

Abstract
Top-down mass spectrometry (MS) analyzes intact proteins at the proteoform level, which allows researchers to better understand the functions of protein modifications. Recently, top-down proteomics has increased in popularity due to advancements in high-resolution mass spectrometers, increased efficiency in liquid chromatography (LC) separation, and advances in data analysis software. Some unique protein proteoforms, which have been distinguished using top-down MS, have even been shown to exhibit marked variation in biological function compared to similar proteoforms. However, the qualitative identification of a particular proteoform may not be enough to determine the biological relevance of that proteoform. Quantitative top-down MS methods have been notably applied to the study of the differing biological functions of protein proteoforms and have allowed researchers to explore proteomes at the proteoform, rather than the peptide, level. Here, we review the top-down MS methods that have been used to quantitatively identify intact proteins, discuss current applications of quantitative top-down MS analysis, and present new areas where quantitative top-down MS analysis may be implemented.
Introduction
Modern MS-based proteomics methods fall generally into two categories: bottom-up proteomics and top-down proteomics. Bottom-up proteomics differ from top-down techniques in the requirement for protein digestion prior to LC-MS separation and analysis.1 Typically, bottom-up proteomics methods use individual proteases or protease mixtures to selectively cleave proteins at multiple amino acid sites to produce a mixture of small peptides. Optimum peptide length for bottom-up MS is considered to be 6–50 amino acid residues for effective computational analysis; trypsin digestion is often used as it produces peptides with an average length of 14 amino acids.1,2 Digestion of proteins into small peptide fragments offers multiple advantages including increased separation efficiency, limited number of charges on each peptide, and increased sample homogeneity which are all beneficial for MS detection. Indeed, for much of history mass spectrometry-based proteomics has only been possible using bottom-up analysis methods due to the limitations of MS instrumentation which was not capable of the isotopic peak resolution required to calculate accurate protein masses. As such, many MS techniques to quantify peptides have been developed. However, bottom-up proteomics has a key limitation: when proteins are digested into small peptides, information about the proteoform that has biological activity can be lost such as location of post-translational modifications (PTMs), number of PTMs, and endogenous proteolysis.
Top-down proteomics methods analyze intact proteins that have not been digested, which allows for the detection of the biologically active forms of the proteins, including location and identity of PTMs, and provides a more in-depth understanding of the action of specific proteoforms in vivo. The advent of highly sensitive, high-resolution mass spectrometers and an increase in efficiency of separation techniques has paved the way for high-throughput top-down MS proteomics to be feasible.3 For example, Fourier transform (FT) MS instruments such as the Fourier transform ion cyclotron resonance (FTICR) MS and orbitrap MS were among the first to be used to achieve isotopic resolution of intact proteins4 and are still among the highest resolution MS instruments available. Furthermore, application of multidimensional separation techniques such as the coupling of orthogonal separation platforms including RPLC,5,6 capillary electrophoresis (CE),7,8 or size-based separations9–11 increase separation efficiency so that deep proteoform characterization can be achieved for intact proteins. Multiple recent reviews have been written that focus on the improvement of MS instrumentation and separation techniques to improve methods for sample preparation, separation, ionization, data acquisition, and data processing.12–14 Additionally, the Consortium for Top-Down Proteomics published a set of best practices for the analysis of intact proteins.15 These technological advances in high-throughput top-down proteomics have allowed researchers to take the next step to perform quantitative analysis on complex mixtures of proteins at the proteoform level.
The ability to identify and relatively quantify intact proteins at the proteoform level has enabled the investigation into how differing proteoforms are involved in biological pathways, determine the effect of disease states on the proteome, and potentially aid in the discovery of disease biomarkers. There are generally three quantitative approaches that have been successfully applied in quantitative top-down proteomics analysis: label-free quantitation, metabolic labeling, and chemical labeling (Fig. 1). Label-free quantitation is a relative quantitation method that directly compares proteoform intensity between LC-MS runs. Metabolic labeling techniques supplement cell cultures with isotopically labeled compounds so that they express isotopically labeled proteins. Samples from the labeled media are mixed with samples grown using typical media and the ratio of labeled and unlabeled protein is used for relative quantitation. Chemical labeling methods utilize isotopically labeled chemical tags that react with specific amino acid residues on proteins. The labeled samples are mixed, and the ratio of the labels is used to relatively quantify proteoforms from different samples.
Fig. 1.
