

Abstract
Background
The GGGGCC (G4C2) repeat expansion in the human open reading frame 72 on chromosome 9, C9orf72, is the most common cause of amyotrophic lateral sclerosis (ALS). Studies in transgenic mouse models have linked the pathogenic mechanism of G4C2 repeat expansion to RNA foci or the accumulation of unnatural dipeptide repeats in neurons. However, only one of the existing transgenic mouse lines developed typical ALS.
Methods
C9orf72 knockin rats were generated by knockin of 80 G4C2 repeats with human flanking fragments within exon1a and exon1b at the rat C9orf72 locus. Protein expression was detected by western blot. Motor coordination and grip force were measured using a Rotarod test and a grip strength test. Neurodegeneration was assessed by Nissl staining with cresyl violet.
Results
C9orf72 haploinsufficiency reduced C9orf72 protein expression 40% in the cerebrum, cerebellum and spinal cords from knockin rats (P < .05). The knockin (KI) rats developed motor deficits from 4 months of age. Their falling latencies and grip force were decreased by 67% (P < .01) and 44% (P < .01), respectively, at 12 months of age compared to wild‐type (WT) mice. The knockin of the hexanucleotide repeat expansion (HRE) caused a 47% loss of motor neurons in the spinal cord (P < .001) and 25% (5/20) of female KI rats developed hind limb paralysis at 13 to 24 months.
Conclusion
Motor defects in KI rats may result from neurotoxicity caused by HRE and the resulting reduction in C9orf72 protein due to haploinsufficiency. These KI rats could be a useful model for investigating the contributions of loss‐of‐function to neurotoxicity in C9orf72‐related ALS.
Keywords: amyotrophic lateral sclerosis, C9orf72, GGGGCC repeat expansion, knockin, rat
1. INTRODUCTION
The GGGGCC (G4C2) repeat expansion within the first intron of the human C9orf72 gene is the most common cause of amyotrophic lateral sclerosis (ALS), accounting for more than 30% of familial ALS and 5% of sporadic ALS cases. 1 , 2 , 3 , 4 The C9orf72 hexanucleotide expansion may lead to pathogenesis through sense and antisense RNA transcripts from the C9orf72 repeat expansion. It is thought that these RNAs may sequester RNA‐binding proteins in RNA foci, which, when translated, result in accumulation of unnatural dipeptide repeat (DPR) proteins in the brain. 5 , 6 , 7 , 8 , 9 A number of transgenic animal models have been employed in studies, including drosophila, 10 zebra fish 11 , 12 , 13 and mice. 14 , 15 , 16 , 17 In the transgenic drosophila and zebra fish models, a gain‐of‐function of C9orf72 repeat expansion caused an accumulation of DPR proteins, or a combination of RNA foci and DPR proteins that was sufficient to promote neurodegeneration, motor neuron loss and muscle atrophy. 10 , 11 , 12 However, the data from four reports on BAC transgenic mouse models were equivocal. RNA foci and repeat‐associated non‐ATG (RAN) proteins were found in all of them, but in two of the studies the mice did not develop the neurodegenerative or behavioral features of ALS/FTD. 14 , 15 One of them showed the neurodegenerative and behavioral features of FTD, but no motor neuron damage or behavioral features of ALS. 16 Only one study showed both motor deficits and neurodegenerative features of ALS/FTD. 17 In ALS patients, the hexanucleotide expansion was found to alter the expression ratio of C9ORF72 protein isoforms. 18 , 19 , 20 , 21 C9orf72 is a multifunctional protein that regulates autophagy, cell membrane and vesicular trafficking, and lysosomal biogenesis through interaction with small Rab GTPases, cofilin and the guanine nucleotide exchange protein SMCR8. 22 , 23 , 24 , 25 , 26 , 27 The data suggest that loss of C9ORF72 function might sensitize neurons to the formation of RNA foci or expression of DPR proteins and that the combination of loss‐ and gain‐of‐function is an essential factor in the pathogenesis of ALS/FTD caused by the C9orf72 hexanucleotide expansion.
