

Abstract
Several studies have suggested a role for the mammalian target of rapamycin (mTOR) in skeletal development and homeostasis, yet there is no evidence connecting mTOR with the key signaling pathways that regulate skeletogenesis. Herein we identified a PTH/PTHrP-SIK3-mTOR signaling cascade essential for skeletogenesis. While investigating a new skeletal dysplasia caused by a homozygous mutation in the catalytic domain of SIK3, we observed decreased activity of mTORC1 and mTORC2 due to accumulation of DEPTOR, a negative regulator of both mTOR complexes. This SIK3 syndrome shared skeletal features with Jansen metaphyseal chondrodysplasia (JMC), a disorder caused by constitutive activation of the PTH/PTHrP receptor. JMC derived chondrocytes showed reduced SIK3 activity, elevated DEPTOR and decreased mTORC1 and mTORC2 activity, indicating a common mechanism of disease. The data we demonstrate that SIK3 is an essential positive regulator of mTOR signaling that functions by triggering DEPTOR degradation in response to PTH/PTHrP during skeletogenesis.
Keywords: SIK3, mTOR, PTH1R, DEPTOR, skeletal dysplasia, Jansen’s metaphyseal chondrodysplasia, metaphyseal dysplasia Csukasi-Krakow type
One sentence summary:
Disruption of a PTH/PTHrP-SIK3 pathway impairs degradation of DEPTOR, affects mTORC1/mTORC2 signaling, and leads to abnormalities in skeletal development
INTRODUCTION
Multiple signaling pathways act in concert to regulate skeletogenesis, including WNT, TGF-β/BMP, Hedgehog, Fibroblast Growth Factor and Parathyroid Hormone/ Parathyroid Hormone-like peptide PTH/PTHLH (or PTHrP) among others (reviewed in 1). PTH/PTHLH governs chondrocyte proliferation and hypertrophy through the binding of PTH or PTHrP to a common receptor, Parathyroid Hormone Receptor-1 (PTH1R). Pthlh knockout mice show decreased chondrocyte proliferation, premature chondrocyte maturation and accelerated bone formation (2, 3). Similarly, in Pth1r knockout mice proliferative chondrocytes prematurely leave the cell cycle and differentiate (4), demonstrating the importance of this pathway in regulating coordinated chondrocyte proliferation and differentiation. Further, activation of PTH1R, a G-protein-coupled receptor initiates a cascade of events that result in the inhibition of two key transcription factors, MEF2 and RUNX2, additional regulators of chondrocyte hypertrophy (5, 6). In humans, heterozygosity for an activating mutation in PTH1R produces metaphyseal chondrodysplasia Jansen Type (JMC) (7), a disorder characterized by hypercalcemia with suppressed concentration of PTH and PTHrP, short bowed limbs, and a distinctive radiographic pattern (8).
The mechanistic target of rapamycin (mTOR) is an evolutionary conserved serine-threonine kinase that integrates diverse environmental stimuli, including nutrients and growth factors, and translates them into a wide variety of central cellular responses (9). mTOR forms the catalytic subunit of two functionally distinct multi-protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1, composed of the proteins RAPTOR, PRAS40, mLST8/GβL and DEPTOR, plays a major role in the regulation of cell growth and proliferation in response to nutrients, amino acids, growth factors and energy sufficiency. The best characterized substrates of mTORC1 are the ribosomal S6 kinases (S6Ks). mTORC2 is less characterized than mTORC1; it is involved in cell survival in response to growth factors, mostly by promoting the activation of AKT. mLST8/GβL and DEPTOR are found in both mTOR complexes, whereas RICTOR, mSIN1 and PROTOR are unique components to mTORC2 (reviewed in 10). mTORC1 and mTORC2 signaling are essential for chondrocyte proliferation and differentiation, as well as bone development. Mouse mutants that lack Raptor or Rictor show delayed chondrocyte hypertrophy and bone formation, (11, 12) and recently, mTORC1 activity was shown to coordinate chondrocyte proliferation and hypertrophy in part through activation of PTHrP transcription (13).
Salt-inducible kinase 3 (SIK3) is a member of the 5’-AMP-activated serine-threonine protein kinase (AMPK) family that comprises 14 members (14, 15). In mammals, the SIK subfamily is composed of SIK1, SIK2 and SIK3, kinases that share a conserved amino-terminal catalytic domain but differ in sequence and function at their carboxyl-terminal (15). SIKs have been mostly characterized in relation to the regulation of hepatic glucose metabolism following their phosphorylation and activation by LKB1, a master kinase for the AMPK family and a negative regulator of mTORC1 signaling (16–18). Although the function of SIK3 is less precisely understood, it has been shown to be important for normal skeletal development; Sik3 knockout mice show growth plate abnormalities associated with delayed chondrocyte hypertrophy and primary bone formation (19). Although there is no evidence of a connection between SIK3 and PTH/PTHrP signaling, deletion of SIK2 in osteocytes demonstrated that SIK2 is required for the cellular response to PTH; PTH signaling inhibits SIK2 by triggering its phosphorylation (20). Conversely, SIK3 deficient osteocytes showed normal responsiveness to PTH, suggesting that, at least in osteocytes, PTH specifically targets SIK2 (20). To date, mutations in SIK2 and SIK3 have not been associated with human disease, whereas heterozygosity for mutations in SIK1 are associated with early infantile epileptic encephalopathy (21).