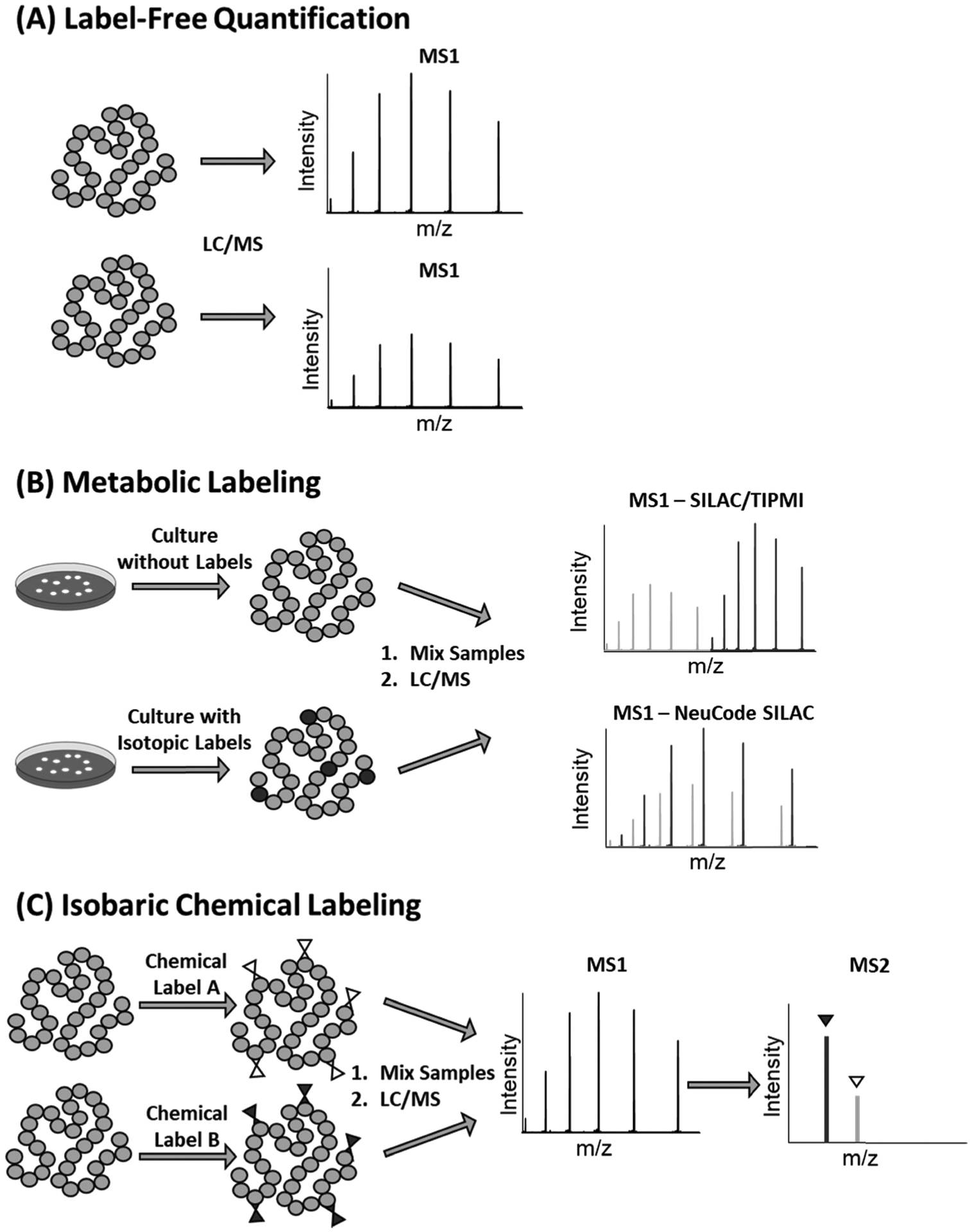
Schematics of labeling and data analysis for (A) label-free quantitation, (B) metabolic labeling, and (C) isobaric chemical labeling.
In this review, we will discuss the fundamentals of these quantitative top-down MS methods, the details of the application of these methods, and literature examples of their use to study intact proteins in biological research (Table 1).
Table 1.
Advantages and limitations of top-down quantitative proteomics techniques
| Category | Method | Pros | Cons | Quantitation level |
|---|---|---|---|---|
| Label-free quantitation | Label-free quantitation |
|
|
MS1 |
| Metabolic labeling | SILAC |
|
|
MS1 |
| NeuCode SILAC |
|
|
MS1 - high resolution instrumentation required | |
| TIPMI |
|
|
MS1 | |
| Chemical labeling | TMT/iTRAQ |
|
|
MS2 |
| pIDL |
|
|
MS2 - high resolution instrumentation required |
Label-free quantitation
Label-free quantitation, when used for MS analysis of intact proteins, relatively quantifies proteoforms by direct comparison between individual LC-MS runs. As such, no isotopic labels or mass tags are required for relative quantitation and sample handling is limited, making label-free quantitation the easiest and most cost-efficient quantitative proteomics method to implement in the laboratory. Furthermore, label-free quantitation can be applied to any protein solution or complex sample and is not limited to those that can be cultured in the laboratory.
In order to compare results between MS runs, the instrument response to a proteoform must be reliably quantified, and quantification often occurs at the MS1 level (Fig. 1A). However quantitation of instrument response to intact proteins can be challenging because ions with different charge states and different m/z values co-exist for a single proteoform when electrospray ionization (ESI) techniques are used.16 To aid in relative quantitation of individual proteoforms, several approaches have been developed to quantify proteoforms across the entire LC-MS peak at the MS1 level.17–19 Label-free quantitation of proteoforms generally occurs in three steps: determination and identification of mass features, calculation of intensity, and statistical analysis. Mass features represent the putative proteoforms present in an LC-MS run (i.e., at the MS1 level). Mass features can be determined using various deconvolution algorithms including MS-Deconv+, ProMex, X-tract, ICR-2LS, etc. Mass features are then compared to identified proteoform databases using mass tolerance and/or normalized retention time for proteoform identification. Relative quantitation is done using the intensities of detected and identified mass features; for example, ProMex and Quantitation Mass Targets (QMTs) combines the intensity data for each charge state of a proteoform across the entire elution time span to generate a final cluster of intensities or a single intensity value. IPQuant, however, utilizes the m/z calculated for each mass feature to produce extracted ion chromatograms (EICs). The area under the curve is then calculated to determine the intensity of a proteoform.19 Once intensity has been determined for each mass feature, the intensities are often normalized according to the total ion chromatogram intensity for each LC-MS run, and various statistical analysis methods such as t-test and ANOVA can be applied to determine the statistical significance.
