

Abstract
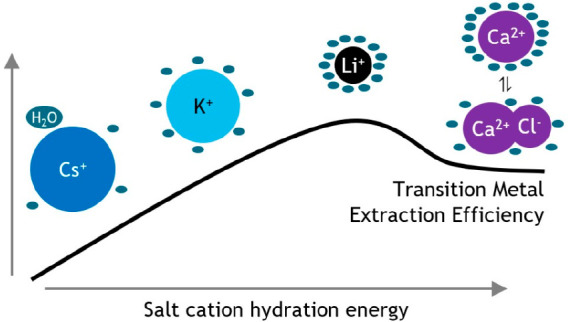
The addition of a nonextractable salt has an important influence on the solvent extraction of metal ions, but the underlying principles are not completely understood yet. However, relating solute hydration mechanisms to solvent extraction equilibria is key to understanding the mechanism of solvent extraction of metal ions as a whole. We have studied the speciation of Co(II), Zn(II), and Cu(II) in aqueous solutions containing different chloride salts to understand their extraction to the basic extractant methyltrioctylammonium chloride (TOMAC). This includes the first speciation profile of Zn(II) in chloride media with the three Zn(II) species [Zn(H2O)6]2+, [ZnCl3H2O]−, and [ZnCl4]2–. The observed differences in extraction efficiency for a given transition metal ion can be explained by transition metal ion hydration due to ion–solvent interactions, rather than by ion–solute interactions or by differences in speciation. Chloride salting agents bearing a cation with a larger hydration Gibbs free energy reduce the free water content more, resulting in a lower hydration for the transition metal ion. This destabilizes the transition metal chloro complex in the aqueous phase and increases the extraction efficiency. Salting agents with di- and trivalent cations reduce the transition metal chloro complex hydration less than expected, resulting in a lower extraction efficiency. The cations of these salting agents have a very large hydration Gibbs free energy, but the overall hydration of these salts is reduced due to significant salt ion pair formation. The general order of salting-out strength for the extraction of metal ions from chloride salt solutions is Cs+ < Rb+ < NH4+ ≈ K+ < Al3+ ≈ Mg2+ ≈ Ca2+ ≈ Na+ < Li+. These findings can help in predicting the optimal conditions for metal separation by solvent extraction and also contribute to a broader understanding of the effects of dissolved salts on solutes.
Short abstract
Addition of a nonextractable salt influences the stability and solvent extraction efficiency of metal complexes. Cations of different chloride salts reduce the solution free water content as a function of their increasing hydration energy and decreasing tendency for ion pair formation with chloride anions. These ion−solvent interactions reduce the hydration of metal complexes, increasing their distribution ratios. These effects influence aqueous transition metal complexes more than direct ion−solute interactions and changes in complex speciation.
1. Introduction
Solvent extraction (liquid–liquid extraction) is often used in industry for the separation and purification of metals.1 In this process, an aqueous phase comprising a mixture of metal ions is mixed with an immiscible organic phase comprising an extractant, a diluent, and sometimes a modifier. The extractant plays a key role as it is an organic ligand that selectively coordinates to the targeted metal ion, transferring it to the organic phase. The three main classes of extractants are (1) acidic extractants (e.g., dialkyl phosphorus acids or carboxylic acids),2,3 (2) solvating extractants (e.g., ketones or organophosphorus esters),4,5 and (3) basic extractants (e.g., protonated amines or quaternary ammonium salts).6,7
For the solvating and basic extractants, the addition of a salt or acid to the aqueous phase is required to efficiently extract metal ions to the organic phase. To have metal ions extracted by solvating extractants, a neutral metal complex should be formed by balancing the charge of the metal ion with counteranions provided by the added salt or acid.8 The formed metal–ligand ion pairs can then be solvated in the organic phase by the solvating extractant molecules in the first or second coordination sphere. The extraction efficiency is determined by the ability to form metal–ligand ion pairs in the aqueous phase, the ability to form a stable metal-extractant complex in the organic phase, and by the capacities of both phases to solvate the extractable metal complexes.
Recently, we have shown that solvent extraction of metal ions by basic extractants functions similarly to solvating extractants when only considering the aqueous phase: the least hydrated metal complex is extracted preferentially.9 The complexation of salt or acid anions to a metal cation results in metal–ligand complexes with different charge densities. The most efficient extraction can be found at the salt or acid concentration where the metal complex with the lowest charge density is preferably formed. This effect generally results in a maximum extraction efficiency at intermediate salt or acid concentrations. Nevertheless, a significant difference between solvating and basic extractant can be observed in the organic phase or at the interface. While the solvating extractant only reacts with the metal salt to form an adduct, the basic extractant anion reacts with the metal salt to form negatively charged metal complexes that electrostatically interact with the basic extractant cation
| 1 |
where [QX] is a basic extractant, and the overbars represent organic species.
However, not only the metal-coordinating ability of the salt or acid anions plays an important role in the extraction behavior of metal ions to solvating and basic extractants. Besides complexation, the choice of salt cation and anion has major effects on compounds in aqueous solution, as first described by Hofmeister in 1888.10 He devised a series that ranks ions according to their influence on the solubility of proteins in aqueous solutions. Salting-in ions enhance the dissolution of a protein, while salting-out ions decrease the solubility of a protein.
Since then, the effects of salts on a whole range of solute properties have been studied in a variety of domains, including biological systems,11−13 geochemistry,14,15 and chemical separations.16−20 In recent years, a renewed interest in salting-in/out effects has led to extensive research in the molecular mechanisms behind the observed effects of dissolved salts on other solutes. Both ion–solute and ion–solvent interactions must be taken into account to explain the observations, but the relative importance of both interaction types varies in different cases.21−23
Although the effects of metal complexation on solvent extraction of metals are rather well understood, the underlying principles governing the salting effects on solvent extractions are not completely clear yet.20,24,25 Some studies have already been performed, predicting salting effects on the extraction of metal ions from nitrate media by solvating extractants.26,27 These studies link salt activity coefficients to the extraction efficiency of a metal ion by a solvating extractant. However, they are unable to explain the observed effects when nitrate salts of di- and trivalent cations are used or when the ionic strength is higher than 5 molal. Efforts have already been made to elucidate the mechanisms behind salting effects of salt anions in solvent extraction systems with basic extractants but less so for the salt cations.16,17,19 However, relating the underlying mechanisms of solute hydration to solvent extraction observations is key to understanding solvent extractions of metal ions as a whole.28 Changing the cation and the concentration of a salting agent are also two rather simple ways to fine-tune the extraction efficiency of metal ions and optimize the separation of metals from a complex feed solutions. Therefore, a better understanding of the origin of the salting effect of cations on the solvent extraction of metal ions is necessary to predict the optimal conditions for metal ion extractions and separations with solvating and basic extractants.
To assess the underlying mechanisms of salting effects on the extraction of metal ions, we studied the speciation of Co(II), Zn(II), and Cu(II) and their extraction to methyltrioctylammonium chloride (TOMAC) in the presence of different chloride salting agents. This paper links metal-complex and salting agent hydration and association to the trends in solvent extraction of transition metals by basic extractants, and it also aims to contribute to a broader understanding in the effects of dissolved salts on solutes.
2. Experimental Section
2.1. Chemicals
HNO3 (65 wt %), NaCl (99.99%), LiCl (99.9%), NaOH (0.1 M), HCl (∼37 wt %), AlCl3·6H2O (>99%), CaCl2·2H2O (>99%), and toluene were purchased from VWR (Leuven, Belgium). CuCl2 (99.995%) and Zn(ClO4)2·6H2O (99.7%) were purchased from Sigma-Aldrich (Overijse, Belgium). The copper, cobalt, scandium, indium, and zinc aqueous standards (1000 mg L–1 in 3–5% HNO3), CoCl2·6H2O (>98%), ZnCl2 (>98%), and MgCl2·6H2O (>99%) were obtained from Chem Lab (Zedelgem, Belgium). Methyltrioctylammonium chloride (TOMAC, 98%) was purchased from J&K Scientific (Lommel, Belgium). RbCl (>99.8%) and NaClO4·H2O (>99%) were purchased from Acros Organics (Geel, Belgium). Water was always of ultrapure quality, deionized to a conductivity of <0.055 μS cm–1 (298.15 K) with a Merck Millipore Milli-Q Reference A+ system. All chemicals were used as received, without any further purification.
2.2. Solvent Extraction of Metals and Quantification
Single element solvent extractions (Co(II), Cu(II), or Zn(II)) were performed with 1.0 mL of aqueous and organic phase in glass vials with a volume of 4 mL. The metal concentration in the aqueous phase was kept constant by adding a fixed volume of an aqueous metal stock solution (1.7 mol L–1 CoCl2, 1.6 mol L–1 CuCl2, or 1.5 mol L–1 ZnCl2 in water) to an aliquot of a highly concentrated acid or salt solution (HCl, LiCl, NaCl, RbCl, MgCl2, CaCl2, or AlCl3), which was subsequently diluted with a certain volume of ultrapure water to a total volume of 10 mL. The final concentrations of Co(II), Cu(II), or Zn(II) in the aqueous samples were 17.0 mmol L–1, 7.9 mmol L–1, and 30.6 mmol L–1, respectively. The salt solutions were prepared working close to the solubility limit. The exact salt concentrations were calculated based on the densities of the prepared solutions to avoid weighing errors due to the uptake of water by the hygroscopic salts.30 The concentrations of the salt solutions of AlCl3 were calculated based on the mass of the dissolved solid because of the unavailability of data concerning the density–concentration relationship. HCl (0.05 mol L–1) was added to the AlCl3 salt solution to avoid hydrolytic precipitation of Al(III), and 0.0225 mol L–1 HCl was added to all Zn(II) aqueous phases to avoid hydrolytic precipitation of Zn(II) during extraction.