Herein we describe a previously uncharacterized recessively inherited skeletal disorder sharing numerous radiographic similarities to JMC. The disorder resulted from homozygosity for a missense mutation in the catalytic domain of SIK3. Loss of SIK3 kinase activity triggered accumulation of the mTOR negative regulator DEPTOR, leading to downstream impairment of mTORC1/mTORC2 activity. Further, constitutive activation of the PTH1R as seen in JMC inhibited SIK3 activity and caused similar downregulation of mTOR signaling, through increased DEPTOR amounts. Our results support a common mechanism of disease in two differing metaphyseal chondrodysplasias and demonstrate a key role for a newly described PTH1R-SIK3-DEPTOR pathway in skeletal development.
RESULTS
A novel skeletal disorder is caused by a recessively inherited mutation in SIK3
We identified siblings (International Skeletal Dysplasia Registry numbers R07–429A and R07–429B) with radiographic characteristics similar to dominantly inherited JMC but with unique features that did not match any of the currently classified skeletal disorders. The affected siblings came from a consanguineous family, suggesting a recessively inherited disorder due to homozygosity for a mutation, identical by descent. The clinical and radiographic phenotypes are detailed in Table S1. In addition to the skeletal phenotype, the siblings manifested significant developmental delay with brain MRI abnormalities, a severe unclassified immunodeficiency, and normal parathyroid hormone concentration with mild hypercalcemia. Radiographic findings included widened/flared metaphyses with irregular ossifications, motheaten long bones, fragmentation of the proximal metacarpals, rounded vertebral bodies, and a distinctive transverse gap seen in the tibias (Figure 1A–F, TableS1). Affected individual R07–429A had a more severe phenotype, particularly in her immune system, and died of an Epstein-Barr virus induced small muscle cancer at 10 years of age. The affected sibling, R07–429B, is still alive at age 14.
Figure 1.

Homozygosity for a missense mutation in SIK3 produces a novel skeletal disorder. (A-F) Radiographs of R07–429A and R07–429B showing a large separation of the irregular epiphyses from metaphyses, widened/flared metaphyses with irregular ossifications and irregular ossification front (A-B), brachydactyly with irregular metacarpal and phalangeal metaphyses and delayed epiphyseal ossification (C-D), absence of pubic bone ossification (E), delayed ossification of the basal occipital bone and subjectively increased density of the skull (F). (G-I) SIK3 protein and mRNA amounts in control and patient fibroblasts. Western blots showing SIK3 protein concentrations in patient derived fibroblasts compared to control cells (G-H). SIK3 gene expression as determined by qPCR (I). Graphs represent average of three independent experiments ± SE. Statistically significant changes are indicated (Student’s t-test, * p<0.05).
Because the radiographic phenotype overlapped with JMC the coding exons of PTH1R were sequenced and no mutations were identified. Exome sequencing was carried out and the data filtered under a recessive model. This analysis identified homozygosity for a missense variant c.385C>T in exon 2 of SIK3 (NM_025164), predicting the amino acid substitution p. R129C (Figure S1A). The unaffected brother and parents were heterozygous for the change. Fibroblast cells derived from the two patients were used to assess the effect of the SIK3 variant on mRNA and protein amounts. The expression of the gene was unaffected but SIK3 protein was decreased more than 60% in cells derived from both affected individuals (Figure 1G–I), indicating that the variant compromises the stability and/or synthesis of the protein. Further supporting the human phenotypic and molecular findings, Sik3 knock-out mice show skeletal defects (19).
SIK3R129C shows decreased kinase activity
To determine if the mutation affected SIK3 kinase activity, we first performed a 3D in silico homology modeling of SIK3 catalytic domain and found that mutated Arg129 adjoins Asp130, a conserved residue directly involved in substrate binding (Figure 2B–C). The position of Arg129 in the structure, along with its evolutionary conservation (Figure 2A), indicated its importance for maintaining the catalytically active conformation of the kinase. The p.R129C substitution exchanges a large positively charged amino acid for a small neutral amino acid, possibly altering local conformation in the vicinity of the substrate binding site, and/or interfering with proper positioning of the substrate binding site with respect to the activation loop (Figure 2B–C).
Figure 2.