After proteoforms have been quantified for individual spectra, the mass and elution time can be used to match and compare proteoforms between LC-MS runs.19 However, in some label-free experiments, the need for relative comparison of proteoforms across LC-MS runs is unnecessary because the ratio of different proteoforms is directly compared in a single MS run rather than comparison of the same proteoform in different LC-MS runs.20–22 This comparison is often done at the MS1 level using the relative ratio of different post translationally modified proteoforms.22 In addition, mixed integer linear optimization (MILP), was developed by the Garcia group to identify and relatively quantify intact proteoforms with a large number of PTMs within the same LC-MS run.23 MILP compares ratios of fragment ions at the MS2 level for quantification of proteoform expression ratios. This label-free quantitation method was used to study expression ratios of highly modified histone proteins.20,21
When top-down label-free quantitation is implemented well, proteoform intensities can be compared across LC-MS runs and have been shown to have relatively high reproducibility with a coefficient of variation range from 8–20% for a simple mixture of standard proteins.24 The disadvantage of label-free quantitation is that run-to-run instrument variation strongly impacts the comparative analysis of the peaks. This means that the success of data analysis using label-free quantitation depends directly on the efficiency and reproducibility of the LC-MS data collection, which makes comparing a large number of runs challenging. Additionally, sample variation due to inconsistent sample handling and human error further decreases the reproducibility of this method. Label-free quantitation is also difficult to combine with 2D separation as it is challenging to accurately determine the sum intensities of proteins that span multiple fractions in the 1st dimension.13
Metabolic labelling
Metabolic labeling approaches were some of the first to be successfully applied to quantitative top-down MS-based proteomics studies. Metabolic labeling approaches are characterized by the isotopic labeling of proteins in vitro for comparative quantification of proteoforms expressed by cells cultured under different conditions.25 Metabolic labeling methods are popular in quantitative proteomics due to their ability to directly probe the protein state in vivo. There are three metabolic labeling techniques that have been applied to top-down MS: stable isotope labeling by amino acids in cell culture (SILAC),26–28 neutron encoding (NeuCode) SILAC,29,30 and tunable intact protein mass increases (TIPMI).31
SILAC is a metabolic labeling method where cells are cultured in media containing isotopically labeled amino acids to express ‘heavy’ (isotopically labeled) proteins. The amino acids in the media are incorporated into the proteins of the cell over several generations of cell cultures to produce proteins that are labeled to a high degree (495%).32 The labeled sample is combined with a sample that was grown in ordinary media to express ‘light’ (unlabeled) proteins for comparative quantitative analysis (Fig. 1B). SILAC growth media generally contains a labeled essential amino acid and may include carbon-13 or nitrogen-15 labeled lysine, arginine, leucine, methionine, or proline.33 Nonessential carbon-13 labeled tyrosine has also been introduced into SILAC growth media to study tyrosine kinases using SILAC.34 The mass difference between the ‘light’ and ‘heavy’ species is detected and proteoforms are quantified at the MS1 level by comparing the ratio of the labeled and unlabeled proteins. One of the disadvantages of traditional SILAC is the lack of multiplexing capabilities, as SILAC is generally only used to compare two experimental conditions. However, SILAC experiments have been performed with 5 isotopically distinct forms of arginine to study adipose tissue over several days of cell growth to display effective SILAC multiplexing.35
Neutron encoding (NeuCode) SILAC has more recently been introduced as a modified form of SILAC that incorporates the theory behind isobaric chemical tagging methods to allow for higher levels of SILAC multiplexing.36 In NeuCode SILAC, cells are cultured in media containing different isotopologues of the same amino acid with the same calculated mass. NeuCode SILAC utilizes the difference in mass defects of the various isotopologues of the isotopically labeled amino acids to distinguish between ‘heavy’ and ‘light’ proteoforms that have overall mass differences on the order of mDas.36 Comparative quantitation of the ‘heavy’ and ‘light’ proteoforms occurs at the MS1 level (Fig. 1B); however, as the mass differences of the isotopologues are very small, high resolution MS instrumentation is required to resolve the mDa mass differences induced using NeuCode labeled media. NeuCode SILAC addresses the lack of multiplexing capabilities of traditional SILAC and is potentially capable of very high levels of multiplexing. In fact, the multiplexing capability of NeuCode SILAC is more limited by the resolving power of modern MS instrumentation than the availability of amino acid isotopologues.37 The utility of Neucode SILAC in quantitative top-down MS has been demonstrated in the literature.29,30 The practicality of top-down Neucode SILAC has also been increased by the introduction of an open-source software package, Proteoform Suite, to assist in quantification of intact Neucode SILAC labeled proteins.38
Tunable intact protein mass increases (TIPMI) differs from SILAC techniques in that carbon-13 labeled sugar or deuterated water is spiked into media or feedstock to isotopically label proteins.31 As such, this method can be used to study animal models as well as cell and tissue cultures. Quijada et al. found that spiking carbon-13 labeled glucose into a nutrient rich media (2% w/v) increased the abundance of carbon-13 from a natural abundance of 1.1% to 9.2% resulting in a protein mass increase of approximately 0.4% which is analyzed via MS. Furthermore, the concentration of labeled glucose could be varied to produce different levels of isotopic labeling to produce multiplexed samples. As with SILAC methods, comparative quantitation occurs at the MS1 level. Currently, there are no publicly available computational analysis programs for top-down TIPMI and manual data analysis for complex samples is complicated and time consuming.