Methyltrioctylammonium chloride (TOMAC) was diluted with toluene to create an organic phase with a concentration of 0.2 M. All extractions were performed by shaking at 2000 rpm for 30 min at room temperature, and phase separation was accomplished by centrifugation for 2 min at 5000 rpm. The metal concentration in the aqueous phase before and after extraction was measured using inductively coupled plasma atomic emission spectroscopy (ICP-OES). Distribution ratios (D) were calculated with the following formula
| 2 |
where cM,aq and cM,org are the equilibrium metal concentrations in the aqueous and organic phase after extraction, respectively. The concentration of metal in the organic phase was calculated via the mass balance. The experimental error was calculated based on triplicate measurements and was less than 5%. Error bars on graphs were omitted to increase legibility.
2.3. Instrumentation and Analysis Methods
Raman spectra of 0.05 mol L–1 aqueous ZnClO4 solutions in different LiCl concentrations were recorded using a confocal Horiba Jobin Yvon LabRam HR Evolution Raman microscope. HCl (0.02 mol L–1 ) was added to all samples to prevent hydrolysis of Zn(II). The spectrometer is equipped with an 1800 grooves/mm holographic grating with a confocal attachment, a Peltier-cooled electron multiplying CCD (SIN-EM FIVIS) for detection, and an Olympus BX41 microscope. For excitation, a green line (λ = 532.0 nm) from a solid-state laser (LAS-532-100-HREV) operating at 14 mW was employed. Raman spectra of liquid samples were collected at room temperature in a glass cuvette (4 mL) using a ×10_VIS_LWD microscope objective to focus the laser beam at the center of the cuvette. Each spectral scan was collected with an acquisition time of 10 s using 30 accumulations. To enhance the spectra between 300 and 500 cm–1, an acquisition time of 30 s, 300 accumulations, and an electron multiplying gain of 250 were used. Solutions with the same composition, except for the presence of ZnClO4, were measured and subtracted from the spectra of the ZnClO4-containing samples to correct for the background signal. The spectra were processed with the LabSpec6 software from Horiba and the Origin 2018b software from OriginLab. The perchlorate peak at 462 cm–1 was used as an internal standard to normalize all spectra. The processed spectra contained a significant amount of noise, due to the low intensity of the Raman signal of the measured Zn(II) complexes. Thus, the processed spectra were smoothed using the locally weighted least-squares (LOESS) method.
UV–vis absorption spectra of the aqueous CoCl2 solutions were measured with an Agilent Cary 6000i spectrophotometer and Cary WinUV software in a quartz Suprasil cuvette with a path length of 1.0 cm. Baseline corrections were carried out with solutions with the same composition as the sample but without CoCl2.
Metal ion concentrations were determined by ICP-OES with a PerkinElmer Avio 500 spectrometer equipped with an axial/radial dual plasma view, a GemCone High Solids nebulizer, a baffled cyclonic spray chamber, and a demountable quartz torch with a 2.0 mm internal diameter alumina injector. Samples, calibration solutions, and quality control solutions were diluted with HNO3 (2 vol %). All ICP-OES spectra were measured in triplicate. Calibration curves were made using a solution of 0.1, 1, and 10 mg L–1 of the corresponding metal from a standard solution. Quality checks were performed with 5 mg L–1 metal solutions. In(III) or Sc(III) was added and applied as an internal standard only if the quality checks failed because of matrix effects.
Densities of the acid and salt solutions were measured with an Anton Paar DMA 4500 M densitometer. Nemus Life Thermo Shakers TMS-200 were used for the extraction experiments. A Heraeus Labofuge 200 centrifuge was used to accelerate phase separation.
2.4. Speciation Calculations
The UV–vis absorption spectra of Co(II) were analyzed following the Multivariate Curve Resolution-Alternating Least Square (MCR-ALS) technique using the MCR-ALS GUI 2.0 toolbox working in Matlab R2018b software.32 Chemical constraints were fixed to a minimum: (1) the total amount of the metal was normalized, (2) the independent UV–vis absorption spectra do not have negative absorbance values, and (3) the concentrations of the species cannot be negative. Principal component analysis (PCA) (using the same toolbox in Matlab) was performed prior to the MCR-ALS study to determine how many different species were present in the solutions, and the results were compared with the literature.
The results of the MCR-ALS analysis are displayed as the UV–vis absorption spectra of the independent metal species present in all samples containing that metal ion and as the mole fraction of all species in function of the acid or salt concentration. The mole fraction of a metal species (M2+) is expressed as follows
| 3 |
where m is the maximum amount of chlorides coordinated to M2+, and n is the number of moles of a given species.
Note that this mathematical treatment of the data is not based on any chemical model. Therefore, ascribing a UV–vis absorption spectrum to a certain metal complex is done by considering the change in the concentration percentages as a function of the acid or salt concentration and/or by comparison with previously published UV–vis absorption spectra.33
3. Results and Discussion
3.1. Extraction of Zn(II) from Monovalent Salting Agents
The extraction efficiency of 30.6 mmol L–1 Zn(II) from different aqueous chloride media to 0.2 mol L–1 TOMAC in toluene decreases when going from small to large alkali cations.9 The effect of the different alkali salts on the extraction of Zn(II) was interpreted by linking metal complex hydration to metal complex extraction. At high salt concentrations (above 2 mol L–1), more anions than the oxidation state of Zn(II) are coordinated to the aqueous metal ion, resulting in a negatively charged chloro complex. This would decrease the extraction efficiency of the Zn(II) complexes, as its higher charge density results in a better hydration and thus better stabilization in the aqueous phase. However, alkali salt cations with a high charge density would be significantly hydrated and thus decrease the amount of free water molecules available for the hydration of the anionic Zn(II) complex at high salt concentrations. The less hydrated anionic Zn(II) complexes are then extracted rather efficiently as their stabilization in the aqueous phase is reduced significantly. This qualitative description of the effect of salt cation hydration on the extraction of metal ions is based on the extraction of Zn(II) from an incomplete series of alkali salts to TOMAC and lacks a quantitative speciation profile of Zn(II) in chloride media.9
Both RbCl and FrCl were lacking as alkali chloride salting agents in the extraction series reported before.9 A first step to better understand the salting effects of salt cations is the completion of the Zn(II) extraction series. It is extremely difficult to create the same extraction curve with FrCl as the most stable isotope (233Fr) has a half-life of 22 min and occurs only in trace quantities in the decay chain of 235U.29 Therefore, only the extraction of 30.6 mmol L–1 Zn(II) from RbCl to 0.2 mol L–1 TOMAC in toluene was added (Figure 1). The extraction curve of Zn(II) from RbCl fits in the series of salting agents with alkali cations based on cation charge density as previously reported, when the correction on the ionic radius of NH4+ of Sidey on the Shannon value is taken into account.9,30,31 However, a more quantitative description of the salting effect of cations can be given by looking at the standard state hydration Gibbs free energies (ΔGhyd,exp0) of the alkali cations taken from ref (32) (Table 1).
Figure 1.
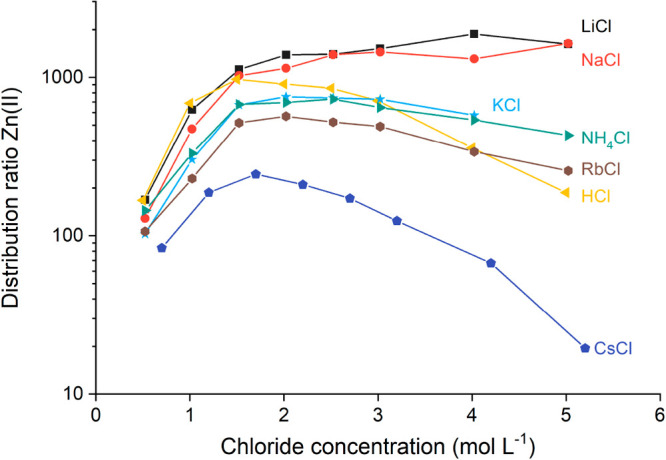
Extraction of Zn(II) from an aqueous phase with RbCl by 0.2 mol L–1 TOMAC dissolved in toluene, added to previously published results.9 The zinc concentration in the aqueous feed was 30.6 mmol L–1. The X-axis shows total chloride concentration in the aqueous phase. Error bars on graphs were omitted to increase legibility.
