma, creating a semipermeable barrier that restricts passive diffusion of large molecules and plasma proteins larger than albumin (67 kD) (1). Endothelial cells in a monolayer form endothelial cell–cell contact via anchoring adherens junctions and occluding tight junctions (2). These junctions, a characteristic feature of the continuous endothelium, consist of various membrane-spanning adhesive proteins such as VE-cadherin (vascular endothelial cadherin), which prevents the paracellular transport of proteins of the size of albumin and larger in the normal endothelium (3). The transport of albumin through the endothelium is attributed to a transcellular pathway via caveolae, the 40- to 80-nm diameter, omega-shaped invaginations that form a linear array along the apical and basal plasma membranes (4, 5). Caveolae internalize cargo such as albumin via receptor-mediated uptake and fluid-phase endocytosis in a dynamin-2–dependent manner (6–8). The internalized caveolae migrate through the cytoplasm in an actin-dependent manner, ultimately fusing with the abluminal membrane to deposit their contents into tissue (9, 10), a process termed caveolae-mediated transcytosis (11, 12).
The caveolar membrane recruits a variety of proteins such as caveolin-1, caveolin-2 and cavins that affect caveolae formation and shape and signaling effectors such as eNOS (endothelial nitric-oxide synthase) that are required for normal vascular function (13–16). Loss of caveolin-1 by genetic deletion or mutations causes flattening of caveolae and leads to vascular remodeling, aberrant filling of pulmonary arteries, pulmonary hypertension, and lung fibrosis (17–19). Endothelial transcytosis is significantly reduced in cav1−/− mice, which is consistent with depletion of caveolae (11). Loss of albumin binding protein (e.g., glycoprotein-60 or albondin) and LDL receptor (low-density lipoprotein receptor) also reduce the uptake and transport of albumin and other proteins via transcytosis (20–23).
The necks of flask-shaped caveolae in the continuous endothelium (with notable exceptions of the brain and skeletal muscle) are spanned by a 5- to 7-nm diaphragm (24). The role of caveolar diaphragms in caveolae formation, morphology, and caveolae-mediated transport remains poorly understood (25). The only identified protein component of these diaphragms is PV1 (plasmalemmal vesicle–associated protein-1), a 60-kD protein that generates highly ordered oligomers responsible for the formation of the diaphragm (26–28). Germline deletion of PV1 was embryonically lethal in mice bred on a single background and was less lethal (25% of expected offspring reaching term) in mice bred on a mixed background (27, 29). Mice that survived on a mixed background exhibited striking defects in vascular development and accumulation of proteins such as albumin and IgG in fenestrated organs (27). It appears that caveolae regulate the stability of PV1 protein by as-yet-unknown mechanisms, as genetic deletion of caveolin-1 resulted in the loss of PV1 in lung endothelial cells (30). Thus, previous studies have not investigated the role of PV1 in regulating changes in the structure of caveolae and permeability properties of the endothelium. In the present study, using adult mice in which PV1 was conditionally deleted in endothelial cells, we observed the loss of caveolar neck–associated diaphragms and marked increase in lung vascular permeability to fluid and albumin. We also observed surprisingly enhanced uptake of albumin-gold tracer in the bulb region of caveolae compared with wild-type (WT) mice as well as varying amounts of caveolar swelling, indicative of increased uptake of albumin and water in absence of diaphragm spanning the caveolar neck. Finally, we observed that PV1 expression was markedly reduced in the endotoxin model of acute lung injury (ALI) in mice, a condition known to increase the permeability of the lung endothelium. These results together show that PV1, a critical component of the diaphragm at caveolae necks, serves as a key restrictive barrier for the uptake of albumin and a determinant of transendothelial permeability of lung microvessels.
Methods
Mice
Animals were bred and maintained in a pathogen-free setting at the University of Illinois at Chicago after approval by the Institutional Animal Care and Use Committee. C57BL/6J mice were purchased from Jackson Laboratory (no. 000664). PV1fl/fl (PV1 flanking/flanked by LoxP) mice (provided by Radu Stan) (27) were crossed with endothelial Cdh5-CreERT2 (endothelial-specific vascular cadherin–Cre-estrogen receptor 2) mice (provided by Ralf Adams), which on tamoxifen delivery will activate Cre recombinase activity specifically in endothelial cells. Tamoxifen (no. T5648; Sigma) was dissolved in corn oil (10 mg/ml) and delivered via intraperitoneal injection daily for 5 consecutive days. Mice were allowed to rest for 2 or 4 weeks before experimentation.
Measurement of Pulmonary-Vessel Filtration Coefficient
Mice were anesthetized and prepared for experimentation as described previously (31). Lung weight gain was measured after a step increase (10 cm H20) in venous pressure for 20 minutes. The rate of the lung weight gain was normalized to the lung dry weight and pressure change, thus providing the microvascular filtration coefficient (ml × min−1 × cm H20 × g of dry weight−1).
Evans Blue-Albumin Uptake and Albumin Permeability-Surface-Area Measurements
Evans blue-albumin (25 mg/kg) or radiolabeled albumin (1 μCi; [125I]albumin tracer; AnazaoHealth) was injected into anesthetized mice and allowed to circulate in the bloodstream for 30 minutes. Mouse lungs were perfused via the right ventricle with Dulbecco’s PBS for 2 minutes and were subsequently extracted, weighed, and homogenized in 1 ml of Dulbecco’s PBS. Homogenized samples were combined with 2 ml of formamide and placed at 60°C for 24 hours. The Evans blue dye concentration in supernatants was measured spectrophotometrically at absorbances of 620 and 740 nm. For mice exposed to radiolabeled albumin, blood was withdrawn from the inferior vena cava before lung perfusion. Radioactivity was subsequently counted in the indicated organs and blood samples.
Transmission EM
Colloidal gold–albumin (Au-albumin) tracer was prepared as previously described (31). Mice were anesthetized followed by cannulation of the pulmonary artery and left atrium. Lungs were first perfused with Hanks’ balanced salt solution (no. 14025092; Gibco) for 5 minutes. Tracer solution was subsequently perfused into the lung and collected from the left atrium for a period of 15 minutes. Lungs were washed with Hanks’ balanced salt solution for 5 minutes, fixed via perfusion with 2.5% glutaraldehyde, 4% paraformaldehyde, 0.1 M HEPES, and 2 mM CaCl2. Lungs were minced into 1-mm pieces and subsequently fixed as previously described (31).
IB
Freshly isolated endothelial cells (32) were homogenized in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors. Samples were subsequently separated via SDS-PAGE, transferred onto 0.45-μm nitrocellulose membranes (no. 1620115; BioRad), and incubated in primary antibodies overnight. Membranes were incubated with secondary antibodies the next day, followed by detection with chemiluminescent reagents. Western blot quantification was performed using ImageJ software.