Here, we generated a rat model by knockin of the hexanucleotide repeat expansion (HRE) at the C9orf72 locus. The HRE inhibited expression of C9ORF72 protein and the rats showed the progressive motor deficits and neurodegenerative characteristics of ALS.
2. MATERIALS AND METHODS
2.1. Generation of HRE knockin rats at C9orf72 loci
An 80‐copy C9orf72 HRE was kindly provided by Dr Charlet‐Berguerand N from the Department of Neurobiology and Genetics, University of Strasbourg, Illkirch, France. Exon1a and exon1b and the flanking sequences of the human C9orf72 HRE were ligated with the HRE according to the human DNA sequence of the locus. The 80 repeats of the C9orf72 HRE, exon1a and exon1b, and the flanking human sequences were confirmed by DNA sequencing. The knockin rats were generated by using CRISPR/cas9 following the procedure described previously. 28 Briefly, we designed two pairs of synthetic oligonucleotides for gRNA targeting to exon1 of the C9orf72 locus: sgRNA 1 (agtcccacgaggaaacagcgg) and sgRNA 2 (aggcaaccagagcgctggcgg). The microinjection mixture containing Cas9 protein (30 ng/μL), sgRNAs (10 ng/μL of each) and the donor DNA with the HRE and flanking sequences (10 ng/μL) was microinjected into the male pronucleiin fertilized eggs and transferred to pseudopregnant SD rats. The knockin pups were genotyped by PCR to verify the presence of the (GC)80 repeats. The wild‐type (WT) genotype primers were 5′TGTAGTGTCAGCCATCCCAATTG and 5′GGAACAGTGTGACTAGAAATTTATCCACC, which resulted in a 1084 bp DNA fragment. The right arm region primers were 5′TAAGAACTTAACAGATGACAGTTGCTGG and 5′TTCTTGTTCACCCTCAGCGAG, which yielded a 965 bp DNA fragment. The left arm region primers were 5′TTTACTTTCCCTCTCATTTCTCTGACC 5′GGAACAGTGTGACTAGAAATTTATCCACC, which produced an 809 bp DNA fragment. Only the pups carrying the (GC)80 repeats and complete right and left arm sequences were selected to breed as (GC)80 repeat knockin rats (KI). All rats used in this study were maintained on a Sprague‐Dawley background and bred at an AAALAC‐accredited facility. The procedures were approved by the Animal Care and Use Committee of the Institute of Laboratory Animal Science of the Peking Union Medical College (ZLF18003). The C9orf72 protein in WT and KI rats was detected by western blot using mouse monoclonal anti‐C9orf72 antibody (1:1000, Proteintech), visualized using anti‐mouse HRP‐conjugated secondary antibodies (1:10 000, Santa Cruz Biotechnology) and quantitated by densitometry using Image J software.
2.2. Motor coordination and gripping force testing
The Rotarod device (DXP‐3, Institute of Materia Medica, Beijing) was used to test impaired muscle and motor coordination and gripping force of the limbs. The test was performed using the rod according to published procedures. 29 Each rat was placed on the rod, which was rotating at a constant speed of 30 rpm, and the latency of falling was recorded. Seven trials were performed with a 5‐minute interval between trials. The first four trails were considered as adaptive training. The latency of falling from the final three trials was averaged and used as the measure for impaired muscle and motor coordination. The grip test was performed using a procedure modified from a reported method. 30 The rat was placed on a plate so that it gripped the metal grid of the Bioseb Grip Test device (bio‐gs3) with its forepaws (or with all four paws) in a comfortable way. The rat was pulled horizontally and the peak force was recorded before the rat released its grip. Three measurements were averaged and used as the measure of grip force for forepaws or all four paws. The gripping force of the hind limbs was calculated by subtracting the forepaw force from the force of the four paws. Finally, the grip strength of WT and KI rats at 4, 8 and 12 months of age was normalized to their corresponding average body weights at the given time point.