Table 1. Distribution Ratios for Extraction of Zn(II) by 0.2 mol L–1 TOMAC in Toluene from 30.6 mmol L–1 ZnCl2 in HCl or a Different Monovalent Salt and the Salt Cation Standard State Gibbs Free Energy of Hydration (ΔGhyd,exp0)32.
| cation | DZn(II) at 4 mol L–1 chloride | ΔGhyd,exp0 (kJ mol–1) |
|---|---|---|
| H+ | (3.6 ± 0.2) · 102 | –1050 |
| Li+ | (1.9 ± 0.1) · 103 | –475 |
| Na+ | (1.3 ± 0.1) · 103 | –365 |
| K+ | (5.8 ± 0.3)· 102 | –295 |
| NH4+ | (5.4 ± 0.3)· 102 | –285 |
| Rb+ | (3.4 ± 0.2) · 102 | –275 |
| Cs+ | (7.5 ± 0.4) · 101 | –250 |
The hydration energy of a compound is the energy released when a compound or ion is hydrated in water. This is a direct measure of the compound’s or ion’s tendency to interact with water, with a more negative value of ΔGhyd,exp0 indicating a stronger interaction. Li+ has the most negative ΔGhyd,exp of the alkali cations, showing that its interaction with water in an aqueous phase is strongest and solvent extraction of metals from an aqueous phase with LiCl would be most efficient. The extraction of Zn(II) from solutions of alkali chloride salts and ammonium chloride perfectly matches with the ΔGhyd,exp0 values of the corresponding cations.
The extraction of Zn(II) from HCl media seems to be an outlier when comparing the ΔGhyd,exp0 of a proton to that of the alkali cations. Competitive extraction between the metal complex and HCl with the formation of [Q][HCl2] is often proposed to explain the decrease in extraction efficiency of metal complexes at higher HCl concentrations.33,34 However, this explanation seems unsatisfactory as association between HCl molecules and the basic extractant does not seem to exist.9 Although not associating with the basic extractant, HCl is present in the organic phase and thus changes the composition and properties of the organic phase. This is not observed for the alkali ions and might (partially) explain the observed extraction behavior of metal complexes from HCl, but additional research on the nature of HCl in the organic phase is required to quantify these effects.
When considering the aqueous phase, the deviation of the extraction of Zn(II) from HCl media from the trend of the extraction from alkali salts might be explained by the significantly different H–Cl and HCl–water bond character compared to the Li–Cl bond. To elaborate on this, the structure of LiCl and HCl in concentrated solutions can be compared. Both the lithium cation and the proton associate with chloride ions at high salt or acid concentrations but in a different way. The Li–Cl bond character is more ionic with the formation of a contact ion pair with a bond length of about 2.7 Å, while the H–Cl bond has a significant covalent character with a bond length of about 1.6 Å.35,36 Furthermore, the proton is covalently bonded to one or two water molecules, resulting in the formation of Cl–H–OH2 or Cl–H2O–H–OH2 molecules.36,37 This might lead to a very different solvation of the metal complexes formed at high LiCl or HCl concentration. With LiCl, there are only a few free water molecules left in the solution to hydrate the metal complex, leaving the complex rather unstabilized in the aqueous phase, and thus the complex is efficiently extracted to the organic phase.35 HCl is also strongly hydrated based on the ΔGhyd,exp0, but the metal complex might also be solvated by the covalently bonded HCl entities that also provide a stabilizing effect on the metal complex in the aqueous phase.
As previously proposed, the extraction efficiency of a metal ion can be increased by lowering the hydration of the metal ion.9 This can be accomplished by creating metal–ligand complexes with a lower charge density or by adding a salt with a strongly hydrated cation. A speciation profile of aqueous Zn(II) chloro complexes would allow for clearly distinguishing between effects of Zn(II) speciation and Zn(II) complex hydration. Although a great amount of research has already been performed on the speciation of aqueous Zn(II) chloro complexes, no consistent speciation profile could be constructed based on these literature data.38−47 The main problems are the difficulty in correcting for increasing ionic strength when formation constants are employed and the unavailability of direct measurements of Zn(II)–chloride speciation at low Zn(II) concentration relevant for fundamental solvent extraction studies.
3.2. Speciation of Zn(II) in Aqueous Chloride Solutions at Room Temperature
The complexation of chloride anions to Zn(II) in aqueous solutions has already been intensively investigated using potentiometry and distribution ratios. In these studies, formation constants for ZnCl+, ZnCl2, ZnCl3–, and ZnCl42– are reported.38,39 These values are given for infinite dilution, and significant deviations can be expected when using formation constants to create a speciation profile of Zn(II) at higher ionic strengths. Also, no direct evidence is given for the existence of all above-mentioned Zn(II)–chloride species in the reports about formation constants. It seems logical that chlorides coordinate stepwise to Zn(II), but not all Zn(II)–chloride complexes might be energetically stable and/or favorable. Thus, methods are necessary to directly determine the speciation of Zn(II) at all relevant chloride concentrations. Both X-ray absorption spectroscopy (XAFS) and Raman spectroscopy can be used to detect different Zn(II) species in water and aqueous chloride solutions, and ab initio calculations can be used to determine the conformation of the most energetically favorable Zn(II) species. However, it does not seem possible to get all necessary information for the construction of a speciation profile from one single experimental technique.
In the near edge region of the XAFS spectra (XANES), Liu et al. could only identify two distinct contributions of Zn(II) species to the spectra, being an octahedral and a tetrahedral contribution.40 They could not identify the exact number of different octahedral and tetrahedral species or their structures. Analysis of the extended X-ray absorption fine structure (EXAFS) of Zn(II)–chloride XAFS spectra is rather difficult, as there is a significant error on the determined coordination numbers of Zn(II)–O and Zn(II)–Cl in a single structure. This error logically increases when a mixture of different Zn(II)–chloride species is present with unknown composition. This results in a significant difference in Zn(II)–chloride speciation deduced from EXAFS spectra.41−43
Raman spectroscopy is an interesting experimental technique to study the speciation of Zn(II) as it is rather accessible and can provide quantitative information on the first coordination sphere. However, the Raman effect of Zn(II) species is weak. This can be seen in a number of literature reports, where the experimental Zn(II) concentration for Raman studies is rather high (>1 mol L–1).44−47 Such high Zn(II) concentrations cannot be used to construct a speciation profile of Zn(II) that can be compared with the solvent extraction experiments from Figure 1, as the Zn(II):chloride ratio would be significantly lower and the ionic strength of the solutions would deviate notably for the Raman studies. It is also impossible to significantly increase the Zn(II) concentration for the extraction studies as loading effects and deviations from ideality in the organic phase will obscure the fundamental principles.
The Raman spectra of Zn(II) in chloride media have already been studied intensively in the past, and peak positions of a number of Zn(II) species have been determined (Table 2). However, it seems difficult to clearly assign Raman peak positions to all five Zn(II) species based on the currently available Raman studies alone, as only one peak is observed between 250 and 320 cm–1. This peak is deconvoluted to determine the contribution of ZnCl+, ZnCl2, ZnCl3–, and ZnCl42–. However, it is not clear that all these species are formed, as the observed Raman peak around 280 cm–1 can be reconstructed with contributions from less than these four species. In Raman studies of Zn(II) in bromide solutions, more clearly separated peaks are observed that make it easier to determine the Zn(II)–bromide speciation, compared to the Zn(II)–chloride speciation in chloride solutions.44,46 This suggests that there are indeed several different Zn(II)–bromide species. The absence of such different features in the Raman spectra of Zn(II) chloride solutions might suggest that it is not correct to explain the Zn(II)–chloride Raman spectra with all four Zn(II)–chloride species.
Table 2. Literature Data on Raman Spectra of Zn(II) Species in Chloride Solutions.
Ab initio calculations can shed more light on the stability and structure of all proposed Zn(II) species in chloride solutions. The most rigorous calculations that could be found in the literature determined the free energy of replacement of water molecules with chloride anions around Zn(II) based on gas phase free energies calculated by second-order Møller–Plesset perturbation theory (MP2) and solvation energies.48 Pye et al. found the following most stable geometries for all possible Zn(II) species: [Zn(H2O)6]2+, [ZnCl(H2O)5]+, [ZnCl2(H2O)4], [ZnCl3(H2O)]−, and [ZnCl4]2–.48 They conclude that the octahedral mono- and dichloro Zn(II) species are not energetically favored. The preference for tetrahedral Zn(II)–chloride complexes can be rationalized as follows. First, Zn(II) is the smallest divalent transition metal ion and therefore the hardest according to the HSAB theory. Chloride is also a rather hard anion, resulting in a strong Zn(II)–chloride bond compared to other transition metal–chloride bonds. Therefore, Zn(II) will exchange water molecules for chloride anions easier. Second, due to the small size of the Zn(II) ion, crowding effects become important when water is replaced by larger chloride anions. The octahedral Zn(II) complexes with some chlorides in their first coordination sphere are therefore less stable than tetrahedral complexes with a lower coordination number.
Recent findings on Co(II) speciation in chloride solutions can also be explained using the same principles. Uchikoshi et al. used UV–vis absorption spectra and EXAFS measurements on Co(II) in HCl solutions to determine the speciation of Co(II) in aqueous solution.49,50 They concluded that only [Co(H2O)6]2+, [CoCl(H2O)5]+, and [CoCl4]2– existed in solution, contrary to older reports of stability constants on the intermediate species.49 Thus, Co(II) seems to prefer more the octahedral structure with five to six water molecules in its first coordination sphere. This can be explained as follows. First, Co(II) is a larger transition metal ion than Zn(II). Therefore, Co(II) is less hard according to the HSAB theory, and its bonds with chloride anions are less strong. Second, the larger ionic radius of Co(II) results in less crowding effects when a water molecule is replaced by a chloride ion in the octahedral structure. However, replacing more than one water molecule with a chloride seems to destabilize the octahedral structure to such an extent that these octahedral complexes are not energetically favored any longer.