Statistics
Results were analyzed using GraphPad Prism Software. The Student’s t test was used for experiments involving two experimental groups. One-way ANOVA with post hoc Tukey’s multiple-comparisons test was used for experiments involving three or more experimental groups. Two-way ANOVA with repeated-measures and post hoc multiple-comparisons test was used to compare the basal weight change between genotypes over the indicated number of days.
Results
Endothelial-Specific Deletion of PV1 in Mice Increases Lung Transendothelial Albumin and Fluid Permeability
To determine the role of PV1 in adult mice, we generated PV1iΔEC (endothelial-specific PV1 knockout) adult mice using PV1 floxed mice expressing tamoxifen-inducible Cre driven by Cdh5 promoter (Figure 1A). Control (PV1fl/fl; Cdh5-CreERT2−/−) and PV1iΔEC mice received tamoxifen from 8 to 10 weeks of age to delete PV1. Deletion of PV1 after tamoxifen injections was validated by Western blot analysis of the lungs (Figure 1B) and by isolated cultured endothelial cells from these mice (see Figure E1 in the data supplement). PV1iΔEC mice showed prominent development of ascites (Figures 1C and E2) and a 45% increase in fluid mass as reflected in their body weight at 5 days after tamoxifen, which partially recovered over 7 days (Figure 1D). Ascites fluid contained protein, lipids, and electrolytes, suggesting that loss of PV1 increased filtration of plasma into extravascular spaces (Table E1).
Figure 1.
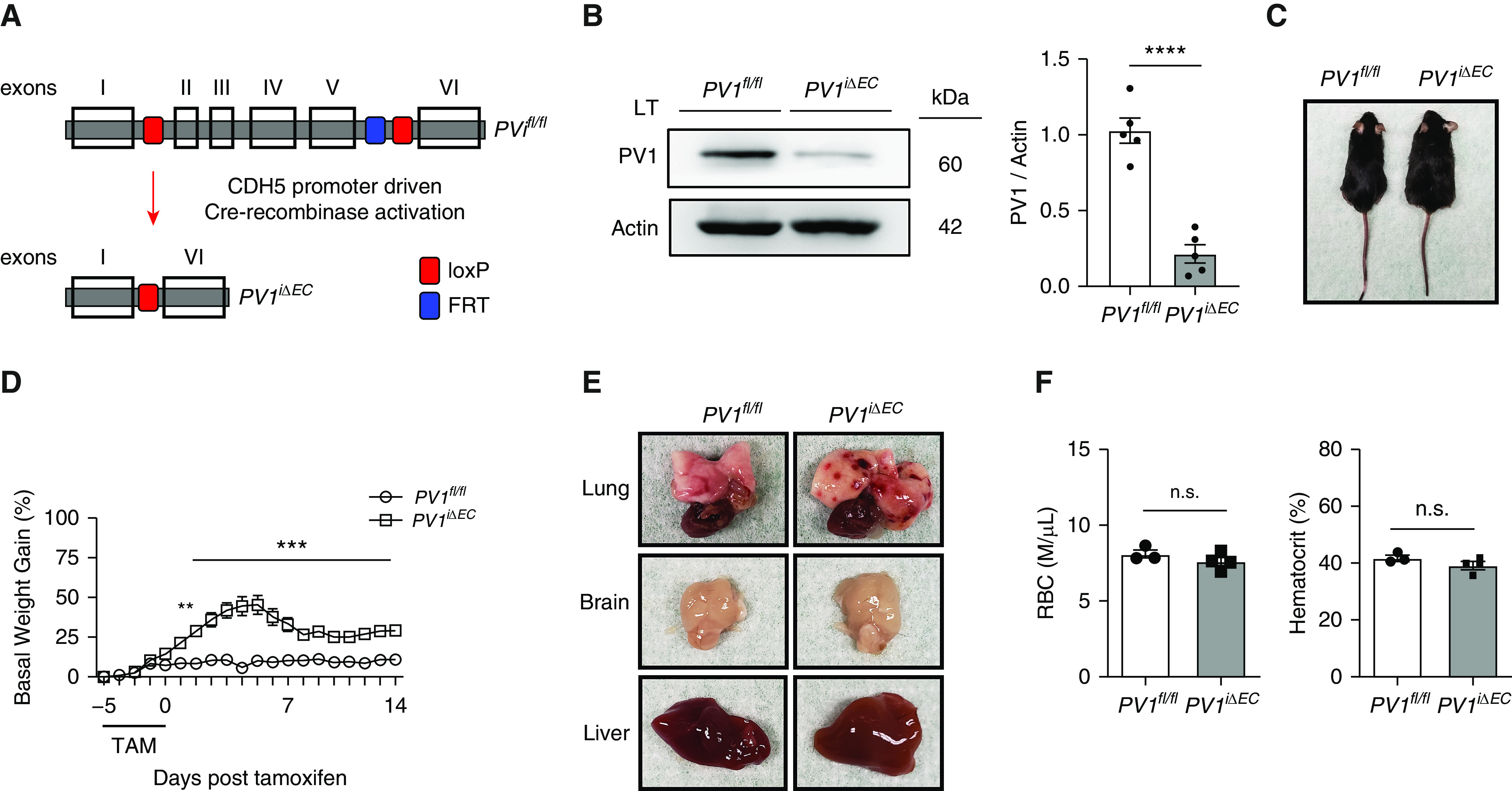
Endothelial cell–specific PV1 (plasmalemmal vesicle–associated protein-1) deletion in mice causes fluid extravasation and capillary hemorrhages. (A) Generation of PV1iΔEC (endothelial-specific PV1 knockout) mice. Transparent boxes with black borders represent exons. The blue and red boxes represent the FRT and loxP sites, respectively. (B) Western blot analysis of PV1 protein in whole-lung lysates after tamoxifen administration; n = 5 mice per group. (C) Ascites in PV1iΔEC mice after tamoxifen administration. (D) Time course of basal-weight change in control and PV1iΔEC mice; n = 12 mice/group. (E) Presence of petechiae on the pleural surface of PV1iΔEC lungs and absence of petechiae in PV1iΔEC brain and liver; n = 5 mice/group. (F) Quantification of circulating RBCs and hematocrit in PV1iΔEC mice; n = 3–4 mice/group. Data are shown as the mean ± SEM. **P < 0.01, ***P < 0.001, and ****P < 0.0001. CDH5 = endothelial-specific vascular cadherin; FRT = flippase recognition target; loxP = locus of X over P1; LT = lung tissue; n.s. = not significant; PV1fl/fl = PV1 flanking/flanked by LoxP; RBC = red blood cells; TAM = tamoxifen.