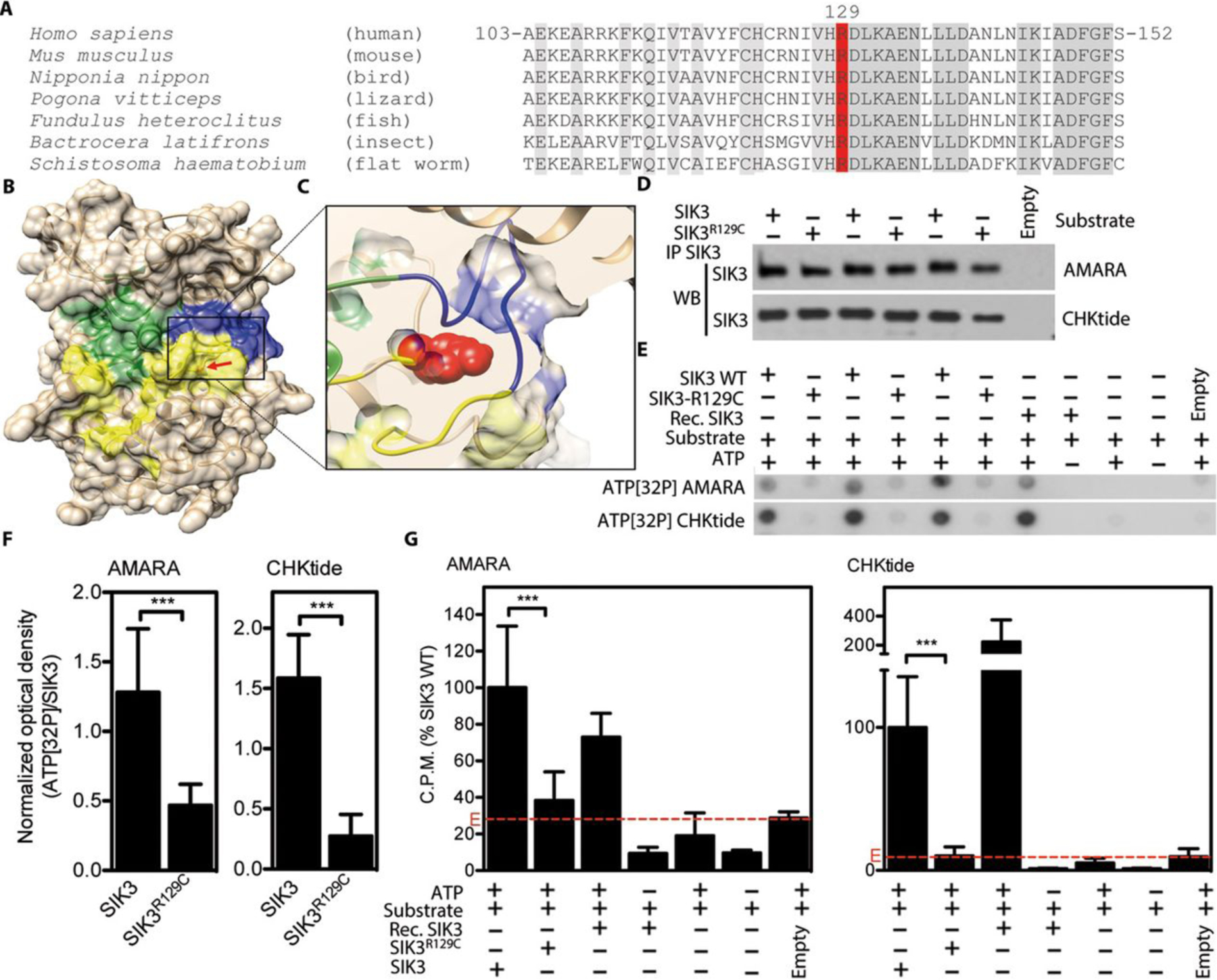
Impaired kinase function in SIK3R129C mutant. (A) Alignment of SIK3 orthologs demonstrating evolutionary conservation of p.R129 (conserved residues in grey; R129 in red). (B) 3D homology model of SIK3 kinase domain with indicated ATP binding site (green), substrate binding site (yellow) and activation loop (blue); arrow indicates position of R129. (C) Zoomed in view of the mutant p.R129 (red) surroundings. (D) C-terminally FLAG-tagged wildtype (WT) and R129C SIK3 variants were transfected into HEK293T cells and purified by FLAG immunoprecipitation (IP). The quantities of three independent transfections for each SIK3 variant were determined by western blot (WB). Labeled Empty were cells transfected with empty plasmid. (E-G) Purified SIK3 was used as a kinase in cell-free kinase assays with radioactive ATP and AMARA or CHKtide peptide as substrates. Phosphorylation signal was determined by ATP[32P] autoradiography of kinase reaction spotted on blotting paper (E, F), or by scintillation (G). Recombinant active SIK3 was used as a positive control for kinase activity, while samples with ATP omitted served as negative controls. (F) The ATP[32P] signal shown in (E) was normalized to concentration of immunoprecipitated SIK3 in each reaction and plotted. Data represent compilation of three (AMARA) and four (CHKtide) experiments, with each individual sample measured twice. Statistically significant changes are indicated (Student’s t-test, *** p<0.001). (G) ATP[32P] scintillations expressed as percentages of signal obtained in kinase assays with WT SIK3. The signal obtained in immunocomplexes isolated from cells transfected with empty plasmid instead of SIK3 (E, red lines) represent the background activity. Note the significantly impaired kinase activity of SIK3-R129C, compared to WT SIK3.
To test this prediction, we compared the catalytic activity of the mutant SIK3 (SIK3R129C) with wildtype SIK3 (WT SIK3). WT SIK3 and SIK3R129C constructs were expressed in HEK293T cells, immunoprecipitated, and used in cell-free kinase assays with AMARA or CHKtide peptides as substrates (Figure 2D). The SIK3-mediated phosphorylation of both AMARA and CHKtide was significantly decreased in the presence SIK3R129C compared with WT SIK3 (Figure.2E–G). These data demonstrated that the p.R129C mutation led to impairment of SIK3 Ser/Thr kinase activity.