After identification of the Zn(II) species present in aqueous chloride solutions, a speciation profile was constructed from Raman measurements on aqueous samples with similar compositions to the samples for the extraction studies depicted in Figure 1. The processed spectra of 0.05 mol L–1 ZnClO4 in 0 to 10 mol L–1 LiCl and 0.02 mol L–1 HCl can be found in Figure 2A. A longer, more sensitive Raman measurement was performed in the spectral region around the peak of [Zn(H2O)6]2+ to enhance the quality of that region, as [Zn(H2O)6]2+ has a very weak Raman signal (Figure 2B). ZnClO4 was used instead of ZnCl2 as the Zn(II) source to avoid the presence of a stoichiometric amount of chloride in the first solution, and as perchlorate can be used as an internal standard to perform a quantitative analysis with all spectra. Only a very small amount of HCl (0.02 mol L–1) is present in the solution with the lowest chloride concentration to avoid hydrolysis of Zn(II). Less than 1% conversion of [Zn(H2O)6]2+ can be expected due to the presence of 0.02 mol L–1 HCl, based on association constants from the literature and an extrapolation of the constructed speciation profile.38,39 This has no significant influence on the constructed speciation profile of Zn(II) and avoids the use of strongly oxidizing and potentially explosive HClO4. The peak at 462 cm–1 corresponds to perchlorate, [Zn(H2O)6]2+ can be found at 388 cm–1 (Table 2), and [ZnCl3(H2O)]− and [ZnCl4]2– have a peak located closely together at 285 and 280 cm–1, respectively (Table 2).51 The feature at 110 cm–1 is sometimes attributed to [ZnCl4]2–, but this peak is located at very low Raman shifts where the background signal quickly increases due to elastic scattering of the incident beam. This makes it very difficult to quantify the peaks of low intensity in the region below 150 cm–1.
Figure 2.
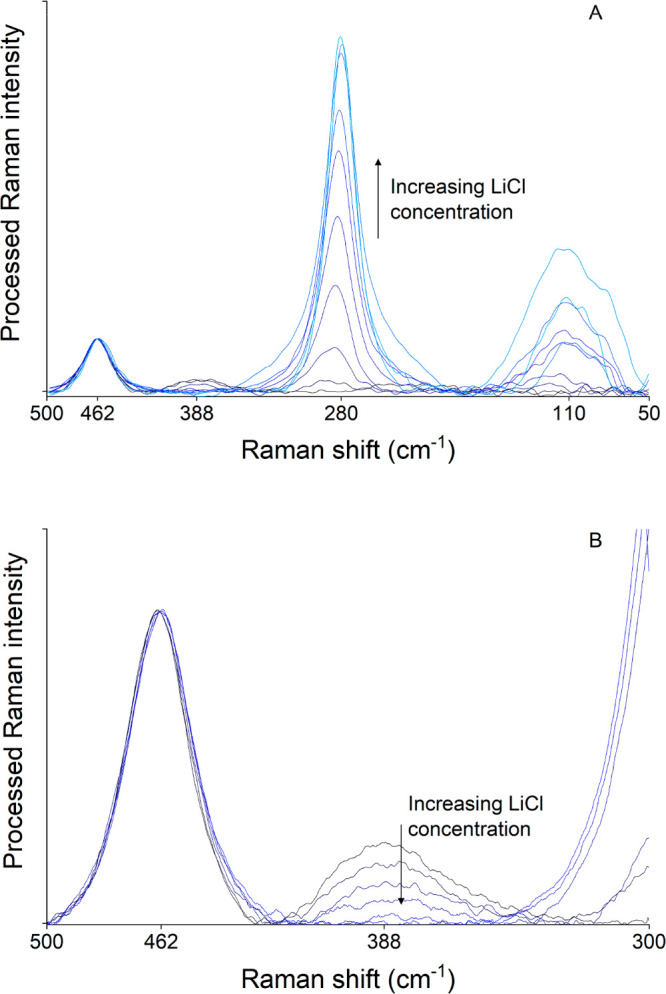
Raman spectra of 0.05 mol L–1 ZnClO4 in different aqueous LiCl solutions with 0.02 mol L–1 HCl. The LiCl concentration increases from 0 (black spectrum) to 10 mol L–1 (lightest blue spectrum). (A) All Raman features of the Zn(II) complexes and (B) the Raman peak of [Zn(H2O)6]2+ recorded with more sensitive experimental parameters.
Manual analysis of the Raman spectra was performed to determine a speciation profile, as the quality of the spectra did not allow for PCA-MCR-ALS analysis such as used to determine Co(II) speciation (vide infra). The intensity of the peak maximum at 388 cm–1 from Figure 2B for the spectrum of the ZnClO4 solution with 0.02 mol L–1 HCl is used as reference for a solution with all Zn(II) as [Zn(H2O)6]2+. The mole fraction of [Zn(H2O)6]2+ in all other samples was calculated based on the intensity of the peak maximum of the 0.02 mol L–1 HCl solution.
The contribution to the Raman spectra of the other two Zn(II) species ([ZnCl3(H2O)]− and [ZnCl4]2–) can then be found in the peak close to 280 cm–1. It would be impossible to determine the mole fraction of both Zn(II) species only using the intensity of the peak, as the Raman intensity of pure [ZnCl3(H2O)]− is unknown. In contrast, the Raman intensity of pure [ZnCl4]2– can be determined by assuming that Zn(II) is only present as [ZnCl4]2– at the highest LiCl concentrations (10 mol L–1). This assumption is justified by the Zn(II)–chloride association constant available in the literature, the match between the experimental peak position and the position of the [ZnCl4]2– peak found in the literature (Table 2), and the very small difference in Raman peak intensity of Zn(II) at 4.0 mol L–1 and 10.0 mol L–1 chloride.38 This thus gives two equations with three unknowns
| 4 |
| 5 |
where xi is the mole fraction of species i, IRaman is the intensity of the experimental Raman peak around 280 cm–1, and IZnCl3– and IZnCl42– are the Raman intensities of the pure components. As xZn2+ and IZnCl42– are already determined, xZnCl3–, xZnCl42–, and IZnCl3– should still be calculated.
The problem at hand can be resolved by also considering the Raman shift of the experimental peak around 280 cm–1. The spectrum from the solution with 10 mol L–1 LiCl shows that the peak of [ZnCl4]2– is centered around 280 cm–1. The [ZnCl3(H2O)]− species is presumably the main contributor to the peak at a similar position in the spectrum of the 1.0 mol L–1 LiCl solution. Thus, the peak of the [ZnCl3(H2O)]− species is centered around 285 cm–1, which closely resembles the peak position of this species found in the literature (Table 2). Now a third equation can be added where xZnCl3– and xZnCl42– are the only unknowns
| 6 |
where SRaman is the Raman shift of the experimental peak around 280 cm–1, and SZnCl3– and SZnCl42– are the Raman shifts of the pure components at 285 and 280 cm–1, respectively. Together with eqs 4 and 5, this results in a system of equations that has one defined solution. The speciation profile of Zn(II) in aqueous LiCl media—calculated as described above—is depicted in Figure 3.
Figure 3.
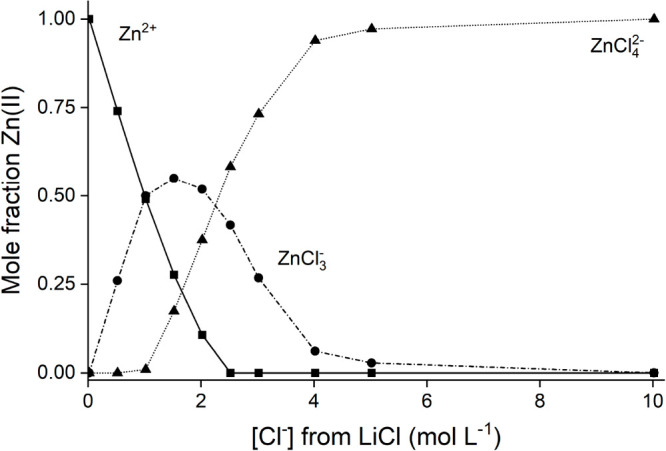
Speciation profile of 0.05 mol L–1 ZnClO4 in aqueous LiCl solution calculated from Raman spectra.
This speciation profile of Zn(II) at low Zn(II) concentrations is believed to be the best possible with the available analytical techniques. However, it should be noted that an assumption is made in the construction of the speciation profile that inherently introduces a small error in the speciation profile. A weighted average equation is used to calculate the contribution of both tetrahedral Zn(II) species to the intensity of the experimental peak (eq 5). A small error is introduced here, as the x-positions of both Zn(II) species do not completely coincide.