We next focused on the effects of PV1 deletion on the lungs. Strikingly, examination of lungs from PV1iΔEC mice revealed numerous petechiae on the pleural-surface microvessels, indicative of red-blood-cell (RBC) extravasation (Figure 1E). Vascular abnormalities were not observed in the other organs examined. To determine whether these findings were associated with loss of endothelial cells, we evaluated VE-cadherin protein concentrations in PV1iΔEC lungs and found no significant difference (Figures E3A and E3B). We next determined the RBC count and hematocrit in PV1iΔEC blood samples and observed no significant change as compared with WT mice (Figure 1F). Endothelial PV1 deletion also did not alter leukocyte numbers (Figures E4A–E4E). We observed the control and PV1iΔEC mice for up to 4 months after tamoxifen administration and did not observe any differences in mortality during this period (Figure E5).
Compared with those of control animals, mouse lungs from PV1iΔEC mice exhibited a 17.6% increase in lung wet weight 2 weeks after the last tamoxifen injection (Figure 2A). Extravascular lung-water content in PV1iΔEC mice was also elevated as compared with control animals (Figure 2B). We interrogated lung transvascular fluid-flux changes by measuring the lung capillary filtration coefficient (Kfc), which reflected transendothelial flux of water. Lungs from PV1iΔEC mice exhibited a 1.6-fold increase in Kfc fluid permeability as compared with control animals (Figure 2C). To quantify protein transport across the lung endothelium, we used Evans blue-dye albumin tracer (25 mg/kg) via intravenous injection and observed that PV1iΔEC mice exhibited a 68.9% increase in albumin uptake in tissue (Figure 2D). Using [125I]albumin tracer, we also observed that endothelial PV1 deletion increased the lung transendothelial albumin permeability × surface area product, a direct measure of lung vascular albumin permeability (33), by 8.3-fold (Figure 2E). By way of comparison, in brain microvessel endothelial cells that show far fewer caveolae (34) and no evidence of caveolar diaphragms (35, 36), we found that endothelial cell–specific deletion of PV1 failed to increase brain transendothelial albumin permeability (Figure 2F). Interestingly, increased transendothelial permeability of albumin was also seen in multiple fenestrated organs after endothelial cell–specific deletion of PV1 (Figures E6A–E6D), indicating that PV1 to varying degrees also controlled transendothelial albumin permeability in other vascular beds.
Figure 2.

Endothelial cell–specific PV1 deletion induces edema genesis and increased lung endothelial permeability to fluid and albumin. (A) Edema formation in PV1iΔEC lungs assessed by wet-to-dry ratio; n = 4 mice/group. (B) Elevation of extravascular lung water, which estimates lung interstitial and/or alveolar fluid, in PV1iΔEC mice; n = 4–7 mice/group. (C) Determination of microvascular capillary filtration coefficient, which is a measure of permeability to fluid; n = 4 mice/group. (D) Assessment of Evans blue-albumin (EBA) uptake in mouse lungs; n = 3 mice/group. (E) Elevation of lung albumin permeability × surface area (PS) product in PV1iΔEC mice, which represents the probability of albumin permeability across the endothelium; n = 5 mice/group. (F) Albumin PS-product measurement in the brain, which reveals no significant difference in uptake; n = 5–6 mice/group. Data are shown as the mean ± SEM. *P < 0.05 and **P < 0.01.
Endothelial PV1 Deletion Increases Caveolae Size and Uptake of Albumin
Because the above results showed increased transendothelial permeability of albumin in PV1iΔEC mouse lungs, we next determined whether the loss of the PV1 diaphragm in caveolae was itself responsible the uptake of the albumin tracer in caveolae. Here, we used the 6- to 9-nm–diameter Au-albumin tracer and performed transmission EM measurements. We quantified Au-albumin tracer amounts in plasma membrane–attached caveolae in PV1iΔEC mice and control mice. PV1 deletion and absence of diaphragm increased the total number of albumin particles, primarily localized in the bulb region of caveolae (Figure 3A). An increased amount of albumin tracer was found in the bulb region of caveolae in PV1iΔEC mice relative to the neck region as compared with control mice (Figure 3A). Studies in lungs from PV1iΔEC mice also showed thickened basal lamina and dilation of perivascular space as compared with control mice (Figure E7A), indicative of tissue edema formation. PV1iΔEC alveoli, however, appeared intact, and there was no other apparent disruption of endothelial cells (except the absence of diaphragms) or lung epithelial cells. The interendothelial junctions also appeared similar in PV1iΔEC mice as compared with WT mice (Figure E7B). Besides PV1 deletion in endothelial cells preventing the formation of diaphragms (Figures 3B and 3C), the only other change was an increase in the neck diameters (Figures 3D and 3E), bulb diameters of caveolae (Figures 3D and 3F), and bulb depth (Figures 3D and 3G), all indicative of gross structural changes in the caveolar shape.
Figure 3.