SIK3 regulates mTOR activity
As previously mentioned, SIK3 is a member of the AMPK family of proteins. Because AMPK inhibits mTORC1 through direct phosphorylation of TSC2 (22) and Raptor (23), we tested if SIK3 also regulated mTOR signaling. We used patient cells to measure the phosphorylation of S6K1 and S6, two downstream effectors of mTORC1 and AKT, a readout for mTORC2 complex activity under serum starvation (inactivation of mTOR). As shown in Figure 3A we observed a clear decrease in the phosphorylation of S6K1, S6 and AKT in the patient cells compared to control (Figure 3A, S2A), indicating that SIK3 is a positive regulator of mTORC1 and mTORC2 activity, and that mTOR signaling in impaired in SIK3R129C cells.
Figure 3.

SIK3 deficient cells show decreased mTORC1 and mTORC2 activity due to accumulation of DEPTOR. (A) Control and patient fibroblasts were starved O/N before protein extraction. Western blots showing amounts of indicated proteins in patient and control cells. (B) Co-immunoprecipitation of SIK3 with different mTORC1/2 components. C-terminally FLAG-tagged wildtype (SIK3) and R129C SIK3 variants were transfected into HEK293T cells and purified by FLAG immunoprecipitation (IP). Co-immunoprecipitation (Co-IP) of endogenous RICTOR, RAPTOR and GβL was detected by western blot. (C) C-terminally FLAG-tagged wildtype (SIK3) and R129C SIK3 variants and N-terminally HA-tagged DEPTOR were co-transfected into HEK293T cells and purified by HA IP. Co-IP of SIK3 was detected by western blot. (D) Control and patient derived fibroblasts were starved O/N and then treated with 10% FBS for 4 h, 7 h and O/N. Western blots of starved and serum stimulated control and patient cells showing amounts of indicated proteins. (E) Western blots of control fibroblasts transfected with 10 nM siRNA (siRNA) and control siRNA (Control). After 48 h cells were starved O/N and then treated with 10% FBS for 4 h and O/N. Western blots showing amounts of indicated proteins. (F) N-terminally HA-tagged DEPTOR was transfected into control and patient fibroblasts. 24 h after transfection, cells were starved O/N and then either harvested or treated with 10% serum plus 15 μM MG132 for 10 h. DEPTOR was immunopurified using HA beads and its interaction with endogenous β-TRCP was detected by western blot. EV (Empty vector) was used as a negative control. WCL (Whole cell lysates).
We next investigated whether SIK3 directly interacted with components of the mTORC1 and mTORC2 complexes. Westernblot analysis of WT SIK3 immunoprecipitated from transfected HEK293T cells detected a clear co-immunoprecipitation of RICTOR (mTORC2) and GβL (mTORC1/2) and a weak interaction with RAPTOR (mTORC1) (Figure 3B). SIK3R129C also interacted with these respective proteins. Because HEK293T cells have very low expression of the mTORC1/2 component DEPTOR (24), we co-transfected DEPTOR with WT SIK3 and SIK3R129C and showed that SIK3 can also interacted with DEPTOR (Figure 3C).
Impaired proteasome DEPTOR degradation in SIK3 deficient cells
Because SIK3 deficiency induced a decrease in mTORC1 and mTORC2 activity we reasoned that one of the components common to both complexes (GβL and/or DEPTOR) might be responsible for the reduced activity on substrates. Therefore, we measured GβL and DEPTOR protein amounts in mutant and control cells under three different conditions, i.e. serum starvation (to inactivate mTOR) and 30 minutes of serum or insulin treatment (to activate mTOR). Under all of these conditions there was a greater accumulation of DEPTOR in mutant versus control cells, whereas no change was observed in GβL concentration (Figure 3A, S2A–S3).
DEPTOR is a negative regulator of mTORC1 and mTORC2 and is phosphorylated and degraded after several hours of serum stimulation (24). We therefore investigated DEPTOR amounts in the patient cells after longer exposures to serum and found that DEPTOR remained higher in patient versus control cells, after four and seven hours of treatment but completely degraded after overnight exposure (Figure 3D, S2B). These results demonstrate that SIK3 contributes to DEPTOR degradation upon serum stimulation and while delayed in mutant cells ultimately is degraded because DEPTOR is also phosphorylated by other kinases, including mTOR (25–28).
To confirm that SIK3 promoted mTOR activity by triggering DEPTOR degradation we measured the phosphorylation of S6K1, S6 and AKT in the same prolonged exposures to serum (four hours, seven hours and overnight treatments) and found that the lower amounts of pS6K1, pS6 and pAKT observed in mutant cells under starvation and after 30 minutes of serum and insulin treatments similarly recovered under these conditions, showing that even in mutant cells, progressive degradation of DEPTOR occurred over time restoring mTOR signaling (Figure 3D, S2B, S3).
To further confirm these findings, knock-down of SIK3 utilizing siRNA also induced an accumulation of DEPTOR and a decrease in pAKT, pS6K1 and pS6 under starvation, replicating the results observed in patient cells, whereas no difference was observed after four hours or overnight serum exposure (Figure 3E, S2C).