2.3. Tissue collection and histology for motor neuron counting
Thirteen‐month‐old rats were anaesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde. Three rats were used from each group. The lumbar spinal cords were dissected out, post‐fixed overnight in 4% paraformaldehyde and embedded in gelatin blocks. Sections of 40 μm thickness (ten from each rat) were prepared using a cryotome (CM3050S, Leica). The sections were rehydrated at room temperature for 5 minutes, stained for 2 hours in a solution of 1% cresyl violet and then washed in distilled water until the color faded to lavender. The stained sections were dehydrated and mounted with DPX. The motor neurons (MNs) were observed under microscope (MZ16F, Leica) and photographed. Only the MNs with diameters larger than 20 μm and with a polygonal shape and prominent nucleoli were counted to assess the MN loss in KI rats.
2.4. Identification and quantitation of peripheral blood lymphocytes by flow cytometry
Peripheral blood from WT and KI rats was collected and red blood cells were lysed using a commercial lysing reagent (BD Biosciences, San Jose, CA, USA). Lymphocytes were isolated by filtration through a sterile nylon mesh (pore size 70 μm) and stained for 30 minutes at 4°C with the following fluorophore‐conjugated antibodies: PE‐CD3 (G4.18), APC‐CD4 (OX35) and FITC‐Granulocyte (HIS48). All antibodies were obtained from eBiosciences (San Diego, CA, USA). Flow cytometry analysis of lymphocytes from peripheral blood was performed as previously reported 31 on a FACS Aria II (BD Biosciences). Data were analyzed by FlowJo software.
2.5. Statistical analysis
The data for motor neuron counts, Rotarod and grip force tests and flow cytometry were analyzed by unpaired Student's t test for two groups and one‐way analysis of variance (ANOVA) for multiple groups. The data were expressed as the means ± SEM from individual experiments. A P value of <.05 was considered significant. Statistical analysis was performed using GraphPad Prism software.
3. RESULTS
3.1. Knock in of HRE inhibited C9orf72 expression
Based on the normal rat genomic sequence (pubmed.gov), the C9orf72 locus in rats consists of a single exon1 rather than being separated into exon1a and exon1b by G4C2 repeats, as in the human C9orf72 locus (Figure 1A). To knock the G4C2 repeats into the rat C9orf72 locus, the whole C9orf72 exon1 was replaced by a 1047 bp human DNA fragment containing eighty repeats of the hexanucleotide sequence, GGGGCC (GGGGCC80) plus exon1a and 1b and the flanking sequences (Figure 1B). The homozygotes of KI rats showed normal body weight and reproductive capacity. Western blots indicated that the inserted HRE inhibited C9orf72 protein expression by 40% in the central nervous system (CNS) including the cerebrum, cerebellum and spinal cord (Figure 1C,D; n = 3, female, P < .05). This HRE‐related C9orf72 haploinsufficiency may be a pathogenic factor in ALS patients. 18 , 19
FIGURE 1.