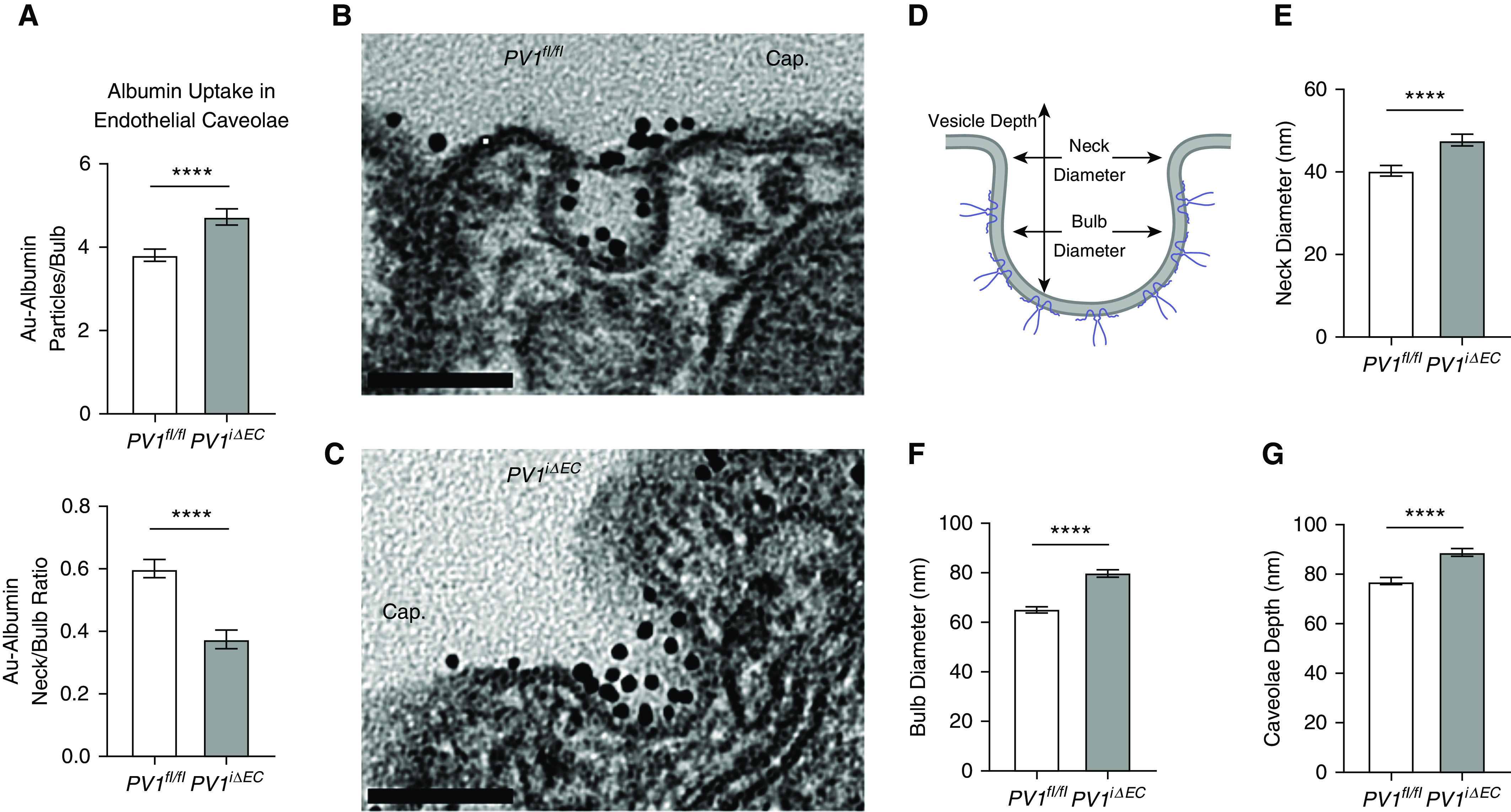
Endothelial PV1 deletion induces caveolar bulb swelling and increased uptake of albumin in bulb domain in endothelial cells. (A) Increased accumulation of albumin in the bulb region of caveolae. The neck-to-bulb albumin ratio (a relative ratio of albumin distribution in the vesicle) was significantly reduced in PV1iΔEC caveolae, whereas the absolute number of bulb particles significantly increased. (B and C) Presence and absence of caveolar diaphragms in control and PV1iΔEC lung endothelia, respectively. Scale bars, 0.10 μm. (D) Illustration of caveolae, highlighting the caveolar neck and bulb regions. Caveolae depth was determined by measuring the length from the lowest point in the bulb to the highest point in the caveolae neck region. At least 100 caveolae/group were selected for morphometric analysis. (E–G) Assessment of caveolae neck width (E), caveolae bulb width (F), and caveolae depth (G) in PV1iΔEC endothelial cells. Experiments were performed with three mice per group. Data are shown as the mean ± SEM. ****P < 0.0001. Cap = capillary lumen.
To assess changes in caveolae numbers between the two groups, we next counted caveolae in PV1iΔEC mouse lung endothelial cells and control endothelial cells (Figures 4A and 4B). We observed 52% increase in tracer-filled endocytic (internalized; membrane-detached) vesicles (Figure 4C). When normalized to the total number of endocytic caveolae present in each sample, the percentage of endocytic caveolae carrying the Au-albumin tracer in PV1iΔEC was significantly increased compared with control mice (Figure 4C). Concurrently, we observed a 26% reduction in luminal caveolae carrying Au-albumin (Figure 4D). When normalized to the total number of membrane-attached caveolae present in each sample, the percentage of membrane caveolae carrying the tracer in PV1iΔEC was significantly increased as compared with control mice (Figure 4D). PV1 deletion, however, did not affect the total caveolae number (Figure 4E). The overall number of internalized Au-albumin tracer particles in PV1iΔEC lung endothelial cells was nearly double that observed in control-mouse lung endothelial cells (Figure 4F). We also noted that caveolae clusters were present in greater abundance in PV1iΔEC endothelial cells (Figures E8A and E8B). Our analysis thus revealed a significant increase in caveolae clusters present both at the endothelial cell membrane and in the cytoplasm. These data suggest that PV1 deletion promotes caveolar fusion and clustering of endocytic vesicles.
Figure 4.
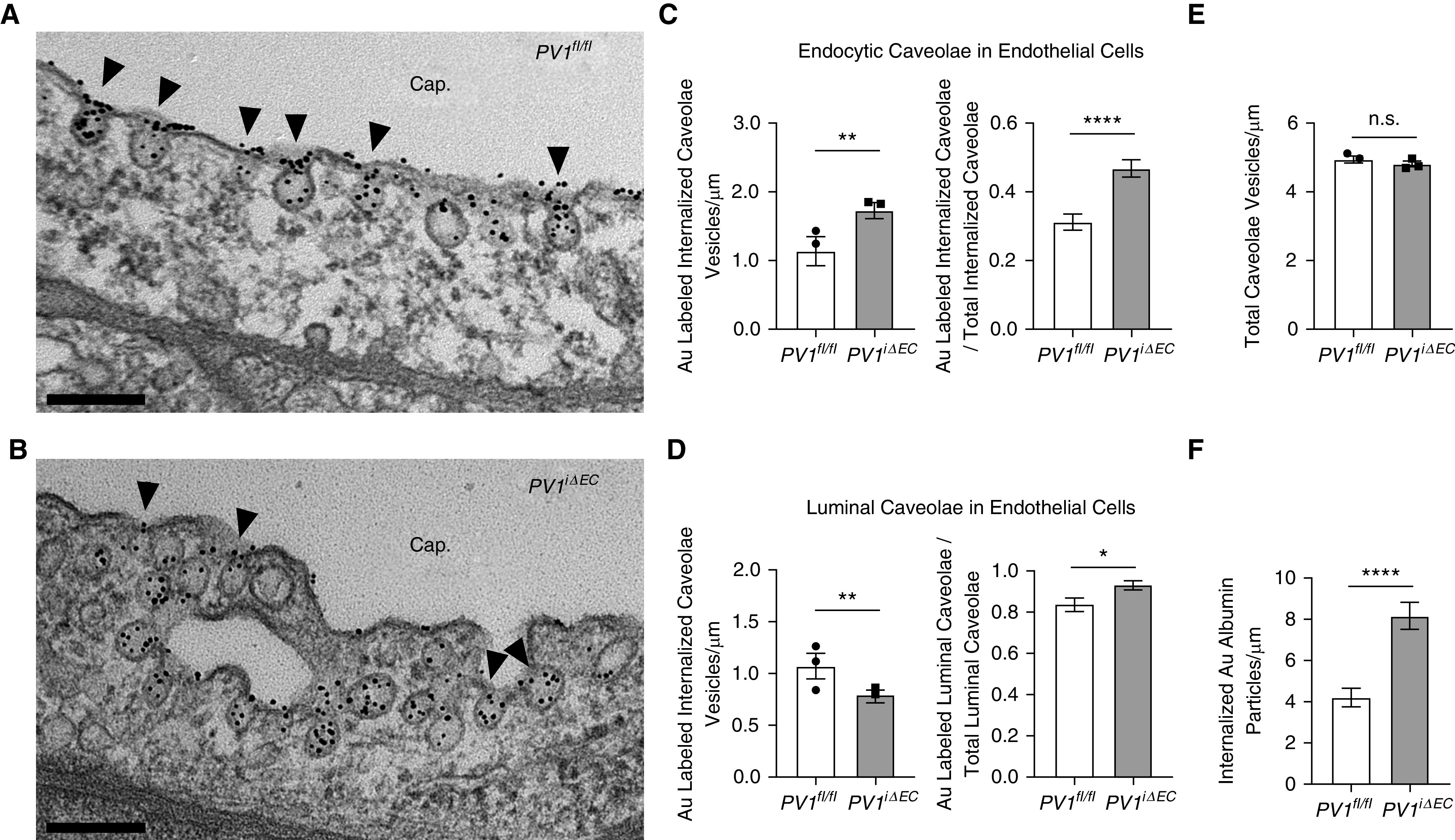