Phosphorylated DEPTOR is recognized by the F-box protein β−TRCP, for subsequent ubiquitination and proteasome degradation by the SCF E3 ubiquitin ligase complex (25–27). To determine if SIK3 is necessary for DEPTOR interaction with β−TRCP, we transfected control and patient cells with HA-tagged DEPTOR, immunoprecipitated DEPTOR, and detected its interaction with endogenous β−TRCP. The lack of SIK3 kinase activity in the patient cells decreased the interaction between DEPTOR and β−TRCP (Figure 3F), demonstrating that SIK3 contributes to the phosphorylation-dependent DEPTOR degradation by the proteasome.
SIK3 is co-expressed with mTOR components in the growth plate
Based on the clinical phenotype, we explored the role played by SIK3 in skeletal development in relation to mTOR components. Expression patterns of SIK3, pS6 and DEPTOR were determined in P1 and P21 mouse cartilage growth plates by immunohistochemistry. At P1, both SIK3 and pS6 were expressed in early proliferating and pre-hypertrophic chondrocytes (Figure 4B, D, E, G arrows). At P1 DEPTOR was primarily expressed in resting and proliferating chondrocytes (Figure 4C, F). At P21, SIK3, DEPTOR, and pS6 showed overlapping expression patterns particularly in proliferating and pre-hypertrophic chondrocytes (Figure 4I–K, M–O) indicating an ongoing role for SIK3-mTOR activity in chondrocyte behavior in the maturing growth plate.
Figure 4.
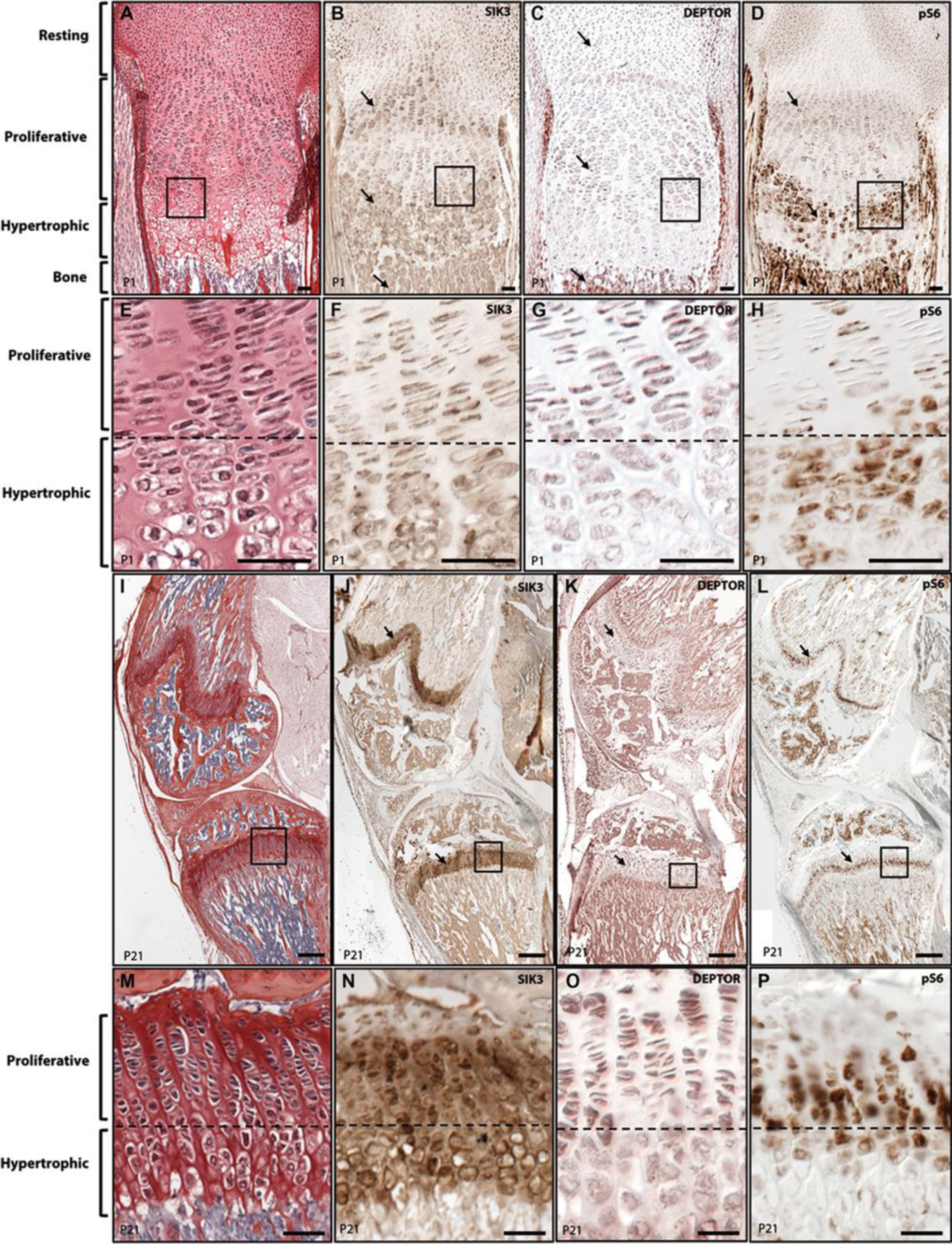
SIK3 is co-expressed with mTOR components in the growth plate. Immunolocalization of SIK3, DEPTOR and pS6 in P1 (A-G) and P21 (H-O) mouse growth plates. (A) Picrosiriuos Red-Haematoxylin staining showing the different differentiation stages of P1 cartilage growth plate chondrocytes. (B-D) Immunolocalization of SIK3 (B) DEPTOR (C) and pS6 (D) at P1. (E-H) Magnification of A-D figures. (I) Picrosiriuos Red-Haematoxylin staining of P21 growth plate (J-L) Immunolocalization of SIK3 (J) DEPTOR (K) and pS6 (L) at P21. (M-P) Magnification of I-L figures.