As the isotopically labeled species used to label the proteins must be tightly controlled to mediate the level of incorporation, the primary drawback of metabolic labeling techniques is that the samples must be cultured or grown in the laboratory. This means that samples which cannot be grown in the lab, such as clinical samples, cannot be metabolically labeled. Furthermore, incomplete metabolic labeling increases the complexity of MS spectra which decreases signal to noise ratio and complicates data analysis.
Chemical labeling
Though chemical labeling techniques are some of the most frequently used quantitative methods for bottom-up MS quantitation, only behind label-free quantitation, they are the newest addition to the quantitative top-down analysis methods.39 Chemical labeling techniques covalently modify peptides at specific amino acid residues, generally lysine residues, and the n-terminal domain. The lack of application of chemical labeling techniques to top-down MS analysis is due in part to the complication of data analysis caused by incomplete/nonspecific labeling, amino acid conversion, and side reactions.40 These challenges exist in bottom-up proteomics when chemical labeling methods are used, but are only worsened by analysis of long peptides/intact proteins. Additionally, current chemical labeling techniques are often not conducive to intact protein labeling because proteins precipitate under the labeling conditions. Currently, pseudoisobaric dimethyl labeling (pIDL) and tandem mass tag (TMT) labeling have been successfully applied to quantitative top-down proteomics; the use of these methods in top-down proteomics will be discussed here.
Pseudoisobaric labeling (pIDL) reacts lysine residues and N-terminal amines with various isotopologues of formaldehyde and sodium cyanoborohydride to dimethylate lysine residues to produce an isotopically unique group.41 By varying the identity and number of carbon-13 and hydrogen-2 isotopes on the reactants, 8-plex multiplexing can be accomplished using commercially available isotopes of formaldehyde and cyanoborohydride. Like NeuCode SILAC, pIDL relies on the mass defect of different isotopologues to differentiate labeled proteins, so this method requires contemporary high-resolution MS instrumentation for data analysis. Product ions with labeled lysine residues are identified in the MS2 and the ratio is determined for relative quantification of the intact protein (Fig. 1C). Fang et al. demonstrated the application of pIDL combined with top-down MS analysis on a myoglobin standard protein as well as hepatocellular carcinoma and normal hepatocellular cell lines.42,43
TMT and iTRAQ are chemical labeling methods that label specific chemical groups with isotopically labeled mass tags. These mass tags consist of four distinct chemical groups: mass reporter, linker, normalizer group, and reactive group.39 The reactive group, generally a hydroxysuccinimide ester, reacts with primary amines on the lysine residues and N-terminus of proteins. The mass tags all have the same mass, but hydrogen, carbon, or nitrogen isotopes are distributed across the mass reporter and normalizer groups. The linker is a cleavable portion of the mass tag between the mass reporter and normalizer group and is the site of cleavage during MS2 dissociation. As the mass of intact proteins is consistent before MS2 dissociation, quantitation is performed at the MS2 level. The mass reporter portions of the mass tags are cleaved from the peptides during MS2 dissociation and produce strong MS2 signals relative to their concentrations (i.e., the concentration of the bound protein in the sample). Relative quantitation is performed by mixing samples with unique mass tags at known ratios and the intensities of the mass tags are used to compare the concentrations of the intact protein.
Hung and Foley demonstrated the ability of TMT labeling to be effective in labeling intact proteins for top-down analysis in a simple mixture of standard proteins.40 Furthermore, Yu et al. have applied TMT labeling to complex cell lysates and, through the implementation of a high molecular weight cutoff filter, have minimized the precipitation under labeling conditions. While iTRAQ labeling of intact proteins has never been used for relative quantitation by MS, it has been used to label a simple mixture of intact proteins.44
Current and prospective applications
Determination of the identity and quantity of protein proteoforms by MS has become increasingly common as MS technology has improved and PTMs have been shown to affect the biological function of proteins. For example, heat shock protein 90 (Hsp90) is known to possess a variety of different PTMs including phosphorylation, acetylation, oxidation, etc., which have varying effects on the association of Hsp90 with client proteins.45 Practical quantitative top-down proteomics has been applied recently to quantify proteoform expression in human disease and aging, biological response to stimuli, and microbiology. Here we discuss some examples of the use of quantitative top-down proteomics techniques in these areas of research as well as prospective applications of these techniques.