Endothelial cell–specific PV1 deletion increases endocytosis of albumin. At least 75 electron micrographs/group were used for counting caveolae. (A and B) The luminal caveolar (indicated by arrowheads) abundance in control animals (A) and PV1iΔEC mice (B), respectively. Scale bars, 0.2 μm. (C) The total number of endocytic vesicles, together with the proportion of tracer-filled endocytic vesicles relative to total endocytic vesicles, was markedly increased in PV1iΔEC endothelial cells. (D) The number of vesicles at the luminal surface was significantly decreased, whereas the proportion of tracer-filled luminal caveolae relative to total luminal vesicles increased in PV1iΔEC endothelial cells. (E) The number of total caveolae (luminal, endocytic, and abluminal) was unchanged in PV1iΔEC endothelial cells. (F) Endothelial PV1 deletion increased the number of internalized Au-albumin particles. Experiments were performed with three mice per group. Data are shown as the mean ± SEM. *P < 0.05, **P < 0.01, and ****P < 0.0001.
Endothelial PV1 Controls Plasma Protein Homeostasis
Because endothelial PV1 deletion in adult mice increased vascular permeability to both fluid and albumin (Figures 2C and 2E), we assessed whether PV1iΔEC mice also exhibited reduced plasma protein concentrations and blood pressure. Analysis of PV1iΔEC mice showed 58% and 84% reductions in plasma albumin concentrations at 2 and 4 weeks after tamoxifen administration, respectively (Figure 5A). PV1iΔEC mice also exhibited 50% and 77% reductions in total protein at 2 and 4 weeks after tamoxifen administration (Figure 5B). PV1iΔEC mice were hypotensive, with systolic pressure reduced by 29% (Figure 5C) and diastolic pressure reduced by 44% 4 weeks after tamoxifen administration (Figure 5D), respectively. We also assessed the protein content in urine and ascites. PV1iΔEC mice did not exhibit elevated urine protein at 4 weeks after tamoxifen administration (data not shown), whereas the ascites protein concentration was 20% of the plasma concentration (data not shown). Thus, the increase in transendothelial albumin permeability in lungs and other organs secondary to endothelial PV1 deletion resulted in loss of albumin and reduction in arterial pressure.
Figure 5.
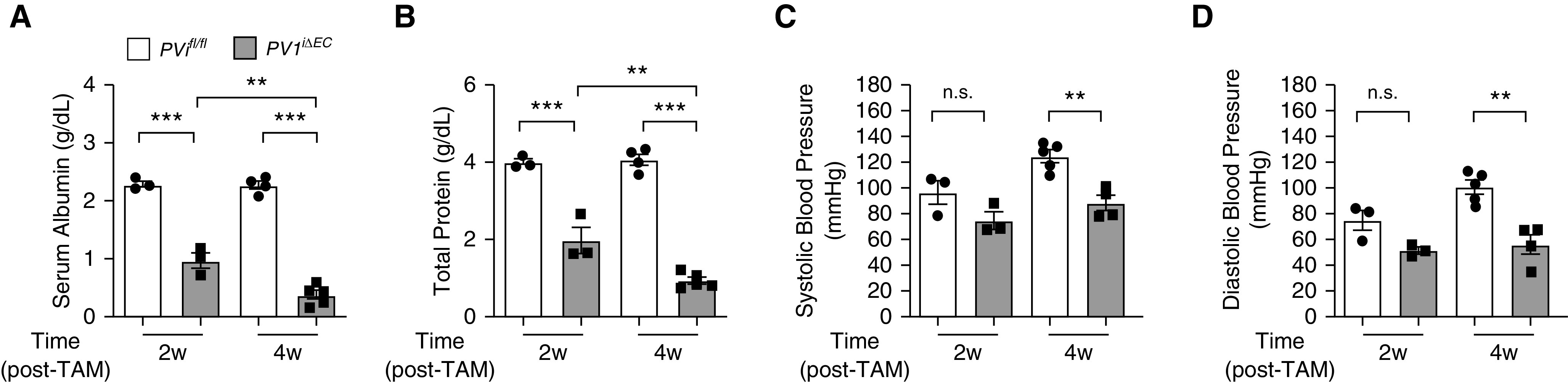
PV1 in the endothelium functions to restrict the loss of plasma proteins. (A and B) Loss of serum albumin and total serum protein in blood harvested from PV1iΔEC mice at 2 and 4 weeks after tamoxifen administration; n = 3–4 mice/group. (C and D) Reduction in systolic and diastolic vascular pressure in PV1iΔEC mice 2 and 4 weeks after tamoxifen administration; n = 3–5 mice/group. Data are shown as the mean ± SEM. **P < 0.01 and ***P < 0.001. w = weeks.