PTH/PTHrP signaling inhibits mTOR activity through SIK3
Because of the similar skeletal abnormalities between the patients with deficient SIK3 and the JMC patients with over activation of PTH1R, we hypothesized that SIK3 was regulated by PTH/PTHrP signaling. Using chondrocytes derived from a patient with JMC (R93–393), we measured the concentration of total cellular DEPTOR and the activation of mTORC1 and mTORC2 substrates under serum starvation and after 4h of serum stimulation. As shown in Figures 5A and S4, DEPTOR protein was elevated in the JMC patient cells compared to the controls under serum deprivation. As seen in SIK3 deficient cells, JMC cells showed persistence of DEPTOR protein after serum treatments relative to controls, indicating that PTH/PTHrP/PTH1R pathway is also involved in DEPTOR degradation. Concordant with this finding, the phosphorylation of S6 and AKT were also decreased (Figure 5A, S4). To determine if PTH/PTHrP/PTH1R might regulate DEPTOR accumulation through controlling the activity of SIK3, we next measured the phosphorylation of SIK3 at a known activating residue (T163) (15, 29) and found it decreased in the JMC patient cells (Figure 5A, S4). Because SIK2 is inhibited by PTH in osteocytes (20); we questioned whether SIK2 phosphorylation was also affected in JMC; JMC chondrocytes showed a clear decrease in pSIK2 at T175, known to be important for its activity (30) (Figure 5A, S4). Treatment of primary chondrocytes with PTHrP showed a decrease in SIK3 activity measured by pSIK3T163, consistent with our results using JMC chondrocytes (Figure S5) and further suggesting that PTHrP-PTHR pathway is a negative regulator of SIK3. No significant change was observed in the activity of SIK2 after the treatment (Figure S5). Histological analysis of a growth plate from a patient with JMC showed a severe disorganization with a hypocellular reserve zone; a reduced proliferative region with clusters of late proliferating and diminished number of hypertrophic chondrocytes that fail to organize into columns; and significant invasion of bone territories (spicules or mineralization front) into the cartilaginous growth plate as observed by intense picrosirius staining (Figure 5B–E). The absence of hypertrophic chondrocytes has also been described in the Sik3 knock-out mouse and the authors concluded that the major effect of Sik3 deficiency in skeletal development was disruption of chondrocyte hypertrophy (19). Immunolocalization of DEPTOR in human cartilage growth plate showed that in control tissues its expression is higher in resting and proliferative chondrocytes decreasing upon commitment to the hypertrophic program (Figure 5F, H). In contrast, JMC patient growth plate showed a constant and higher expression of DEPTOR throughout the poorly organized growth plate (Figure 5G, I), concordant with western blot analyses that showed increased amounts of DEPTOR. Altogether, these results support that in WT chondrocytes, PTH/PTHrP inhibits SIK3 (and SIK2 in osteocytes, but not in chondrocytes) phosphorylation and acts as a negative regulator of mTORC1/2 activity,. In JMC because of constitutive activation of the PTH/PTHrP pathway, there is exaggerated and persistent inhibition of SIK3 leading to DEPTOR accumulation and subsequent decrease in mTOR activity (Figure 5J).
Figure 5.