Disease and aging
Determination of the effect of disease states on proteoform expression can be valuable in the study of disease pathways and the discovery of disease biomarkers. However, it is not always sufficient to qualitatively observe which proteoforms are expressed in diseased proteomes. Quantitative top-down proteomics methods have been applied to offer deeper understanding of diseased proteomes at a proteoform level. For example, the Ge group used label-free top-down proteomics methods to study proteoform expression of cardiac troponin I (cTnI) in heart tissues in early, mid, and late stage chronic heart failure (CHF). They determined that a higher ratio of phosphorylated cTnI, compared to nonphosphorylated cTnI, was observed in CHF samples, which may serve as an indicator of cardiac health. Therefore, quantitation of phosphorylated cTnI can be used as a diagnostic method for CHF.22 The Ge group further used top-down label-free quantitation methods to examine the skeletal muscle of aging rats to study the molecular mechanism of sarcopenia.46 The Kelleher group utilized top-down label-free quantitation methods to study regulatory mechanisms in human colorectal cancer cells.47 They developed a strategy to enrich KRAS proteins so they could determine the effect of mutations on KRAS proteoform expression. They found that a mutated DLD-1 cell line, with a G13D mutation, expressed a unique KRAS4b proteoform when compared with the wild type, which suggests that post-translational modifications of KRAS proteins affect cell growth and proliferation pathways. Additionally, quantitative top-down proteomics methods have been used to study Parkinson’s disease,48 autoimmune diseases,49 and the maturation of human pluripotent stem cell-derived cardiomyocytes.50
Saliva
Saliva is a valuable source to search for known and potential biomarkers because it is easier and less invasive to collect that other biological samples like blood or cerebral spinal fluid.51 Top-down MS has been applied to quantitatively study the salivary proteome in search of potential biomarkers that could lead to noninvasive methods for early disease diagnosis and disease monitoring.19 In fact, quantitative top-down proteomics have revealed differentially expressed proteins and potential biomarkers in saliva samples from patients afflicted with schizophrenia and bipolar disorder,52 multiple sclerosis,53 edentulism,54 and early onset Alzheimer’s disease as it is associated with down syndrome.55
Cellular response
The study of the cellular response to external stimuli at the proteoform level is critical to understanding how specific proteoforms are involved in the initiation of cellular pathways and mechanisms, and may also offer insight into the effects of chemical treatment on the proteome (e.g., the effect of pharmaceuticals on cellular proteomes). Quantitative top-down proteomics methods have been used to study highly modified proteins such as histones,21 the effect of mechanical stimulation on mice,56 exercise on humans,57 and heat on disulfide bonds of b-lactoglobulin.58 For example, the Young lab utilized MILP to accurately quantify highly modified proteoforms such as histone proteins to study the effect of SUV4–20 methyltransferase inhibition on breast cancer cell lines.21 Furthermore, they studied histone acetylation upon addition of sodium butyrate and discovered a highly specific hierarchical order of acetylation that occurred exclusively on histone proteins with a pre-existing H4K20me2 modification.20 The importance of the order of acetylation, while it had been previously hypothesized, could not previously be observed using bottom-up MS or antibody-based methods.
Microbiology
The study of intact microbiological proteomes can offer valuable information on the pathways and mechanisms of infection and disease and help inform treatment options. For example, Chamot-Rooke et al. used top-down MS to study phosphorylation of type IV pili in Neisseria meningitidis.59 They identified a phosphoglycerol transferase responsible for the transfer of a phosphoglycerol PTM to Ser93 of the PilE protein. The addition of this PTM allows the pili to release from a bacterial aggregate where replication occurs, which allows the bacteria to infect new hosts. Ansong et al. used quantitative top-down MS proteomics to study proteoform expression of Salmonella typhimurium under normal and infection-like conditions.60 They found the first instance of a condition dependent PTM switch, from an S-glutathionylation under basal growth conditions to S-cysteinylation under infection-like conditions, which may inform cellular response in redox regulation.
Future directions and prospectives
Top-down proteomics methods have seen a rise in popularity as MS technology has improved; however, there are still drawbacks to the use of top-down compared to bottom-up methods. These drawbacks primarily exist in two areas: (1) large, highly charged proteins are difficult to detect as a result of MS m/z limitations and low signal intensity due to charge envelope broadening. These limitations lead to only the lowest molecular weight and most abundant proteoforms being identified. (2) Coelution of intact proteins after LC separation also complicates MS spectra and decreases ion intensity making the detection of high molecular weight and low abundance proteoforms difficult.16 Fortunately, MS technology and methods that seek to improve qualitative protein identification of intact proteoforms have been successful. For example, parallel ion parking and ion/ion proton transfer reactions methods were designed to improve the signal-to-noise ratio of low intensity MS signals by mediating the number and level of charge states produced during the ESI process so that the protein may be analyzed within the available m/z range.61,62 This also decreases spectral complexity and increases the signal-to-noise ratio of proteins in complex samples. Another emerging MS technology that may be applied to quantitative top-down MS is data-independent analysis (DIA). DIA contrasts with data dependent analysis (DDA) in that specific precursor masses are not chosen for fragmentation.63 Rather, repeating m/z ranges are sequentially isolated and sent for fragmentation and MS detection during the full length of separation.63 Using DDA, multiple parent ions may be selected for fragmentation and MS spectra may be very complicated; thus, many software packages have been developed to analyze mass spectra produced using DIA methods.64–66 Application of parallel ion parking, ion/ion transfer, and DIA to top-down proteomics could help overcome some of the difficulties in envelope broadening, spectral complexity, and detection of low abundance proteoforms that have been inherent to top-down proteomics.
A further decrease in the spectral complexity caused by coelution of proteoforms for MS analysis has been achieved by increased separation efficiency. Specifically, highly efficient and multidimensional separation approaches have been developed and implemented to better separate intact proteins and increase qualitative proteoform identification and quantitative analysis.5–11 Increasing separation efficiency in quantitative top-down studies should lead to deeper and more accurate proteoform identification and quantitation. However, multidimensional separation techniques do present a disadvantage, particularly in quantitative methods, in that proteins may elute over multiple 1st-dimension fractions and become difficult to accurately quantify. Label-free quantitation methods are especially unsuitable for multidimensional separation as quantitation must occur between LC-MS runs for individual samples. The increased number of LC-MS runs for a single sample in multidimensional separation increases the opportunity for run-to-run variability and decreases the accuracy of label-free quantitation methods. Moreover, one proteoform can elute across different fractions which complicates quantitation of proteoforms because multiple second dimension LC runs must be combined. Metabolic and chemical labeling methods, however, are much more suitable for quantitative analysis in multidimensional separation as the labeled proteoforms from different samples can be combined prior to separation and directly compared within the same LC run.