Endotoxin Downregulates PV1 Expression in Mouse Lungs
Previous studies have suggested that caveolae-mediated transport contributes to the pathophysiology and mortality rate of ALI (37, 38). Because the results above showed that PV1 regulated the filling of caveolae with albumin and caveolae shape, we determined PV1 expression in mouse models with ALI. A recent study published by our group has investigated the endothelial translatome in mice exposed to LPS during injury and recovery phases (39). We analyzed data from this study (http://www.rehmanlab.org/ribo) and found that endotoxin reduced ribosome-associated PV1 mRNA expression 6 hours after LPS delivery in the lungs, suggesting that endothelial cells respond to inflammation by downregulating PV1 transcription and/or translation. Given these findings, we assessed PV1 protein expression in an inhaled LPS injury model, which induces vascular inflammation in lungs. In these experiments, WT mice were exposed daily for 1 hour to nebulized LPS for a consecutive 4 days, as described by Oliveira and colleagues (40), and PV1 expression was analyzed in lungs at the indicated times (Figure E9A). PV1 protein concentrations decreased at Day 4 and persisted up to Day 7 after LPS exposure (Figures E9B and E9C). In contrast, we have shown previously that caveolin-1 expression amounts in lungs were reduced at Day 4, but unlike those of PV1, they recovered fully by Day 7 (40). Thus, LPS markedly reduced PV1 expression.
Discussion
The transport of albumin via caveolae-mediated transcytosis in the endothelium is a major means of transendothelial albumin permeability (10, 41). Previous studies have shown that inhibition of transcytosis in endothelial cells reduced transendothelial albumin transport by ∼80% (42). Caveolae therefore substantially contribute to permeability of albumin across the continuous endothelium, which is rich in caveolae (41, 43). In the present study, we observed that genetic deletion in adult mice of PV1 in endothelial cells, the protein composing the diaphragm at the neck region of the caveolae (24), increased transendothelial permeability to albumin and fluid. These studies were made in vivo by measuring the Kfc and the albumin permeability × surface area product in mouse lungs (3), in which both measures of endothelial permeability were markedly increased. Furthermore, we demonstrated that deletion of PV1 caused swelling of caveolae, as evident by the increases in the diameters of caveolae necks and caveolar bulbs, which was coupled to augmented filling with albumin nanoparticles, mostly localized in the caveolae bulbs in the absence of the diaphragm. Finally, we observed a rapid reduction of PV1 expression in mouse lungs after LPS–endotoxin exposure, which may contribute to increased caveolae-mediated transport during endotoxemia (38). These findings thus indicate a novel role for PV1 in regulating caveolae-mediated transport of albumin and fluid across continuous-type endothelium and a possible role of PV1 in the increase in lung vascular permeability seen during endotoxemia.
Loss of the caveolae-associated proteins caveolin-1/2 and cavins affect caveolae formation and blood-vessel organization and also increase the susceptibility to organ failure (17, 18, 44). We demonstrated that caveolin-1 deficiency was associated with abnormalities in pulmonary-artery filling and induced pulmonary hypertension (19). Here, we show that endothelial PV1, the component of the caveolae that forms the neck-spanning diaphragm, has a distinct function. The observation that PV1iΔEC mice rapidly developed protein-rich ascites points to an endothelial barrier–restrictive function of PV1 through its ability to form caveolae neck–associated diaphragms. The diaphragm may reduce the flux of plasma proteins and fluid into caveolae. This function of PV1-generated diaphragms is consistent with our observations of significantly increased transendothelial albumin permeability and edema formation in PV1iΔEC lungs. The loss of plasma albumin in PV1iΔEC mice into tissue and elevated tissue oncotic pressure may be the primary driving force for the transendothelial filtration of water, resulting in ascites and edema formation seen in multiple organs, including the lungs.
The present studies describe an important role for PV1 in the regulation of transendothelial albumin transport. We showed through morphometric analysis that PV1 controlled caveolar shape rather than caveolae formation and abundance. Increased caveolae neck diameter and enlarged caveolae seen in PV1iΔEC mice were correlated with increased uptake of tracer albumin, suggesting that the diaphragm itself is a barrier that restricts uptake of albumin. The mechanism of diaphragm-mediated restriction in the filling of caveolae with albumin is not known. One possibility is that the spoke-like features composing the diaphragms can function as a sieve, limiting the entry of albumin and other large molecules into caveolae. We observed in the PV1iΔEC endothelium that caveolae swelling and filling of albumin in the bulb regions of caveolae occurred in the absence of open adherens junctions. Thus, the increase in transendothelial albumin permeability in PV1iΔEC mice is likely attributable to enhanced caveolae-mediated transport, as opposed to increased permeability via the interendothelial junction. We also noted an increased tendency of caveolae to form clusters in PV1iΔEC endothelial cells, reflecting lower membrane tension due to the increase in internalized caveolae and associated cytoskeletal rearrangements occurring during endocytosis (45, 46). In support of this finding, increased production of intracellular ceramide, which promotes albumin internalization, was associated with formation of caveolae clusters in rat kidney cells (47, 48). Vacuoles present in both control and PV1iΔEC endothelia were far less abundant than caveolae and thus were unlikely to contribute increased endothelial permeability in PV1iΔEC mice.
PV1 has a role in embryonic development (27, 29). Analysis of PV1−/− embryos revealed focal hemorrhaging at E16.5, which preceded mortality at E17.5 (29). In our study, we found that adult PV1iΔEC mice survived for longer than 4 months after tamoxifen administration and remained relatively active despite the loss of serum protein and increased endothelial permeability. Interestingly, gross analysis of PV1iΔEC organs indicated that hemorrhaging occurred in lungs and not in other organs. These findings correlate with the expression profile of PV1 in different organs, with lung endothelial PV1 expression being greater than that of most other organs (24). Our results did not show any changes in VE-cadherin expression, endothelial-junction permeability, or alveolar barrier disruption. The presence of petechial hemorrhages, indicative RBC extravasation into tissue, might reflect transcellular migration of RBCs occurring via the enlarged caveolae in PV1iΔEC mouse endothelium. EM studies in rabbits with thrombocytopenia showed diapedesis of RBCs, which was associated with an elevated number of internalized caveolae-like vesicles (49). In addition, another study showed that caveolin-1 was necessary for leukocyte diapedesis and that it was localized near intracellular ICAM-1–positive microdomains (50). It is therefore possible that the increased internalization of the enlarged caveolae in endothelial cells after loss of PV1 contributes to the transmigration of RBCs seen in the PV1iΔEC mouse endothelium.
Increased endothelial permeability is a feature of ALI, a condition that has an ∼26% mortality rate (51). Caveolae-mediated transcytosis of albumin may contribute to increased transendothelial albumin permeability and edema formation in ALI (37, 38). In a recent study, we analyzed RNA-sequencing data generated from studies involving mice expressing hemagglutinin A tags specifically in endothelial cell ribosomes (39). In these studies, the endothelial translatome was determined at several time points after LPS challenge to uncover differential endothelial responses to endotoxin in the lungs, heart, and brain. Using available data from this study (http://www.rehmanlab.org/ribo), we found that endotoxin reduced PV1 gene expression in the continuous endothelia of lungs and heart at 6 hours after LPS administration, whereas expression in the brain endothelium remained unchanged. Notably, brain endothelial cells expressed relatively little PV1 at baseline and after LPS administration, which is consistent with the results of previous studies in mice (52). In the present study, we found that PV1 protein expression was reduced in the lungs of WT mice after exposure to nebulized LPS. Thus, endothelial cells respond to LPS by downregulating PV1 transcription and expression. The significance of our observation of decreased PV1 expression after endotoxin administration is not clear. It is possible that as with deletion of PV1 in endothelial cells of PV1iΔEC mice, the endotoxin-induced decrease in PV1 may similarly impair the sieve function of the diaphragm and hence contribute increased lung vascular permeability of albumin via transcytosis after LPS challenge.