Nevertheless, the speciation profile shows that the Zn(II) species with the lowest charge density is located between 1.5 and 2.0 mol L–1 LiCl. It can be expected that the speciation profile of Zn(II) in other alkali chloride salts and HCl is similar based on previous studies on Co(II)–chloride and Cu(II)–chloride speciation.9,52,53 The most efficient extraction of Zn(II) to a basic extractant would be observed around 1.5–2.0 mol L–1 chloride for weak salting-out or salting-in agents, and the extraction of Zn(II) would not show a maximum for strong salting-out agents. This can indeed be observed in Figure 1 and clearly shows that a decrease in Zn(II)–chloride hydration caused by salting agent hydration becomes increasingly important when stronger salting-out agents are used.
3.3. Extraction from Di- and Trivalent Salting Agents
So far, only the effect of alkali salting agents on solvent extraction has been investigated. It was shown that in this category, the chloride salt with the cation having the largest ΔGhyd,exp0 (LiCl) is the most efficient salting-out agent (Table 1). However, cations with a higher charge result in chloride salts with an even larger ΔGhyd,exp.32 These cations would be hydrated more than the ions of LiCl, and thus, even more efficient solvent extraction of metals would be expected when di- or trivalent chloride salts are used as salting agents. This assumption was tested by performing extractions with 17.0 mmol L–1 Co(II) or 7.9 mmol L–1 Cu(II) from LiCl, NaCl, CaCl2, MgCl2, and/or AlCl3 to 0.2 mol L–1 TOMAC in toluene. The resulting extraction curves are plotted on a graph with the total chloride concentration in the aqueous phase on the X-axis (Figure 4).
Figure 4.
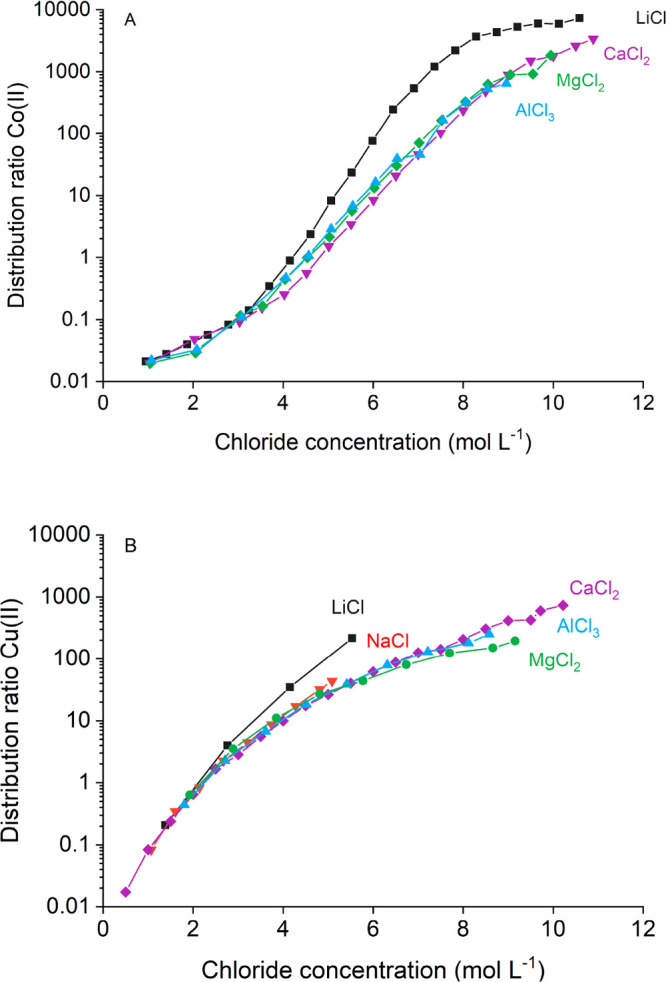
Extraction of Co(II) (A) or Cu(II) (B) from an aqueous phase with different salt content by 0.2 mol L–1 TOMAC dissolved in toluene. Aqueous feed: 7.9 mmol L–1 CuCl2 or 17.0 mmol L–1 CoCl2. Error bars on graphs were omitted to increase legibility.
The extraction of Cu(II) and Co(II) from di- and trivalent salting agents is lower compared to the extraction from LiCl, contrary to what would be expected based on the ΔGhyd,exp0 of the corresponding cations. However, the di- and trivalent salting agents only have half or a third of the cations compared to monovalent salting agents for a given total chloride concentration. Even when the stoichiometry of the salting agents is taken into account by dividing ΔGhyd,exp by the charge of the cation, the corrected ΔGhyd,exp0/z is still larger for the di- and trivalent cations compared to Li+ (Table 3). This seems to contradict the hypothesis that solvent extraction of metals is most efficient from the most hydrated salting agents. Thus, at first, this seems to question the hypothesis that metal complex hydration determines metal complex extractability to a given organic phase.
Table 3. Overview of Distribution Ratios for Extraction of Co(II) or Cu(II) by 0.2 mol L–1 TOMAC in Toluene from 7.9 mmol L–1 CuCl2 or 17.0 mmol L–1 CoCl2 in LiCl or Different Di- and Trivalent Salts and the Salt Cation Standard State Gibbs Free Energy of Hydration (ΔGhyd, exp0)32.
| cation | DCo(II) at 8.5 mol L–1 Cl– | DCu(II) at 5.5 mol L–1 Cl– | ΔGhyd,exp0 (kJ mol–1) | ΔGhyd,exp0/z (kJ mol–1) |
|---|---|---|---|---|
| Li+ | (2.1 ± 0.1) · 103 | (2.2 ± 0.1) · 102 | –475 | –475 |
| Ca2+ | (4.8 ± 0.3) · 102 | (4.0 ± 0.2) · 101 | –1505 | –753 |
| Mg2+ | (5.8 ± 0.3) · 102 | (3.8 ± 0.2) · 101 | –1830 | –915 |
| Al3+ | (5.2 ± 0.3) · 102 | (4.1 ± 0.2) · 101 | –4525 | –1508 |
The di- and trivalent cations are more hydrated in aqueous solutions, but another key aspect plays an important role in the effect of salting agents on solvent extraction of metals. The tendency to form ion pairs is significantly higher for the salts of di- and trivalent metals ions, compared to the salts of monovalent ions.54 This ion pair formation between cations and chloride anions creates ion pairs with a lower charge density and effectively reduces the hydration ability of the salt solution. This leaves more water molecules available to hydrate to extractable metal complexes than could be deduced from the ΔGhyd,exp0 alone, and the more hydrated metal complexes are extracted less efficiently. Also, the more pronounced ion pair formation for salts of di- and trivalent metals ions reduces the amount of chloride anions available for the complexation with metal ion. Thus, a higher total chloride concentration is necessary to form the same amount of metal–chloride complexes when salts of di- or trivalent metal ions are used.
For instance, the association/dissociation reaction of CaCl2 can be written as
| 7 |
The right-hand side of the equilibrium reaction of eq 7 is found at low ionic strength, but the association reactions between Ca2+ and Cl– cannot be neglected at higher ionic strengths (from 1 mol L–1). Equation 7 clearly shows that the charge of the ions is significantly reduced by this association reaction: one +II charged metal ion (Ca2+) and one −I charged chloride ion (Cl–) are replaced by one calcium(II) monochloride cation (CaCl+) with a +I charge.
Equation 7 implies the formation of contact ion pairs (CIP) where the chloride anions can be found in the first coordination sphere of Ca2+. This is an oversimplification, as predominantly solvent-shared ion pairs (SIP, e.g., [Ca(H2O)Cl]+) and solvent-separated ion pairs (SSIP, e.g., [Ca(H2O)(H2O)Cl]+) are formed in solutions of alkali salts, CaCl2, MgCl2, and AlCl3.55−60 CIP formation would decrease the effective hydration most strongly, but also the SIPs and to a lesser extend the SSIPs would be hydrated less than the individual ions without ion pair formation. The presence of a variety of ion pair structures makes it difficult to quantify the effect of ion pair formation on the solvent extraction of metals solely based on the molecular structure. Information on the water activity (aw) and solute activity coefficients of the aqueous phases for solvent extractions can give a more quantitative prediction of these salting effects.
The water activity of aqueous single salts solutions is readily available in the literature. Looking at the water activity of the salts used in this study shows that CaCl2, MgCl2, and AlCl3 solutions have a higher aw compared to LiCl solutions (Figure 5).61,62 This higher aw physically means that there is more water available and/or that the available water is more active to participate in other processes in solution. The aw values of Guendouzi et al. and Richter et al. were converted from mol kg–1 to mol L–1 via the solution densities and put on a common X-scale by using the total chloride concentration, to allow for comparison with the solvent extraction results from Figure 4A. The water activity of CaCl2, MgCl2, and AlCl3 solutions is very similar, showing that the salt hydration in these solutions is also very similar. These observations match closely with the similar extraction efficiency of Co(II) and Cu(II) from CaCl2, MgCl2, and AlCl3 solutions.
Figure 5.