Quantitative top-down MS has been well established in the identification of proteoforms for biomarker discovery and to study biochemical disease pathways as discussed above. Another potential application of quantitative top-down proteomics is in the area of structural biology. Traditionally, structural elucidation of biological macromolecules has been accomplished using nuclear magnetic resonance (NMR),67 X-ray crystallography,68 cryogenic electron microscopy (cryo-EM),69 or a combination of these techniques.70 Computational structural biology has also been widely implemented independently and in conjunction with physical experiments to probe the structure of biological macromolecules.71 The structural biology methods discussed above have a common drawback in that the proteins must be purified to use these methods. In many cases, these proteins must be bacterially expressed and purified to be obtained in a high enough quantity to use these methods, and key PTMs may be missing. MS-based methods for structural biology have been implemented to overcome some of these difficulties. A primary advantage of the use of MS in structural biology is the ability to analyze complex sample matrices by coupling MS methods to high resolution separation techniques. This union decreases sample complexity, allows for the collection of data on multiple peptides/proteins simultaneously, and uses highly sensitive MS detection. For example, MS-based ‘footprinting’ methods including hydrogen deuterium exchange (HDX), hydroxyl radical footprinting (HRF), and chemical crosslinking72,73 have been used to study aspects of protein structure and interactions. These footprinting methods have generally been applied using bottom-up MS methods; however, a recent application of top-down MS to HRF suggested the potential of top-down ‘footprinting’ approaches in structural biology.74
Another area of research where quantitative top-down proteomics may be applied to examine protein structure and interaction is to some recently introduced techniques that probe protein stability called ‘stability proteomics’.73 One of these techniques, thermal proteome profiling (TPP), examines the resistance of a protein to thermal denaturation and has been notably used to determine protein drug targets.75 TPP can be paired with quantitative top-down MS proteomics methods to determine the effect of PTMs on the thermal stability of the proteins. Furthermore, quantitative top-down TPP can be applied to drug target analysis to determine if specific proteoforms are active in drug binding.
Quantitative top-down techniques have the potential to greatly increase the scope of the proteomics field. Already, quantitative top-down techniques have been used to identify biomarkers, study human and bacterial disease pathways, and observe cellular response to stimuli. We believe that further improvement on and application of MS instrument technologies, data collection/analysis techniques, and advanced separation techniques such as those suggested in this review to top-down proteomics will propel the field forward and lead to many new discoveries.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Zhang Y, et al. , Protein Analysis by Shotgun/Bottom-up Proteomics, Chem. Rev, 2013, 113(4), 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burkhart JM, et al. , Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics, J. Proteomics, 2012, 75(4), 1454–1462. [DOI] [PubMed] [Google Scholar]
- 3.Kelleher NL, Top-down proteomics, Anal. Chem, 2004, 76(11), 196A–203A. [PubMed] [Google Scholar]
- 4.Loo JA, et al. , High-resolution tandem mass spectrometry of large biomolecules, Proc. Natl. Acad. Sci. U. S. A, 1992, 89(1), 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiu L, et al. , Effective Protein Separation by Coupling Hydrophobic Interaction and Reverse Phase Chromatography for Top-down Proteomics, Anal. Chem, 2014, 86(15), 7899–7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, et al. , Two-dimensional separation using high-pH and low-pH reversed phase liquid chromatography for top-down proteomics, Int. J. Mass Spectrom, 2018, 427, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCool EN, et al. , Deep Top-Down Proteomics Using Capillary Zone Electrophoresis-Tandem Mass Spectrometry: Identification of 5700 Proteoforms from the Escherichia coli Proteome, Anal. Chem, 2018, 90(9), 5529–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lubeckyj RA, et al. , Large-Scale Qualitative and Quantitative Top-Down Proteomics Using Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry with Nanograms of Proteome Samples, J. Am. Soc. Mass Spectrom, 2019, 30, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JE, et al. , A Robust Two-Dimensional Separation for Top-Down Tandem Mass Spectrometry of the Low-Mass Proteome, J. Am. Soc. Mass Spectrom, 2009, 20(12), 2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucholski T, et al. , A Top-Down Proteomics Platform Coupling Serial Size Exclusion Chromatography and Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, Anal. Chem, 2019, 91(6), 3835–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vellaichamy A, et al. , Size-Sorting Combined with Improved Nanocapillary Liquid Chromatography-Mass Spectrometry for Identification of Intact Proteins up to 80 kDa, Anal. Chem, 2010, 82(4), 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen B, et al. , Correction to Top-Down Proteomics: Ready for Prime Time?, Anal. Chem, 2018, 90(24), 14643. [DOI] [PubMed] [Google Scholar]