In summary, we demonstrate that endothelial PV1 functions in the normal lung to reduce transendothelial albumin permeability through restricting caveolae-mediated uptake of albumin. Studies made in PV1iΔEC mice showed that PV1 regulated the structure of caveolae minimizing the diameters of caveolae necks and the bulb region of caveolae. Deletion of PV1 also increased the uptake of albumin in the bulb region and permeability of albumin across the endothelial barrier. Thus, loss or dysregulated function of PV1 in disease states such as endotoxemia may enhance transendothelial permeability through increased uptake and transport of albumin.
Supplementary Material
Footnotes
Supported by U.S. National Institutes of Health Grants R01 HL045638 (A.B.M.), R01 HL125356 (R.D.M.), T32 HL007829 (E.F.), and UL1 TR002003 (J.H.J.).
Author Contributions: R.D.M. and A.B.M. conceived of the study. J.H.J., R.D.M., and A.B.M. designed the experiments. J.H.J., E.F., and Z.H. performed the experiments and analyzed the data. J.H.J., R.D.M., and A.B.M. wrote the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0102OC on July 14, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Del Vecchio PJ, Siflinger-Birnboim A, Shepard JM, Bizios R, Cooper JA, Malik AB. Endothelial monolayer permeability to macromolecules. Fed Proc. 1987;46:2511–2515. [PubMed] [Google Scholar]
- 2.Komarova YA, Kruse K, Mehta D, Malik AB. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res. 2017;120:179–206. doi: 10.1161/CIRCRESAHA.116.306534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Predescu D, Palade GE. Plasmalemmal vesicles represent the large pore system of continuous microvascular endothelium. Am J Physiol. 1993;265:H725–H733. doi: 10.1152/ajpheart.1993.265.2.H725. [DOI] [PubMed] [Google Scholar]
- 4.Grotte G. Passage of dextran molecules across the blood-lymph barrier. Acta Chir Scand Suppl. 1956;211:1–84. [PubMed] [Google Scholar]
- 5.Lanken PN, Hansen-Flaschen JH, Sampson PM, Pietra GG, Haselton FR, Fishman AP. Passage of uncharged dextrans from blood to lung lymph in awake sheep. J Appl Physiol (1985) 1985;59:580–591. doi: 10.1152/jappl.1985.59.2.580. [DOI] [PubMed] [Google Scholar]
- 6.John TA, Vogel SM, Tiruppathi C, Malik AB, Minshall RD. Quantitative analysis of albumin uptake and transport in the rat microvessel endothelial monolayer. Am J Physiol Lung Cell Mol Physiol. 2003;284:L187–L196. doi: 10.1152/ajplung.00152.2002. [DOI] [PubMed] [Google Scholar]
- 7.Villaseñor R, Lampe J, Schwaninger M, Collin L. Intracellular transport and regulation of transcytosis across the blood-brain barrier. Cell Mol Life Sci. 2019;76:1081–1092. doi: 10.1007/s00018-018-2982-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnitzer JE, Oh P, McIntosh DP. Role of GTP hydrolysis in fission of caveolae directly from plasma membranes. Science. 1996;274:239–242. doi: 10.1126/science.274.5285.239. [DOI] [PubMed] [Google Scholar]
- 9.Stoeber M, Stoeck IK, Hänni C, Bleck CK, Balistreri G, Helenius A. Oligomers of the ATPase EHD2 confine caveolae to the plasma membrane through association with actin. EMBO J. 2012;31:2350–2364. doi: 10.1038/emboj.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milici AJ, Watrous NE, Stukenbrok H, Palade GE. Transcytosis of albumin in capillary endothelium. J Cell Biol. 1987;105:2603–2612. doi: 10.1083/jcb.105.6.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schubert W, Frank PG, Razani B, Park DS, Chow CW, Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem. 2001;276:48619–48622. doi: 10.1074/jbc.C100613200. [DOI] [PubMed] [Google Scholar]
- 12.Jones JH, Minshall RD. Lung endothelial transcytosis. Compr Physiol. 2020;10:491–508. doi: 10.1002/cphy.c190012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verma P, Ostermeyer-Fay AG, Brown DA. Caveolin-1 induces formation of membrane tubules that sense actomyosin tension and are inhibited by polymerase I and transcript release factor/cavin-1. Mol Biol Cell. 2010;21:2226–2240. doi: 10.1091/mbc.E09-05-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen CG, Bright NA, Howard G, Nichols BJ. SDPR induces membrane curvature and functions in the formation of caveolae. Nat Cell Biol. 2009;11:807–814. doi: 10.1038/ncb1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z, Bakhshi FR, Shajahan AN, Sharma T, Mao M, Trane A, et al. Nitric oxide-dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol Biol Cell. 2012;23:1388–1398. doi: 10.1091/mbc.E11-09-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siddiqui MR, Komarova YA, Vogel SM, Gao X, Bonini MG, Rajasingh J, et al. Caveolin-1-eNOS signaling promotes p190RhoGAP-A nitration and endothelial permeability. J Cell Biol. 2011;193:841–850. doi: 10.1083/jcb.201012129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 18.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maniatis NA, Shinin V, Schraufnagel DE, Okada S, Vogel SM, Malik AB, et al. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1−/− mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L865–L873. doi: 10.1152/ajplung.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tiruppathi C, Song W, Bergenfeldt M, Sass P, Malik AB. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J Biol Chem. 1997;272:25968–25975. doi: 10.1074/jbc.272.41.25968. [DOI] [PubMed] [Google Scholar]
- 21.Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, et al. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol. 2000;150:1057–1070. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dehouck B, Fenart L, Dehouck MP, Pierce A, Torpier G, Cecchelli R. A new function for the LDL receptor: transcytosis of LDL across the blood-brain barrier. J Cell Biol. 1997;138:877–889. doi: 10.1083/jcb.138.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraehling JR, Chidlow JH, Rajagopal C, Sugiyama MG, Fowler JW, Lee MY, et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat Commun. 2016;7:13516. doi: 10.1038/ncomms13516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stan RV, Ghitescu L, Jacobson BS, Palade GE. Isolation, cloning, and localization of rat PV-1, a novel endothelial caveolar protein. J Cell Biol. 1999;145:1189–1198. doi: 10.1083/jcb.145.6.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stan RV. Endothelial stomatal and fenestral diaphragms in normal vessels and angiogenesis. J Cell Mol Med. 2007;11:621–643. doi: 10.1111/j.1582-4934.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stan RV. Multiple PV1 dimers reside in the same stomatal or fenestral diaphragm. Am J Physiol Heart Circ Physiol. 2004;286:H1347–H1353. doi: 10.1152/ajpheart.00909.2003. [DOI] [PubMed] [Google Scholar]