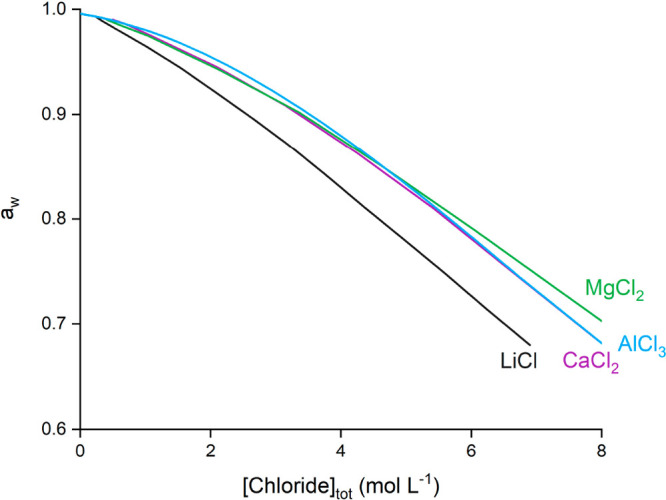
Water activity (aw) of aqueous LiCl, MgCl2, CaCl2, and AlCl3 solutions as a function of the total chloride concentration (based on data reported by Guendouzi et al. and Richter et al.).61,62
3.4. Effect of Mono-, Di-, and Trivalent Salts on Aqueous Speciation of Co(II)
The effect of salt ion association on metal complex formation can be investigated by determining the speciation of a metal ion in different chloride solutions. UV–vis absorption spectroscopy and PCA-MCR-ALS are used to determine the speciation of Co(II) in aqueous solutions at extraction conditions, similarly to what was discussed in a previous paper.9 Only [Co(H2O)6]2+, [CoCl(H2O)5]+, and [CoCl4]2– are found in solution according to speciation analysis by UV–vis absorption spectroscopy and EXAFS analysis from Uchikoshi et al.50 The results of this analysis for the speciation of Co(II) in aqueous LiCl, CaCl2, MgCl2, and AlCl3 are presented in Figure 6.
Figure 6.
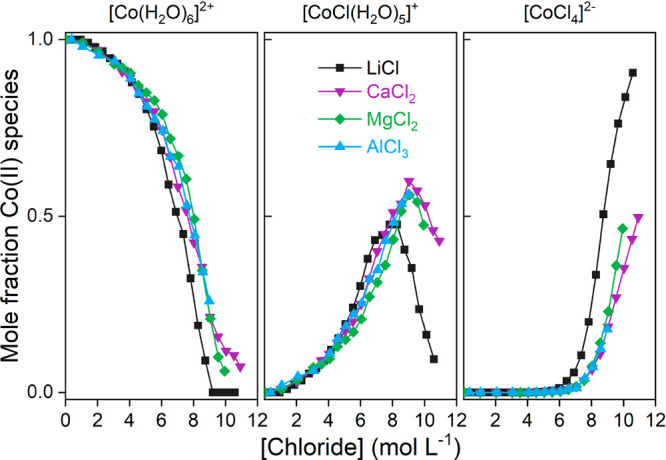
Speciation profile of Co(II) in LiCl, CaCl2, MgCl2, and AlCl3 as a function of the total chloride concentration, calculated with PCA-MCR-ALS from UV–vis absorption spectra.
The shift in Co(II) speciation shows that there are less chlorides available for complexation with Co(II), due to ion pair formation for the salts of di- and trivalent metal ions. However, it is not possible to directly quantify the extent of ion pair formation due to a non-negligible error on the speciation profile and differences in chloride activity for the different salting agents. The mole fraction of [Co(H2O)6]2+ present in solutions of salts of di- and trivalent metals ions stays somewhat higher compared to the LiCl solutions. This will have a negative impact on the extraction efficiency of Co(II), as can be noticed in Figure 4A. The maximum mole fraction of the least hydrated [CoCl(H2O)5]+ complex is shifted about 1 mol L–1 to higher chloride concentrations for the di- and trivalent salts, but the [CoCl(H2O)5]+ complex is overall more present, resulting in no absolute difference in mole fraction at the maximum of [CoCl(H2O)5]+ in LiCl. The higher mole fractions of [CoCl(H2O)5]+ in salts of di- and trivalent metal ions actually suggest that extraction would be more efficient from these salting agents based on Co(II) speciation alone. Also, less [CoCl4]2– is formed in salts of di- and trivalent metal ions compared to LiCl. This again suggests that Co(II) extraction at high chloride concentrations—where the amount of [CoCl4]2– is relevant—should be more efficient from di- and trivalent salting agents. In general, the effects of ion pair formation in di- and trivalent salting agents have a significant impact on the speciation of Co(II), but the observed shift in Co(II) speciation contradicts the lower extraction efficiency of Co(II) from di- and trivalent salting agents compared to the extraction efficiency from LiCl solutions at high chloride concentrations. Therefore, the reduced metal extraction is mainly related to the reduced hydration of the salt ion pairs predominantly formed in solutions of salts of di- and trivalent metal ions.
The findings on salting effects in solvent extractions using basic extractants can also be extended to solvating extractants. As already proposed by other authors: the extraction efficiency of a metal ion by solvating extractants is closely related to the activity coefficients of the cations of the salting agent.26,27 However, it is advisable to consider the activity coefficients of the extractable metal complexes directly. These activity coefficients are influenced by the hydration of the cation of the salting agent, given the anion is the same in all comparisons. The cation hydration depends not only on the ΔGhyd,exp0 of that cation but also on the association behavior of the cation and anion of the salts. Ion association of cations with nitrate ions is generally less important for the investigated salting agents compared to chlorides, but it is still significant for di- and trivalent salts.63
Although the salting effects on transition metal extraction can adequately be explained by ion–solvent interactions, direct salt cation–metal complex interactions probably also play a role in the exact strength of the salting agents. To a certain extent, it can be expected that the cations with the lowest ΔGhyd,exp0 (e.g., Cs+) form ion pairs with the tetrachlorometalate(II) complexes, which might lower the extraction of tetrachlorometalate(II) complexes to basic extractants further.64
4. Conclusions
The effects of dissolved inorganic salts on the solvent extraction behavior of inorganic transition metal complexes have been investigated. It is evident that the type of cation in the chloride salts has a significant influence on the extraction of Cu(II), Zn(II), and Co(II) by the basic extractant methyltrioctylammonium chloride (TOMAC). The observed differences in extraction efficiency for a given transition metal can be explained by the concept of ion hydration, rather than by differences in transition metal speciation. The more a salt reduces the water activity (aw) of a solution, the less other solutes (such as inorganic transition metal complexes) are hydrated and stabilized in that solution. These observations seem to indicate that salting effects in metal ion–solvent extraction processes are mainly related to ion–solvent interactions, rather than to ion–solute interactions.
These findings also imply that solvent extraction techniques can be easily accessible techniques to determine the salting effects of specific salts on other (extractable) solutes. When ion–solvent interactions are most important, one expects that the salting effects can be predicted by ΔGhyd,exp0 values and aw measurements. However, cation–anion association of certain salts must be taken into account. Therefore, aw measurements seem to be preferred for investigation of, for instance, salts of di- and trivalent metal ions. When ΔGhyd,exp values and aw measurements do not correctly predict salting effects, there should be an increased importance of ion–solute interactions, and these specific interactions should be investigated by other techniques.
Acknowledgments
The authors wish to thank Stijn Raiguel (KU Leuven) for the fruitful discussions and Jakob Bussé (KU Leuven) for his assistance with the measurements on the Raman microscope.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors thank the FWO Flanders (projects G0B6918N and I000718N) for financial support. The research was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme: Grant Agreement 694078—Solvometallurgy for critical metals (SOLCRIMET). The contents of this publication are the sole responsibility of the authors and do not necessarily reflect the opinion of the European Union.
The authors declare no competing financial interest.