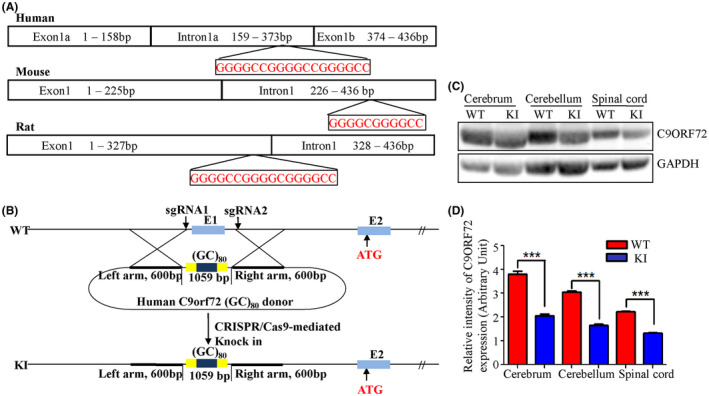
Generation of C9orf72 hexanucleotide repeat expansion knockin rats. The DNA sequence of the C9orf72 exon1 region from human, mouse and rat were compared (A). A 1047 bp human DNA fragment containing 80 G4C2 repeats, exon1a and exon1b, and flanking sequences was used to replace the entire C9orf72 exon1 of the rat using CRISPR/Cas9 (B). The C9orf72 protein expression levels were detected by western blot and quantitated by densitometry using NIH Image J software (C and D, n = 3, 2 males and 1 female, ***P < .001 vs respective controls)
3.2. HRE knockin altered lymphocyte profiles
The KI rats developed visibly shrunken thymi and enlarged cervical lymph nodes from two months of age (Figure 2A,B; n = 8, P < .05).The spleens of KI rats were not obviously changed compared to WT rats (Figure 2C). Flow cytometry analysis for lymphocytes from peripheral blood showed that granulocyte numbers were significantly increased by 81% in the KI rats at 4 months of age compared with same‐age WT rats (Figure 2D; n = 8, 4 males and 4 females, P < .001). The numbers of CD3+ cells were decreased by 21% in KI rats at 4 months of age compared to WT (Figure 2E; n = 8, 4 males and 4 females, P < .01). The CD4 cells were decreased by 32% in the KI rats at 4 months of age compared to WT rats (Figure 2E; n = 8, 4 males and 4 females, P < .001). The B cells and CD8 cells were not changed in the KI rats (data not shown).
FIGURE 2.
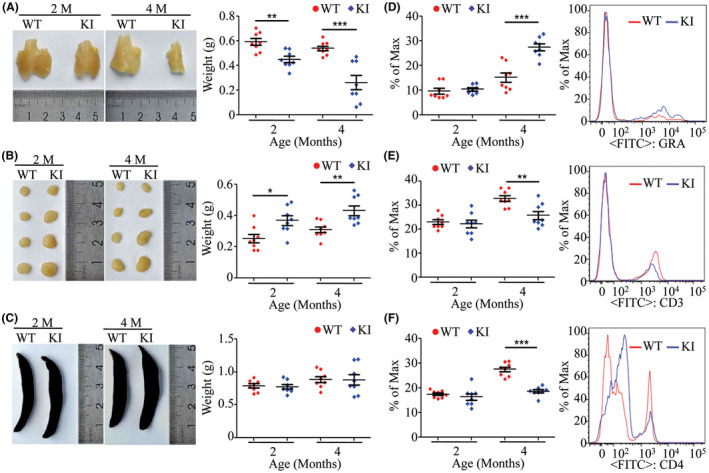
Gross tissue examination and lymphocyte profiles from WT and KI rats. Thymi, cervical lymph nodes and spleens from WT and KI rats at 2 and 4 mo of age were examined and weighed to show shrinkage of the thymus and enlargement of cervical lymph nodes (A‐C, n = 8, 4 males and 4 females). The lymphocytes from peripheral blood of WT and KI rats at 2 and 4 mo of age were analyzed by flow cytometry. The number of granulocytes was significantly increased in the KI rats at 4 mo of age (D, n = 8, 4 males and 4 females). The CD3 and CD4 cells were significantly decreased in the KI rats at 4 mo of age (E and F, n = 8, 4 males and 4 females). *P < .05, **P < .01, ***P < .001
3.3. HRE knockin caused motor deficits and hind limb paralysis
To detect motor deficits, motor coordination and hind limb grip strength of WT and KI rats were tested at 4, 8 and 12 months of age. The motor coordination of WT rats on the Rotarod decreased slightly from 4 to 12 months old (P = .675 by ANOVA), whereas the motor coordination of KI rats decreased significantly over time (P = .0002 by ANOVA). In contrast, the hind limb grip strength normalized to body weight of both WT and KI rats decreased significantly over time (P = .0001 by ANOVA for WT rats, P < .0001 by ANOVA for KI rats). However, the data from two tests in KI rats showed a significant decrease when compared with WT rats at 8 and 12 months of age, suggesting that the KI rats developed progressive motor deficits starting from the age of 4 months and getting worse over time. Specifically, the motor deficit increase with age corresponded to a significant decrease in the latency of falling in the Rotarod test and a decrease in hind limb gripping force. The latency of falling for KI rats was reduced from 25% less than WT latency at 4 months of age (Figure 3A; n = 19 for WT, n = 24 for KI, female P = .07) to 67% at 12 months of age (Figure 3A; n = 16 for WT, n = 22 for KI, female, P < .01). The grip strength of KI rats was reduced by 9.5% compared to that of WT rats at 4 months of age (Figure 3B; n = 19 for WT, n = 24 for KI, female, P = .2), by 33% at 8 months of age (Figure 3B; n = 18 for WT, n = 23 for KI, female, P < .0001) and by 44% at 12 months of age (Figure 3B; n = 16 for WT, n = 22 for KI, female, P < .0001). Finally, hind limb paralysis occurred in 25% of female KI rats (5/20) between the ages of 13 and 24 months (Figure 3C). These data indicated that knockin of the HRE in rats caused not only motor deficits but also hind limb paralysis.