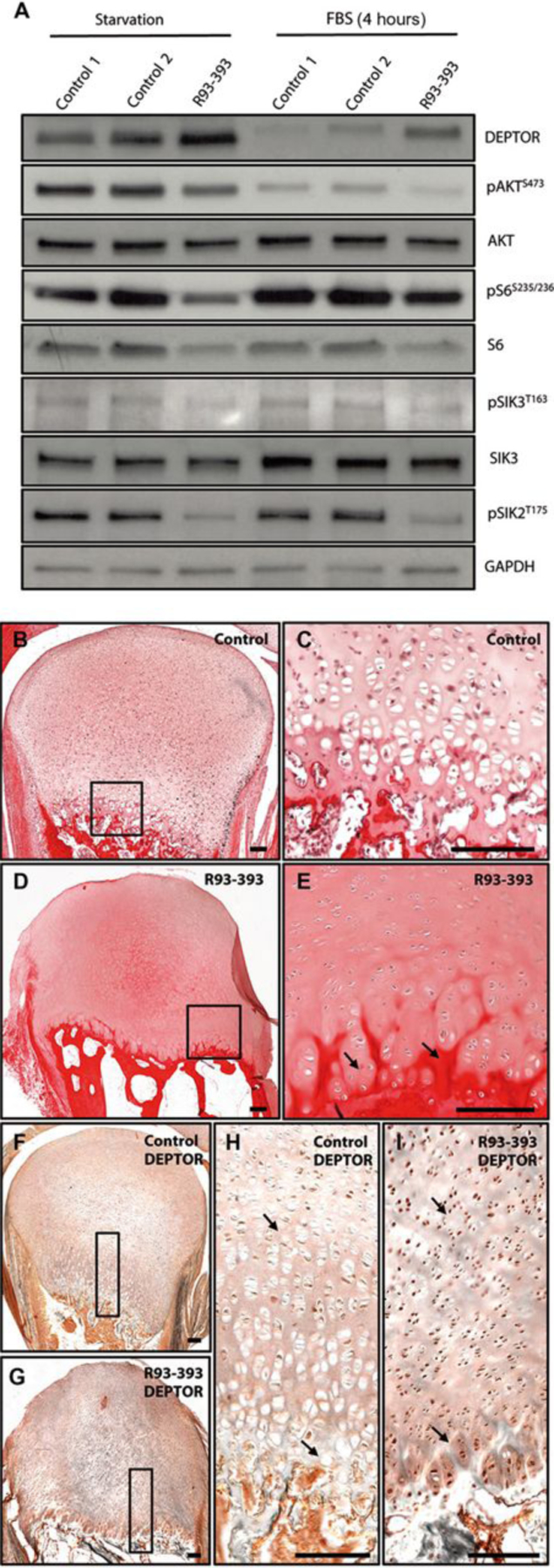
PTH/PTHrP signaling regulates mTOR activity through SIK3 mediated DEPTOR degradation. (A) JMC derived chondrocytes were starved O/N and then treated with 10% FBS for 4 h. Western blots showing amounts of indicated proteins in patient and control cells. (B-E) Picrosiriuos Red-Haematoxylin staining of control (B-C) and JMC patient (D-E). (F-I) Immunolocalization of DEPTOR in control (F, H) and patient growth plates (G, I). (J) Model of action of SIK3. Different upstream signals control SIK3 activity. Under nutrient deficiency DEPTOR inhibits mTORC1 and mTORC2. Serum stimulation activates SIK3 and promotes DEPTOR interaction with β-TRCP inducing its degradation by the proteasome. In the absence of PTH/PTHrP, SIK3 triggers DEPTOR degradation through the same mechanism. PTH/PTHrP binding to its receptor (PTH1R) inhibits SIK3 phosphorylation and therefore its activity, inducing DEPTOR accumulation and subsequent mTOR inhibition.
DISCUSSION
In this study we identify a novel signaling pathway connecting PTH/PTHrP, SIK3, and mTOR and demonstrate that it has a key role in regulation and maintenance of skeletal development. The findings were determined based on uncovering a new recessively inherited skeletal dysplasia similar to autosomal dominantly inherited JMC but with several distinctions. The causative gene encodes SIK3 and the change is within the highly conserved kinase domain, resulting in decreased Ser/Thr kinase activity. Supporting the human phenotypic findings, Sik3 knock-out mice show similar skeleton defects, including metaphyseal expansion of the limb joint region, shortened long bones, hypoplastic pelvis, and delayed membranous ossification of the skull bones (large fontanelle remained open from juvenile stage until adulthood) (19). Addressing whether this family represents an isolated finding, review of the literature identified a report of a one year old male from a consanguineous mating with similar radiographic and clinical findings to our patients, particularly the findings of the frontal bossing, hypertelorism, prominent joints, psychomotor delay, hypercalcemia, a transverse gap in the ulna and significant epiphyseal delay with absent ossification of the pubis, and a highly irregular distorted metaphyses (31). JMC was considered in that report but excluded as final diagnosis based on the unique constellation of findings. Interrogation of the International Skeletal Dysplasia Registry, identified another case with similar findings, particular the finding of a transverse gap in the long bones from another consanguineous mating. Attempts were made to obtain DNA from these cases but failed due to passage of time and loss of contact. However, these other cases support that this new disorder is not isolated solely to this family.
We demonstrate that SIK3 promotes the activity of mTOR, a central regulator of mammalian metabolism and physiology. Furthermore, our data expands the knowledge on how mTOR is rapidly switched from a low to a high activity state through SIK3-dependent DEPTOR degradation in response to nutrient sufficiency. Several lines of evidence support this model. First, SIK3 directly interacts with DEPTOR. Second, mutant SIK3 patient cells show a high accumulation of DEPTOR protein under starvation and after short serum treatments; DEPTOR amounts return to normal after long-term serum treatments, corresponding with recovery of normal activity of mTORC1 and mTORC2. Third, DEPTOR protein in patient cells is incapable of interacting with β−TRCP, the F-box subunit of a SCF E3 ubiquitin ligase complex, further explaining why it remains high in patient cells. Finally, knock-down of SIK3 resulted in increased DEPTOR concentration, phenocopying mutant kinase deficient SIK3. Recently, SIK3 was proposed as a downstream target of mTORC2; however, our results position SIK3 upstream of mTOR, suggesting the existence of some kind of feedback between them. Supporting this hypothesis is that DEPTOR and mTOR also form a feedback loop, mTOR kinase activity is partially responsible for DEPTOR phosphorylation and degradation (25–27). Other kinases that phosphorylate DEPTOR include Casein Kinase 1 (CKI) (25) and the MAPKs p38γ and δ (28); all of them act in cooperation to promote DEPTOR degradation in response to upstream signals. Our work adds a new component to the already complex and tightly regulated mechanism of activation of mTOR.