References
- Solvent Extraction Principles and Practice, Revised and Expanded, 2nd ed.; Rydberg J., Ed.; CRC Press: New York, 2004; 10.1201/9780203021460. [DOI] [Google Scholar]
- Lumetta G. J.; Sinkov S. I.; Krause J. A.; Sweet L. E. Neodymium(III) Complexes of Dialkylphosphoric and Dialkylphosphonic Acids Relevant to Liquid–Liquid Extraction Systems. Inorg. Chem. 2016, 55 (4), 1633–1641. 10.1021/acs.inorgchem.5b02524. [DOI] [PubMed] [Google Scholar]
- Rice N. M. Recent Developments and Potential Uses for Carboxylic Acid Extractants—A Review. Hydrometallurgy 1978, 3 (2), 111–133. 10.1016/0304-386X(78)90015-4. [DOI] [Google Scholar]
- Xie F.; Zhang T. A.; Dreisinger D.; Doyle F. A Critical Review on Solvent Extraction of Rare Earths from Aqueous Solutions. Miner. Eng. 2014, 56, 10–28. 10.1016/j.mineng.2013.10.021. [DOI] [Google Scholar]
- Qi D.Chapter 2 - Extractants Used in Solvent Extraction-Separation of Rare Earths: Extraction Mechanism, Properties, and Features. In Hydrometallurgy of Rare Earths; Qi D., Ed.; Elsevier: 2018; pp 187–389, 10.1016/B978-0-12-813920-2.00002-7. [DOI] [Google Scholar]
- Coleman C. F.; Brown K. B.; Moore J. G.; Crouse D. J. Solvent Extraction with Alkyl Amines. Ind. Eng. Chem. 1958, 50 (12), 1756–1762. 10.1021/ie50588a032. [DOI] [Google Scholar]
- Vander Hoogerstraete T.; Wellens S.; Verachtert K.; Binnemans K. Removal of Transition Metals from Rare Earths by Solvent Extraction with an Undiluted Phosphonium Ionic Liquid: Separations Relevant to Rare-Earth Magnet Recycling. Green Chem. 2013, 15 (4), 919–927. 10.1039/c3gc40198g. [DOI] [Google Scholar]
- Wilson A. M.; Bailey P. J.; Tasker P. A.; Turkington J. R.; Grant R. A.; Love J. B. Solvent Extraction: The Coordination Chemistry behind Extractive Metallurgy. Chem. Soc. Rev. 2014, 43 (1), 123–134. 10.1039/C3CS60275C. [DOI] [PubMed] [Google Scholar]
- Lommelen R.; Vander Hoogerstraete T.; Onghena B.; Billard I.; Binnemans K. Model for Metal Extraction from Chloride Media with Basic Extractants: A Coordination Chemistry Approach. Inorg. Chem. 2019, 58 (18), 12289–12301. 10.1021/acs.inorgchem.9b01782. [DOI] [PubMed] [Google Scholar]
- Hofmeister F. Zur Lehre von der Wirkung der Salze. Naunyn-Schmiedeberg's Arch. Pharmacol. 1888, 24 (4), 247–260. 10.1007/BF01918191. [DOI] [Google Scholar]
- Ball P.; Hallsworth J. E. Water Structure and Chaotropicity: Their Uses, Abuses and Biological Implications. Phys. Chem. Chem. Phys. 2015, 17 (13), 8297–8305. 10.1039/C4CP04564E. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Cremer P. S. Interactions between Macromolecules and Ions: The Hofmeister Series. Curr. Opin. Chem. Biol. 2006, 10 (6), 658–663. 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Baldwin R. L. How Hofmeister Ion Interactions Affect Protein Stability. Biophys. J. 1996, 71 (4), 2056–2063. 10.1016/S0006-3495(96)79404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWalt-Kerian E. L.; Kim S.; Azam Md. S.; Zeng H.; Liu Q.; Gibbs J. M. PH-Dependent Inversion of Hofmeister Trends in the Water Structure of the Electrical Double Layer. J. Phys. Chem. Lett. 2017, 8 (13), 2855–2861. 10.1021/acs.jpclett.7b01005. [DOI] [PubMed] [Google Scholar]
- Flores S. C.; Kherb J.; Cremer P. S. Direct and Reverse Hofmeister Effects on Interfacial Water Structure. J. Phys. Chem. C 2012, 116 (27), 14408–14413. 10.1021/jp3029352. [DOI] [Google Scholar]
- Sun P.; Huang K.; Liu H. The Nature of Salt Effect in Enhancing the Extraction of Rare Earths by Non-Functional Ionic Liquids: Synergism of Salt Anion Complexation and Hofmeister Bias. J. Colloid Interface Sci. 2019, 539, 214–222. 10.1016/j.jcis.2018.12.058. [DOI] [PubMed] [Google Scholar]
- Sun P.; Huang K.; Liu H. Specific Salt Effect on the Interaction between Rare Earth Ions and Trioctylphosphine Oxide Molecules at the Organic–Aqueous Two-Phase Interface: Experiments and Molecular Dynamics Simulations. Langmuir 2018, 34 (38), 11374–11383. 10.1021/acs.langmuir.8b02301. [DOI] [PubMed] [Google Scholar]
- Fowler C. J.; Haverlock T. J.; Moyer B. A.; Shriver J. A.; Gross D. E.; Marquez M.; Sessler J. L.; Hossain Md. A.; Bowman-James K. Enhanced Anion Exchange for Selective Sulfate Extraction: Overcoming the Hofmeister Bias. J. Am. Chem. Soc. 2008, 130 (44), 14386–14387. 10.1021/ja806511b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont D.; Depuydt D.; Binnemans K. Overview of the Effect of Salts on Biphasic Ionic Liquid/Water Solvent Extraction Systems: Anion Exchange, Mutual Solubility, and Thermomorphic Properties. J. Phys. Chem. B 2015, 119 (22), 6747–6757. 10.1021/acs.jpcb.5b02980. [DOI] [PubMed] [Google Scholar]
- Hyde A. M.; Zultanski S. L.; Waldman J. H.; Zhong Y.-L.; Shevlin M.; Peng F. General Principles and Strategies for Salting-Out Informed by the Hofmeister Series. Org. Process Res. Dev. 2017, 21 (9), 1355–1370. 10.1021/acs.oprd.7b00197. [DOI] [Google Scholar]
- Salis A.; Ninham B. W. Models and Mechanisms of Hofmeister Effects in Electrolyte Solutions, and Colloid and Protein Systems Revisited. Chem. Soc. Rev. 2014, 43 (21), 7358–7377. 10.1039/C4CS00144C. [DOI] [PubMed] [Google Scholar]
- Nihonyanagi S.; Yamaguchi S.; Tahara T. Counterion Effect on Interfacial Water at Charged Interfaces and Its Relevance to the Hofmeister Series. J. Am. Chem. Soc. 2014, 136 (17), 6155–6158. 10.1021/ja412952y. [DOI] [PubMed] [Google Scholar]
- Ferreira L. A.; Uversky V. N.; Zaslavsky B. Y. Effects of the Hofmeister Series of Sodium Salts on the Solvent Properties of Water. Phys. Chem. Chem. Phys. 2017, 19 (7), 5254–5261. 10.1039/C6CP08214A. [DOI] [PubMed] [Google Scholar]
- Marcus Y. Effect of Ions on the Structure of Water: Structure Making and Breaking. Chem. Rev. 2009, 109 (3), 1346–1370. 10.1021/cr8003828. [DOI] [PubMed] [Google Scholar]
- Narbutt J.Chapter 4 - Fundamentals of Solvent Extraction of Metal Ions. In Liquid-Phase Extraction; Poole C. F., Ed.; Handbooks in Separation Science; Elsevier: 2020; pp 121–155, 10.1016/B978-0-12-816911-7.00004-9. [DOI] [Google Scholar]
- Jenkins I. L.; McKay H. a. C. The Partition of Uranyl Nitrate between Water and Organic Solvents. Part 6. Salting-out by a Second Nitrate. Trans. Faraday Soc. 1954, 50 (0), 107–119. 10.1039/tf9545000107. [DOI] [Google Scholar]
- Sato T. The Extraction of Uranyl Nitrate by Tributyl Phosphate in the Presence of Some Nitrate Salting-out Agents. J. Inorg. Nucl. Chem. 1960, 16 (1), 156–158. 10.1016/0022-1902(60)80103-0. [DOI] [Google Scholar]
- Moyer B. A.; Sun Y.. Principles of Solvent Extraction of Alkali Metal Ions: Understanding Factors Leading to Cesium Selectivity in Extraction by Solvation. In Ion Exchange and Solvent Extraction; New York: Marcel Dekker, Inc., 1997; Vol. 13, pp 295–391. [Google Scholar]
- Audi G.; Bersillon O.; Blachot J.; Wapstra A. H. The Nubase Evaluation of Nuclear and Decay Properties. Nucl. Phys. A 2003, 729 (1), 3–128. 10.1016/j.nuclphysa.2003.11.001. [DOI] [Google Scholar]
- Shannon R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 32 (5), 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Sidey V. On the Effective Ionic Radii for Ammonium. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72 (4), 626–633. 10.1107/S2052520616008064. [DOI] [PubMed] [Google Scholar]
- Marcus Y. Thermodynamics of Solvation of Ions. Part 5.—Gibbs Free Energy of Hydration at 298.15 K. J. Chem. Soc., Faraday Trans. 1991, 87 (18), 2995–2999. 10.1039/FT9918702995. [DOI] [Google Scholar]
- Sato T.; Watanabe H.; Kikuchi S. Extraction of Some Mineral Acids by High Molecular Weight Quaternary Ammonium Chloride. J. Appl. Chem. Biotechnol. 1975, 25 (1), 63–72. 10.1002/jctb.5020250107. [DOI] [Google Scholar]