FIGURE 3.
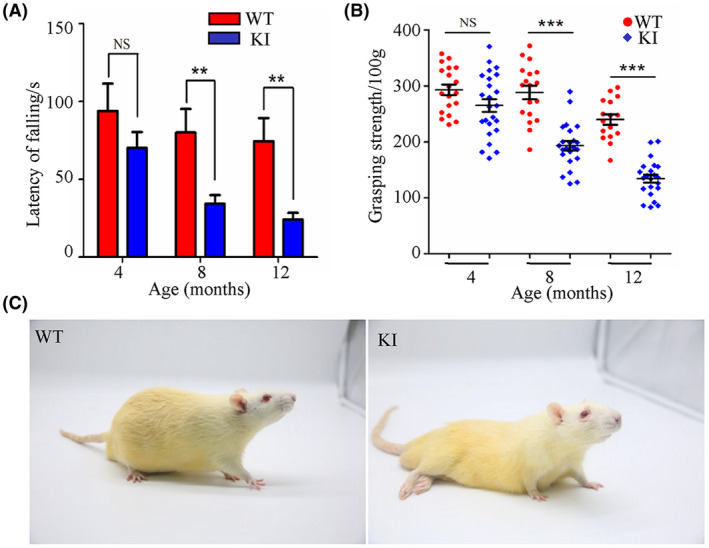
Analysis of motor deficits. The impairment of muscle and motor coordination of KI rats was determined using the Rotarod test at 4, 8 and 12 mo of age (A, n = 16‐19 for WT, n = 22‐24 for KI, female). The grip force of the limbs was measured by the grip test (B, n = 16‐19 for WT, n = 22‐24 for KI, female). The female KI rats developed hind limb paralysis by 13 mo of age (C, n = 25). NS, no significant difference; *P < .05, **P < .01, ***P < .001
3.4. HRE knockin decreased the number of motor neurons in the spinal cord
Sections of lumbar spinal cord tissues from WT and KI rats at 13 months of age were stained with cresyl violet dye, which revealed the Nissl bodies of the motor neurons, and the number of motor neurons was counted in a given area. Neurons in the lumbar spinal cord from WT rats showed a normal shape with the nucleus in the center and purple blue Nissl bodies in the soma, whereas neurons in the KI rats showed eccentric nuclei and lighter staining in the soma, indicating a reduction in Nissl bodies and neuronal vacuolation (Figure 4A; n = 3, female). The HRE knockin reduced the number of motor neurons in the lumbar spinal cord from KI rats by 47% compared to that in WT rats (Figure 4B; n = 3, female, P < .001). These results demonstrated that knockin of the HRE in rats induced severe motor neuron degeneration.
FIGURE 4.