- 27.Stan RV, Tse D, Deharvengt SJ, Smits NC, Xu Y, Luciano MR, et al. The diaphragms of fenestrated endothelia: gatekeepers of vascular permeability and blood composition. Dev Cell. 2012;23:1203–1218. doi: 10.1016/j.devcel.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stan RV, Tkachenko E, Niesman IR. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol Biol Cell. 2004;15:3615–3630. doi: 10.1091/mbc.E03-08-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herrnberger L, Seitz R, Kuespert S, Bösl MR, Fuchshofer R, Tamm ER. Lack of endothelial diaphragms in fenestrae and caveolae of mutant Plvap-deficient mice. Histochem Cell Biol. 2012;138:709–724. doi: 10.1007/s00418-012-0987-3. [DOI] [PubMed] [Google Scholar]
- 30.Tkachenko E, Tse D, Sideleva O, Deharvengt SJ, Luciano MR, Xu Y, et al. Caveolae, fenestrae and transendothelial channels retain PV1 on the surface of endothelial cells. PLoS One. 2012;7:e32655. doi: 10.1371/journal.pone.0032655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedrich EE, Hong Z, Xiong S, Zhong M, Di A, Rehman J, et al. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. Proc Natl Acad Sci USA. 2019;116:12980–12985. doi: 10.1073/pnas.1902165116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Zhang L, Marsboom G, Jambusaria A, Xiong S, Toth PT, et al. Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat Commun. 2019;10:2126. doi: 10.1038/s41467-019-10134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker JC. Acute lung injury and pulmonary vascular permeability: use of transgenic models. Compr Physiol. 2011;1:835–882. doi: 10.1002/cphy.c100013. [DOI] [PubMed] [Google Scholar]
- 34.Chow BW, Nuñez V, Kaplan L, Granger AJ, Bistrong K, Zucker HL, et al. Caveolae in CNS arterioles mediate neurovascular coupling. Nature. 2020;579:106–110. doi: 10.1038/s41586-020-2026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 1995;202:325–332. doi: 10.1002/aja.1002020402. [DOI] [PubMed] [Google Scholar]
- 36.van der Wijk AE, Wisniewska-Kruk J, Vogels IMC, van Veen HA, Ip WF, van der Wel NN, et al. Expression patterns of endothelial permeability pathways in the development of the blood-retinal barrier in mice. FASEB J. 2019;33:5320–5333. doi: 10.1096/fj.201801499RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heckel K, Kiefmann R, Dörger M, Stoeckelhuber M, Goetz AE. Colloidal gold particles as a new in vivo marker of early acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;287:L867–L878. doi: 10.1152/ajplung.00078.2004. [DOI] [PubMed] [Google Scholar]
- 38.Jiao H, Zhang Y, Yan Z, Wang ZG, Liu G, Minshall RD, et al. Caveolin-1 Tyr14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates MyD88-dependent signaling and sepsis-induced lung inflammation. J Immunol. 2013;191:6191–6199. doi: 10.4049/jimmunol.1300873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, et al. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. Elife. 2020;9:e51413. doi: 10.7554/eLife.51413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oliveira SDS, Castellon M, Chen J, Bonini MG, Gu X, Elliott MH, et al. Inflammation-induced caveolin-1 and BMPRII depletion promotes endothelial dysfunction and TGF-β-driven pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2017;312:L760–L771. doi: 10.1152/ajplung.00484.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simionescu N, Simionescu M, Palade GE. Permeability of muscle capillaries to exogenous myoglobin. J Cell Biol. 1973;57:424–452. doi: 10.1083/jcb.57.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Predescu D, Horvat R, Predescu S, Palade GE. Transcytosis in the continuous endothelium of the myocardial microvasculature is inhibited by N-ethylmaleimide. Proc Natl Acad Sci USA. 1994;91:3014–3018. doi: 10.1073/pnas.91.8.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner RC, Chen SC. Transcapillary transport of solute by the endothelial vesicular system: evidence from thin serial section analysis. Microvasc Res. 1991;42:139–150. doi: 10.1016/0026-2862(91)90082-m. [DOI] [PubMed] [Google Scholar]
- 44.Ding S-Y, Liu L, Pilch PF. Muscular dystrophy in PTFR/cavin-1 null mice. JCI Insight. 2017;2:e91023. doi: 10.1172/jci.insight.91023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golani G, Ariotti N, Parton RG, Kozlov MM. Membrane curvature and tension control the formation and collapse of caveolar superstructures. Dev Cell. 2019;48:523–538, e4. doi: 10.1016/j.devcel.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Echarri A, Del Pozo MA. Caveolae: mechanosensitive membrane invaginations linked to actin filaments. J Cell Sci. 2015;128:2747–2758. doi: 10.1242/jcs.153940. [DOI] [PubMed] [Google Scholar]
- 47.Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife. 2013;2:e00926. doi: 10.7554/eLife.00926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuebler WM, Wittenberg C, Lee WL, Reppien E, Goldenberg NM, Lindner K, et al. Thrombin stimulates albumin transcytosis in lung microvascular endothelial cells via activation of acid sphingomyelinase. Am J Physiol Lung Cell Mol Physiol. 2016;310:L720–L732. doi: 10.1152/ajplung.00157.2015. [DOI] [PubMed] [Google Scholar]
- 49.Aursnes I, Pedersen OO. Petechial hemorrhage in the ciliary processes of thrombocytopenic rabbits: an electron microscopic study. Microvasc Res. 1979;17:12–21. doi: 10.1016/0026-2862(79)90003-7. [DOI] [PubMed] [Google Scholar]
- 50.Millán J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- 51.Erickson SE, Martin GS, Davis JL, Matthay MA, Eisner MD NIH NHLBI ARDS Network. Recent trends in acute lung injury mortality: 1996-2005. Crit Care Med. 2009;37:1574–1579. doi: 10.1097/CCM.0b013e31819fefdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bosma EK, van Noorden CJF, Schlingemann RO, Klaassen I. The role of plasmalemma vesicle-associated protein in pathological breakdown of blood-brain and blood-retinal barriers: potential novel therapeutic target for cerebral edema and diabetic macular edema. Fluids Barriers CNS. 2018;15:24. doi: 10.1186/s12987-018-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.