- du Preez J. G. H. Recent Advances in Amines as Separating Agents for Metal Ions. Solvent Extr. Ion Exch. 2000, 18 (4), 679–701. 10.1080/07366290008934703. [DOI] [Google Scholar]
- Pethes I. The Structure of Aqueous Lithium Chloride Solutions at High Concentrations as Revealed by a Comparison of Classical Interatomic Potential Models. J. Mol. Liq. 2018, 264, 179–197. 10.1016/j.molliq.2018.05.044. [DOI] [Google Scholar]
- Fulton J. L.; Balasubramanian M. Structure of Hydronium (H3O+)/Chloride (Cl–) Contact Ion Pairs in Aqueous Hydrochloric Acid Solution: A Zundel-like Local Configuration. J. Am. Chem. Soc. 2010, 132 (36), 12597–12604. 10.1021/ja1014458. [DOI] [PubMed] [Google Scholar]
- Jiang J.-C.; Wang Y.-S.; Chang H.-C.; Lin S. H.; Lee Y. T.; Niedner-Schatteburg G.; Chang H.-C. Infrared Spectra of H+(H2O)5–8 Clusters: Evidence for Symmetric Proton Hydration. J. Am. Chem. Soc. 2000, 122 (7), 1398–1410. 10.1021/ja990033i. [DOI] [Google Scholar]
- Aparicio J. L.; Elizalde M. P. Activity Coefficient Calculations Applied to ZnCl2 in LiCl Media. Distinction between the Real Activity Coefficient and the Effect of Complexation. J. Solution Chem. 1996, 25 (11), 1055–1069. 10.1007/BF00972921. [DOI] [Google Scholar]
- Powell K. J.; Brown P. L.; Byrne R. H.; Gajda T.; Hefter G.; Leuz A.-K.; Sjöberg S.; Wanner H. Chemical Speciation of Environmentally Significant Metals with Inorganic Ligands. Part 5: The Zn2+ + OH–, Cl–, CO32–, SO42–, and PO43– Systems. Pure Appl. Chem. 2013, 85 (12), 2249–2311. 10.1351/pac-rep-13-06-03. [DOI] [Google Scholar]
- Liu W.; Etschmann B.; Foran G.; Shelley M.; Brugger J. Deriving Formation Constants for Aqueous Metal Complexes from XANES Spectra: Zn2+ and Fe2+ Chloride Complexes in Hypersaline Solutions. Am. Mineral. 2007, 92 (5–6), 761–770. 10.2138/am.2007.2225. [DOI] [Google Scholar]
- Mayanovic R. A.; Anderson A. J.; Bassett W. A.; Chou I. XAFS Measurements on Zinc Chloride Aqueous Solutions from Ambient to Supercritical Conditions Using the Diamond Anvil Cell. J. Synchrotron Radiat. 1999, 6 (3), 195–197. 10.1107/S0909049599001727. [DOI] [PubMed] [Google Scholar]
- Dreier P.; Rabe P. EXAFS-Study of the Zn2+ Coordination in Aqueous Halide Solutions. J. Phys. Colloques 1986, 47 (C8), C8-809–C8–812. 10.1051/jphyscol:19868155. [DOI] [Google Scholar]
- Mei Y.; Sherman D. M.; Liu W.; Etschmann B.; Testemale D.; Brugger J. Zinc Complexation in Chloride-Rich Hydrothermal Fluids (25–600°C): A Thermodynamic Model Derived from Ab Initio Molecular Dynamics. Geochim. Cosmochim. Acta 2015, 150, 265–284. 10.1016/j.gca.2014.09.023. [DOI] [Google Scholar]
- Morris D. F. C.; Short E. L.; Waters D. N. Zinc Chloride and Zinc Bromide Complexes—III: Structures of Species in Solution. J. Inorg. Nucl. Chem. 1963, 25 (8), 975–983. 10.1016/0022-1902(63)80031-7. [DOI] [Google Scholar]
- Irish D. E.; McCarroll B.; Young T. F. Raman Study of Zinc Chloride Solutions. J. Chem. Phys. 1963, 39 (12), 3436–3444. 10.1063/1.1734212. [DOI] [Google Scholar]
- Marley N. A.; Gaffney J. S. Laser Raman Spectral Determination of Zinc Halide Complexes in Aqueous Solutions as a Function of Temperature and Pressure. Appl. Spectrosc. 1990, 44 (3), 469–476. 10.1366/0003702904086128. [DOI] [Google Scholar]
- Maeda M.; Ito T.; Hori M.; Johansson G. The Structure of Zinc Chloride Complexes in Aqueous Solution. Z. Naturforsch., A: Phys. Sci. 1996, 51, 63–70. 10.1515/zna-1996-1-210. [DOI] [Google Scholar]
- Pye C. C.; Corbeil C. R.; Rudolph W. W. An Ab Initio Investigation of Zinc Chloro Complexes. Phys. Chem. Chem. Phys. 2006, 8 (46), 5428–5436. 10.1039/b610084h. [DOI] [PubMed] [Google Scholar]
- Uchikoshi M. Determination of the Distribution of Cobalt-Chloro Complexes in Hydrochloric Acid Solutions at 298 K. J. Solution Chem. 2018, 47 (12), 2021–2038. 10.1007/s10953-018-0831-z. [DOI] [Google Scholar]
- Uchikoshi M.; Shinoda K. Determination of Structures of Cobalt(II)-Chloro Complexes in Hydrochloric Acid Solutions by X-Ray Absorption Spectroscopy at 298 K. Struct. Chem. 2019, 30, 945. 10.1007/s11224-018-1245-7. [DOI] [Google Scholar]
- Jones M. M.; Jones E. A.; Harmon D. F.; Semmes R. T. A Search for Perchlorate Complexes. Raman Spectra of Perchlorate Solutions. J. Am. Chem. Soc. 1961, 83 (9), 2038–2042. 10.1021/ja01470a003. [DOI] [Google Scholar]
- Brugger J.; McPhail D. C.; Black J.; Spiccia L. Complexation of Metal Ions in Brines: Application of Electronic Spectroscopy in the Study of the Cu(II)-LiCl-H2O System between 25 and 90°C. Geochim. Cosmochim. Acta 2001, 65 (16), 2691–2708. 10.1016/S0016-7037(01)00614-7. [DOI] [Google Scholar]
- Uchikoshi M. Determination of the Distribution of Cupric Chloro-Complexes in Hydrochloric Acid Solutions at 298 K. J. Solution Chem. 2017, 46 (3), 704–719. 10.1007/s10953-017-0597-8. [DOI] [Google Scholar]
- Johnson K. S.; Pytkowicz R. M. Ion Association of Cl– with H+, Na+, K+, Ca2+, Mg2+ in Aqueous Solutions at 25°C. Am. J. Sci. 1978, 278 (10), 1428–1447. 10.2475/ajs.278.10.1428. [DOI] [Google Scholar]
- Friesen S.; Hefter G.; Buchner R. Cation Hydration and Ion Pairing in Aqueous Solutions of MgCl2 and CaCl2. J. Phys. Chem. B 2019, 123 (4), 891–900. 10.1021/acs.jpcb.8b11131. [DOI] [PubMed] [Google Scholar]
- Wachter W.; Fernandez Š.; Buchner R.; Hefter G. Ion Association and Hydration in Aqueous Solutions of LiCl and Li2SO4 by Dielectric Spectroscopy. J. Phys. Chem. B 2007, 111 (30), 9010–9017. 10.1021/jp072425e. [DOI] [PubMed] [Google Scholar]
- Cauët E.; Bogatko S. A.; Bylaska E. J.; Weare J. H. Ion Association in AlCl3 Aqueous Solutions from Constrained First-Principles Molecular Dynamics. Inorg. Chem. 2012, 51 (20), 10856–10869. 10.1021/ic301346k. [DOI] [PubMed] [Google Scholar]
- Pham V.-T.; Fulton J. L. Ion-Pairing in Aqueous CaCl2 and RbBr Solutions: Simultaneous Structural Refinement of XAFS and XRD Data. J. Chem. Phys. 2013, 138 (4), 044201 10.1063/1.4775588. [DOI] [PubMed] [Google Scholar]
- Dai Q.; Xu J.-J.; Li H.-J.; Yi H.-B. Ion Association Characteristics in MgCl2 and CaCl2 Aqueous Solutions: A Density Functional Theory and Molecular Dynamics Investigation. Mol. Phys. 2015, 113 (22), 3545–3558. 10.1080/00268976.2015.1039618. [DOI] [Google Scholar]
- Callahan K. M.; Casillas-Ituarte N. N.; Roeselová M.; Allen H. C.; Tobias D. J. Solvation of Magnesium Dication: Molecular Dynamics Simulation and Vibrational Spectroscopic Study of Magnesium Chloride in Aqueous Solutions. J. Phys. Chem. A 2010, 114 (15), 5141–5148. 10.1021/jp909132a. [DOI] [PubMed] [Google Scholar]
- Guendouzi M. E.; Dinane A.; Mounir A. Water Activities, Osmotic and Activity Coefficients in Aqueous Chloride Solutions at T= 298.15 K by the Hygrometric Method. J. Chem. Thermodyn. 2001, 33 (9), 1059–1072. 10.1006/jcht.2000.0815. [DOI] [Google Scholar]
- Richter U.; Brand P.; Bohmhammel K.; Könnecke T. Thermodynamic Investigations of Aqueous Solutions of Aluminium Chloride. J. Chem. Thermodyn. 2000, 32 (2), 145–154. 10.1006/jcht.1999.0557. [DOI] [Google Scholar]
- Xu M.; Larentzos J. P.; Roshdy M.; Criscenti L. J.; Allen H. C. Aqueous Divalent Metal–Nitrate Interactions: Hydration versus Ion Pairing. Phys. Chem. Chem. Phys. 2008, 10 (32), 4793–4801. 10.1039/b807090n. [DOI] [PubMed] [Google Scholar]
- Jin G. B.; Lin J.; Estes S. L.; Skanthakumar S.; Soderholm L. Influence of Countercation Hydration Enthalpies on the Formation of Molecular Complexes: A Thorium–Nitrate Example. J. Am. Chem. Soc. 2017, 139 (49), 18003–18008. 10.1021/jacs.7b09363. [DOI] [PubMed] [Google Scholar]