- 13.Schaffer LV, et al. , Identification and Quantification of Proteoforms by Mass Spectrometry, Proteomics, 2019, 19(10), e1800361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catherman AD, Skinner OS and Kelleher NL, Top Down proteomics: Facts and perspectives, Biochem. Biophys. Res. Commun, 2014, 445(4), 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donnelly DP, et al. , Best practices and benchmarks for intact protein analysis for top-down mass spectrometry, Nat. Methods, 2019, 16(7), 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Compton PD, et al. , On the Scalability and Requirements of Whole Protein Mass Spectrometry, Anal. Chem, 2011, 83(17), 6868–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ntai I, et al. , Applying Label-Free Quantitation to Top Down Proteomics, Anal. Chem, 2014, 86(10), 4961–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J, et al. , Informed-Proteomics: open-source software package for top-down proteomics, Nat. Methods, 2017, 14(9), 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, et al. , Quantitative analysis of human salivary glandderived intact proteome using top-down mass spectrometry, Proteomics, 2014, 14(10), 1211–1222. [DOI] [PubMed] [Google Scholar]
- 20.Wang T, et al. , Early butyrate induced acetylation of histone H4 is proteoform specific and linked to methylation state, Epigenetics, 2018, 13(5), 519–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang T, et al. , The histone H4 proteoform dynamics in response to SUV4–20 inhibition reveals single molecule mechanisms of inhibitor resistance, Epigenet. Chromatin, 2018, 11(1), 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, et al. , Top-Down Quantitative Proteomics Identified Phosphorylation of Cardiac Troponin I as a Candidate Biomarker for Chronic Heart Failure, J. Proteome Res, 2011, 10(9), 4054–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiMaggio PA, et al. , A Mixed Integer Linear Optimization Framework for the Identification and Quantification of Targeted Post-translational Modifications of Highly Modified Proteins Using Multiplexed Electron Transfer Dissociation Tandem Mass Spectrometry, Mol. Cell. Proteomics, 2009, 8(11), 2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ntai I, et al. , Applying Label-Free Quantitation to Top Down Proteomics, Anal. Chem, 2014, 86(10), 4961–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du Y, et al. , Top-Down Approaches for Measuring Expression Ratios of Intact Yeast Proteins Using Fourier Transform Mass Spectrometry, Anal. Chem, 2006, 78(3), 686–694. [DOI] [PubMed] [Google Scholar]
- 26.Collier TS, et al. , Top-Down Identification and Quantification of Stable Isotope Labeled Proteins from Aspergillus flavus Using Online Nano-Flow Reversed-Phase Liquid Chromatography Coupled to a LTQ-FTICR Mass Spectrometer, Anal. Chem, 2008, 80(13), 4994–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collier TS, et al. , Quantitative Top-Down Proteomics of SILAC Labeled Human Embryonic Stem Cells, J. Am. Soc. Mass Spectrom, 2010, 21(6), 879–889. [DOI] [PubMed] [Google Scholar]
- 28.Waanders LF, Hanke S and Mann M, Top-down quantitation and characterization of SILAC-labeled proteins, J. Am. Soc. Mass Spectrom, 2007, 18(11), 2058–2064. [DOI] [PubMed] [Google Scholar]
- 29.Rhoads TW, et al. , Neutron-Encoded Mass Signatures for Quantitative Top-Down Proteomics, Anal. Chem, 2014, 86(5), 2314–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shortreed MR, et al. , Elucidating Proteoform Families from Proteoform Intact-Mass and Lysine-Count Measurements, J. Proteome Res, 2016, 15(4), 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quijada JV, et al. , Heavy Sugar and Heavy Water Create Tunable Intact Protein Mass Increases for Quantitative Mass Spectrometry in Any Feed and Organism, Anal. Chem, 2016, 88(22), 11139–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ong S-E, et al. , Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics, Mol. Cell. Proteomics, 2002, 1(5), 376. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, et al. , Quantitative proteomics using SILAC: Principles, applications, and developments, Proteomics, 2015, 15(18), 3175–3192. [DOI] [PubMed] [Google Scholar]
- 34.Ibarrola N, et al. , A Novel Proteomic Approach for Specific Identification of Tyrosine Kinase Substrates Using [13C]-Tyrosine, J. Biol. Chem, 2004, 279(16), 15805–15813. [DOI] [PubMed] [Google Scholar]
- 35.Molina H, et al. , Temporal Profiling of the Adipocyte Proteome during Differentiation Using a Five-Plex SILAC Based Strategy, J. Proteome Res, 2009, 8(1), 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merrill AE, et al. , NeuCode Labels for Relative Protein Quantification, Mol. Cell. Proteomics, 2014, 13(9), 2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hebert AS, et al. , Neutron-encoded mass signatures for multiplexed proteome quantification, Nat. Methods, 2013, 10, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cesnik AJ, et al. , Proteoform Suite: Software for Constructing, Quantifying, and Visualizing Proteoform Families, J. Proteome Res, 2018, 17(1), 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Couto N, et al. , Making sense out of the proteome: the utility of iTRAQ and TMT, New Dev. Mass Spectrom, 2014, 1, 51–79, (quantitative proteomics). [Google Scholar]
- 40.Hung C-W and Tholey A, Tandem Mass Tag Protein Labeling for Top-Down Identification and Quantification, Anal. Chem, 2012, 84(1), 161–170. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Y, et al. , Mass Defect-Based Pseudo-Isobaric Dimethyl Labeling for Proteome Quantification, Anal. Chem, 2013, 85(22), 10658–10663. [DOI] [PubMed] [Google Scholar]
- 42.Fang H, et al. , Intact Protein Quantitation Using Pseudoisobaric Dimethyl Labeling, Anal. Chem, 2016, 88(14), 7198–7205. [DOI] [PubMed] [Google Scholar]
- 43.Liu Z, et al. , Global Quantification of Intact Proteins via Chemical Isotope Labeling and Mass Spectrometry, J. Proteome Res, 2019, 18(5), 2185–2194. [DOI] [PubMed] [Google Scholar]
- 44.Wiese S, et al. , Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research, Proteomics, 2007, 7(3), 340–350. [DOI] [PubMed] [Google Scholar]