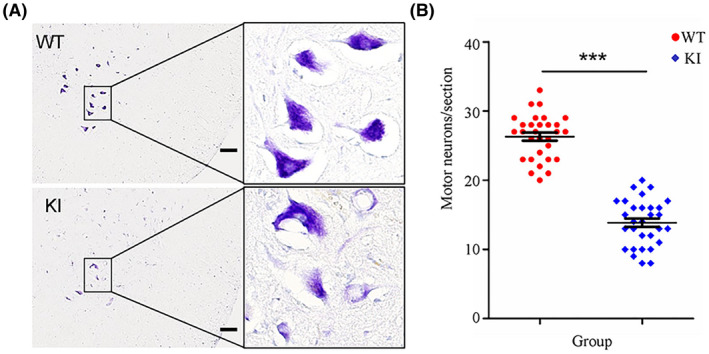
Staining and counting of motor neurons. The lumbar spinal cord tissues of WT and KI rats (n = 3/group) were processed using a standard procedure and sections of lumbar segments at L2 to L4 were stained with cresyl violet. The right ventral horns are shown (A, scale bar = 100 μm). Cells with diameters larger than 20 μm and with a polygonal shape and prominent nucleoli in the ventral horns were counted as motor neurons (B, n = 3 rats/group and 10 serial sections/rat, female). ***P < .001
4. DISCUSSION
C9orf72 HRE‐containing transcripts form nuclear RNA foci that may sequester RNA‐binding proteins 5 , 7 , 32 , 33 or initiate a non‐ATG RAN translation to produce C9orf72 RAN dipeptide‐repeat (DPR) proteins. 9 , 34 , 35 The HRE‐induced RNA foci and DPR proteins associated with the C9orf72 locus are considered to act as one of the gene function mechanisms causing the neurotoxicity seen in ALS. The HRE was also found to block expression of C9orf72 or alter the expression ratios of the translated protein isoforms. This constitutes another possible loss‐of‐function mechanism of neurodegeneration. 18 , 19 , 36 The evidence indicates that the C9orf72 proteins interact with small Rab GTPases, cofilin and SMCR8 and regulate autophagy, cell membrane and vesicle trafficking, and lysosomal biogenesis. 22 , 23 , 24 , 25 , 26 , 27 This suggests that the loss of C9orf72 functionality might exacerbate pathogenesis in combination with the gain‐of‐function factor. The relative contributions of C9orf72 loss‐of‐function and gain‐of‐function to neurotoxicity in ALS is still being debated. The neurodegenerative disorders were not detected in mice with either a neural‐specific knockout of C9orf72 or a systemic C9orf72 knockout. 37 , 38 However, our previous results showed that C9orf72 knockout rats developed progressive motor deficits in the presence of excitotoxicity (unpublished results), suggesting that the absence of functional C9orf72 protein sensitized neurons to excitotoxicity.
Here, we report the generation of a rat model of ALS by knockin of the HRE at the C9orf72 locus. Expression of the C9orf72 protein was inhibited by the HRE knockin and this has also been observed in ALS patients with the HRE 18 , 19 (Figure 1). As evident from other published data for the systemic C9orf72 knockout mouse, 14 abnormalities of the cellular immune system were induced by the reduction of C9orf72 protein in KI rats (Figure 2). The KI rats developed progressive, chronic motor deficits from 4 months of age and hind limb paralysis occurred in female KI rats after 13 months of age (Figure 3). Severe degeneration of motor neurons was also detected in the KI rats (Figure 4). So far, four BAC transgenic mouse models have been generated in different labs and the related ALS phenotypes are summarized and compared with our KI rats (Table 1). The BAC (G4C2) 500 and BAC (G4C2) 100‐1000 transgenic C57BL/6J mice expressed transcripts of the human G4C2 repeats at levels similar to those from endogenous C9orf72 in human brain tissues. Neither of the two mouse models suffered motor neuron loss or motor deficits. 14 , 15 The BAC (G4C2) 450 transgenic C57BL/6J mice expressed the mRNA for the human (G4C2) repeats at a level that was eight times higher than that of endogenous C9orf72 mRNA. This model showed spatial learning defects but no motor deficits. 16 The BAC (G4C2) 500 transgenic FVB/NJ mice expressed the human (G4C2) repeat mRNA at levels similar to those from endogenous C9orf72. These mice showed progressive motor neuron loss, motor deficits and hind limb paralysis. 17 All of the four mouse models produced RNA foci and DPR proteins, but no haploinsufficiency of the human HRE.