- 45.Mollapour M and Neckers L, Post-translational modifications of Hsp90 and their contributions to chaperone regulation, Biochim. Biophys. Acta, Mol. Cell Res, 2012, 1823(3), 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei L, et al. , Novel Sarcopenia-related Alterations in Sarcomeric Protein Post-translational Modifications (PTMs) in Skeletal Muscles Identified by Top-down Proteomics, Mol. Cell. Proteomics, 2018, 17(1), 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ntai I, et al. , Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk, Proc. Natl. Acad. Sci. U. S. A, 2018, 115(16), 4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kellie JF, et al. , Quantitative Measurement of Intact Alpha-Synuclein Proteoforms from Post-Mortem Control and Parkinson’s Disease Brain Tissue by Intact Protein Mass Spectrometry, Sci. Rep, 2014, 4, 5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Z, et al. , Top-down Mass Spectrometry Analysis of Human Serum Autoantibody Antigen-Binding Fragments, Sci. Rep, 2019, 9(1), 2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai W, et al. , Unbiased Proteomics Method to Assess the Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes, Circ. Res, 2019, 125, 936–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malamud D, Saliva as a Diagnostic Fluid, Dent. Clin. North Am, 2011, 55(1), 159–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iavarone F, et al. , Characterization of salivary proteins of schizophrenic and bipolar disorder patients by top-down proteomics, J. Proteomics, 2014, 103, 15–22. [DOI] [PubMed] [Google Scholar]
- 53.Manconi B, et al. , Top-down proteomic profiling of human saliva in multiple sclerosis patients, J. Proteomics, 2018, 187, 212–222. [DOI] [PubMed] [Google Scholar]
- 54.Manconi B, et al. , Top-down HPLC-ESI–MS proteomic analysis of saliva of edentulous subjects evidenced high levels of cystatin A, cystatin B and SPRR3, Arch. Oral Biol, 2017, 77, 68–74. [DOI] [PubMed] [Google Scholar]
- 55.Cabras T, et al. , Significant Modifications of the Salivary Proteome Potentially Associated with Complications of Down Syndrome Revealed by Top-down Proteomics, Mol. Cell. Proteomics, 2013, 12(7), 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moehring F, et al. , Quantitative Top-Down Mass Spectrometry Identifies Proteoforms Differentially Released during Mechanical Stimulation of Mouse Skin, J. Proteome Res, 2018, 17(8), 2635–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurgan N, et al. , Changes to the Human Serum Proteome in Response to High Intensity Interval Exercise: A Sequential Top-Down Proteomic Analysis, Front. Physiol, 2019, 10, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhan L, et al. , Heat-Induced Rearrangement of the Disulfide Bond of Lactoglobulin Characterized by Multiply Charged MALDI-TOF/TOF Mass Spectrometry, Anal. Chem, 2018, 90(18), 10670–10675. [DOI] [PubMed] [Google Scholar]
- 59.Chamot-Rooke J, et al. , Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination, Science, 2011, 331(6018), 778–782. [DOI] [PubMed] [Google Scholar]
- 60.Ansong C, et al. , Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions, Proc. Natl. Acad. Sci. U. S. A, 2013, 110(25), 10153–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chrisman PA, Pitteri SJ and McLuckey SA, Parallel Ion Parking of Protein Mixtures, Anal. Chem, 2006, 78(1), 310–316. [DOI] [PubMed] [Google Scholar]
- 62.Stephenson JL and McLuckey SA, Ion/Ion Proton Transfer Reactions for Protein Mixture Analysis, Anal. Chem, 1996, 68(22), 4026–4032. [DOI] [PubMed] [Google Scholar]
- 63.Gillet LC, et al. , Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis, Mol. Cell. Proteomics, 2012, 11(6), O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rardin MJ, et al. , MS1 Peptide Ion Intensity Chromatograms in MS2 (SWATH) Data Independent Acquisitions. Improving Post Acquisition Analysis of Proteomic Experiments, Mol. Cell. Proteomics, 2015, 14(9), 2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bruderer R, et al. , New targeted approaches for the quantification of data-independent acquisition mass spectrometry, Proteomics, 2017, 17(9), 1700021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenberger G, et al. , Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses, Nat. Methods, 2017, 14, 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wuthrich K, Protein structure determination in solution by nuclear magnetic resonance spectroscopy, Science, 1989, 243(4887), 45. [DOI] [PubMed] [Google Scholar]
- 68.Garman EF, Developments in X-ray Crystallographic Structure Determination of Biological Macromolecules, Science, 2014, 343(6175), 1102. [DOI] [PubMed] [Google Scholar]
- 69.Nogales E, The development of cryo-EM into a mainstream structural biology technique, Nat. Methods, 2015, 13, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cerofolini L, et al. , Integrative Approaches in Structural Biology: A More Complete Picture from the Combination of Individual Techniques, Biomolecules, 2019, 9(8), 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nussinov R, et al. , Computational Structural Biology: Successes, Future Directions, and Challenges, Molecules, 2019, 24(3), 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Calabrese AN and Radford SE, Mass spectrometry-enabled structural biology of membrane proteins, Methods, 2018, 147, 187–205. [DOI] [PubMed] [Google Scholar]
- 73.Kaur U, et al. , Proteome-Wide Structural Biology: An Emerging Field for the Structural Analysis of Proteins on the Proteomic Scale, J. Proteome Res, 2018, 17(11), 3614–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu XR, et al. , A Single Approach Reveals the Composite Conformational Changes, Order of Binding, and Affinities for Calcium Binding to Calmodulin, Anal. Chem, 2019, 91(9), 5508–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Savitski MM, et al. , Tracking cancer drugs in living cells by thermal profiling of the proteome, Science, 2014, 346(6205), 1255784. [DOI] [PubMed] [Google Scholar]