TABLE 1.
Comparison of ALS‐related behavioral features of four published C9 BAC transgenic mouse models and our KI rats
| Model | Size of human C9orf72 DNA fragment | Human DNA transcript | C9orf72 protein levels | Motor neuron loss | Motor deficits | Hindlimb paralysis |
|---|---|---|---|---|---|---|
| A:BAC(G4C2)500 transgenicC57BL/6J 14 | BAC DNA including exon1‐5, 800 G4C2 repeats and 141Kb of promoter region. | Mix of 300‐500 repeats expressed at similar levels to human. | No change of the endogenous C9orf72 protein. | Undetected | Undetected | Undetected |
| B: BAC(G4C2)100‐1000 transgenic C57BL/6J 15 | BAC DNA including 800 G4C2 repeats and complete C9orf72 gene. | Mix of 100‐1000 repeats expressed at similar levels to human. | C9orf72 protein increased from transgenic mRNA. | Undetected | Undetected | Undetected |
| C: BAC(G4C2)450 C57BL6/C3H 16 | BAC DNA including exon1‐5, 450 G4C2 repeats and 141Kb of promoter region. | 450 repeats expressed up to 8‐fold higher than human levels. | No change of the endogenous C9orf72 protein. | Undetected | Undetected | Undetected |
| D: BAC(G4C2)500 transgenic FVB/NJ 17 | BAC DNA including 830 G4C2 repeats and complete C9orf72 gene. | 500 repeats expressed at similar levels to human. | C9orf72 protein increased from transgenic mRNA. | Detected | Detected | Detected |
| E: (G4C2)80 knockin SD rats | DNA including exon1a and exon1b, 80 G4C2 repeats and the flanking regions. | (G4C2)80 was derived by endogenous promotor and assumed to be at similar levels to WT rats. | C9orf72 protein reduced by 50%. | Detected | Detected | 25% in female rats. |
In summary, our HRE knockin rats showed haploinsufficiency of HRE and developed physical defects in balance and muscle function similar to those in ALS. These KI rats could be a useful tool for investigating the relative contributions of C9orf72 loss‐of‐function and gain‐of‐function to neurotoxicity and the mechanism of neurodegeneration in ALS.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
LFZ conceived and designed the experiments, LFZ and LZ wrote and revised the main manuscript text; WD performed the main experiments. CXS supervised the pathological observation. FFG and YWM complete the design and construction of rat model. XG and JL performed the genotyping and management of the knockout rats. WC performed the microinjection of knockout rats. All authors have read and approved the final manuscript.
ACKNOWLEDGMENTS
The present work was supported in part by National Natural Science Foundation of China (81571222), CAMS Innovation Fund for Medical Sciences (CIFMS, 2016‐I2M‐1‐004) and Beijing Municipal Natural Science Foundation (7172135). We appreciated Dr Charlet‐Berguerand N from university of Strasbourg kindly shared the DNA of hexanucleotide repeat expansion with us.
Dong W, Zhang L, Sun C, et al. Knock in of a hexanucleotide repeat expansion in the C9orf72 gene induces ALS in rats. Anim Models Exp Med. 2020;3:237–244. 10.1002/ame2.12129
REFERENCES
- 1. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron. 2011;72(2):257‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron. 2011;72(2):245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol. 2012;11(4):323‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moens TG, Partridge L, Isaacs AM. Genetic models of C9orf72: what is toxic? Curr Opin Genet Dev. 2017;44:92‐101. [DOI] [PubMed] [Google Scholar]
- 5. Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length‐dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cooper‐Knock J, Walsh MJ, Higginbottom A, et al. Sequestration of multiple RNA recognition motif‐containing proteins by C9orf72 repeat expansions. Brain. 2014;137(Pt 7):2040‐2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335‐1338. [DOI] [PubMed] [Google Scholar]
- 9. Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mizielinska S, Gronke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science. 2014;345(6201):1192‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohki Y, Wenninger‐Weinzierl A, Hruscha A, et al. Glycine‐alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol Neurodegener. 2017;12(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaw MP, Higginbottom A, McGown A, et al. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol Commun. 2018;6(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Swaminathan A, Bouffard M, Liao M, et al. Expression of C9orf72‐related dipeptides impairs motor function in a vertebrate model. Hum Mol Genet. 2018;27(10):1754‐1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Rourke JG, Bogdanik L, Muhammad A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peters OM, Cabrera GT, Tran H, et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88(5):902‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD‐linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC‐containing RNAs. Neuron. 2016;90(3):535‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Pattamatta A, Zu T, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90(3):521‐534. [DOI] [PubMed] [Google Scholar]
- 18. van Blitterswijk M, Gendron TF, Baker MC, et al. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015;130(6):863‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu EY, Russ J, Wu K, et al. C9orf72 hypermethylation protects against repeat expansion‐associated pathology in ALS/FTD. Acta Neuropathol. 2014;128(4):525‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waite AJ, Baumer D, East S, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35(7):1779.e5‐1779.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao S, MacNair L, McGoldrick P, et al. Isoform‐specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78(4):568‐583. [DOI] [PubMed] [Google Scholar]
- 22. Corbier C, Sellier C. C9ORF72 is a GDP/GTP exchange factor for Rab8 and Rab39 and regulates autophagy. Small GTPases. 2017;8(3):181‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Webster CP, Smith EF, Grierson AJ, De Vos KJ. C9orf72 plays a central role in Rab GTPase‐dependent regulation of autophagy. Small GTPases. 2018;9(5):399‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sivadasan R, Hornburg D, Drepper C, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19(12):1610‐1618. [DOI] [PubMed] [Google Scholar]
- 25. Aoki Y, Manzano R, Lee Y, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140(4):887‐897. [DOI] [PubMed] [Google Scholar]
- 26. Amick J, Roczniak‐Ferguson A, Ferguson SM. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016;27(20):3040‐3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi Y, Lin S, Staats KA, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma Y, Zhang X, Shen B, et al. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 2014;24(1):122‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bohlen M, Cameron A, Metten P, Crabbe JC, Wahlsten D. Calibration of rotational acceleration for the rotarod test of rodent motor coordination. J Neurosci Methods. 2009;178(1):10‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Papalia I, Tos P, Stagno d'Alcontres F, Battiston B, Geuna S. On the use of the grasping test in the rat median nerve model: a re‐appraisal of its efficacy for quantitative assessment of motor function recovery. J Neurosci Methods. 2003;127(1):43‐47. [DOI] [PubMed] [Google Scholar]
- 31. Bai L, Shi GY, Yang YJ, Chen W, Zhang LF, Qin C. Rehmannia glutinosa exhibits anti‐aging effect through maintaining the quiescence and decreasing the senescence of hematopoietic stem cells. Animal Model Exp Med. 2018;1(3):194‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harley HG, Brook JD, Rundle SA, et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature. 1992;355(6360):545‐546. [DOI] [PubMed] [Google Scholar]
- 33. Almeida S, Gascon E, Tran H, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC‐derived human neurons. Acta Neuropathol. 2013;126(3):385‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gendron TF, Chew J, Stankowski JN, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72‐associated amyotrophic lateral sclerosis. Sci Transl Med. 2017;9(383):eaai7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mori K, Arzberger T, Grasser FA, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881‐893. [DOI] [PubMed] [Google Scholar]
- 36. Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koppers M, Blokhuis AM, Westeneng HJ, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78(3):426‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Rourke JG, Bogdanik L, Yanez A, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351(6279):1324‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]