Deregulation of mTOR activity contributes to many types of diseases including cancer and diabetes (reviewed in 32, 33). Previous work shows that about 28% of multiple myelomas overexpressed DEPTOR (24). To our knowledge, this is the first report linking a genetic disease, and in particular a skeletal dysplasia, with reduced mTOR activity. Although no mutations in any mTOR components have been identified in humans, Raptor, Rictor and TSC1 loss-of-function mice show skeletal defects (11–13), demonstrating the importance of mTOR activity in cartilage and bone formation. We show that SIK3 is present in the growth plate where it is co-expressed with DEPTOR and the mTORC1 substrate S6 in proliferative and early hypertrophic chondrocytes. Previous studies have shown that mTORC1 activity is required for chondrocyte growth but that its inactivation is essential for chondrocyte terminal differentiation, establishing that dynamic regulation of mTORC1 activity is crucial for skeleton development (11, 13). Although mTORC2 signaling is much less characterized in cartilage and bone, high amounts of phosphorylated Akt have been found in proliferative chondrocytes in mice (34). Akt signaling is known to enhance chondrocyte proliferation and delay hypertrophy (35), suggesting that pathogenicity due to diminished SIK3 activity and DEPTOR accumulation on chondrocyte proliferation and differentiation may also result from perturbed mTORC2 activity. Our findings point to the importance of a SIK3-dependent activation of mTORC1 and mTORC2 during chondrocyte development. This activation might also be dependent on the changing upstream signals that surround chondrocytes during differential stages of development.
JMC results from constitutive activation of PTH1R, the PTH/PTHrP receptor (7) and presents with skeletal similarities to those patients with mutant SIK3(8). The findings that DEPTOR is also overexpressed in JMC resulting in lower mTORC1 and mTORC2 signaling and that this correlates with low activity of SIK3 supports that in part a common mechanism of disease is responsible for both skeletal disorders.
While no cartilage growth plate was available for the SIK3 affected patients, the growth plate from the patient with JMC showed grossly abnormal terminal differentiation in the hypertrophic zone with few cells, lack of normal hypertrophy, and poor column formation; supporting the role of DEPTOR-regulated mTOR activity in the development of the human cartilage growth plate. Further, mTORC1 was recently shown to control chondrocyte proliferation and differentiation through the regulation of PTHrP transcription (13). Our results support a model in which active PTH-PTHrP/PTH1R meditated signaling in early proliferative chondrocytes inhibits SIK3 activity leading to an accumulation of DEPTOR and therefore low mTOR activity. Upon commitment to the hypertrophic program, decrease of PTH-PTHrP/PTH1R relieves SIK3 inhibition, triggering DEPTOR degradation and activation of mTOR signaling to promote the initiation of chondrocyte hypertrophy (Figure 5J).
Altogether, our results demonstrate that aberrant PTH/PTHrP-SIK3-mTOR signaling causes similar, but distinct skeletal dysplasias. While there are clinical distinctions based on neurologic and immune systems abnormalities, underscoring the multifunctional role of SIK3, we uncovered a detailed mechanism of overlapping disease in which hyperactivation of PTH1R or SIK3 deficiency leads to decreased mTOR signaling due to impaired DEPTOR turnover. Finally, our results open a door for the development of a new treatment of these and perhaps other skeletal diseases by targeting mTOR components. Furthermore, given the paramount importance of mTOR signaling in cancer, the implications of this work include identifying SIK3 as a new target for cancer drug development, recognizing that one characteristic of some cancers is aberrant concentrations of DEPTOR (24) which could be recovered by modification of SIK3 activity.
MATERIALS AND METHODS
Study design
The objective of this study was to identify the gene responsible for the rare skeletal phenotype observed in two patients. Once the gene was identified we focused on the description of the molecular mechanism underlying the disease. Patients and their unaffected family members were ascertained under an approved human subjects protocol. Clinical information and imaging were obtained from review of all available medical records. All experiments performed in cells were done at least three independent times and all replicates were included in our data analyses. Detailed description of exome analysis and experimental procedures is available in this section.
Cell culture
Dermal fibroblast cultures were established from explanted skin biopsies from the cases (International Skeletal Dysplasia Registry reference numbers R07–429A and R07–429B) and controls. Primary chondrocytes were isolated from distal femurs of the affected individual (International Skeletal Dysplasia Registry reference number R93–393) or age- matched normal controls by incubation of fragmented cartilage with 0.03% bacterial collagenase II. All cells were grown in Dulbecco-Vogt Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS). For serum treatments, cells were starved overnight (no serum) and then treated with the same media supplemented with 10% FBS. For protein analyses, cells were collected in IP Lysis Buffer (Thermo Scientific, 87787) supplemented with proteinase inhibitors.
SIK3 overexpression experiments were performed by introducing a vector containing the SIK3 or SIK3R129C coding sequences tagged with DDK-flag; SIK3-DDK vector was obtained from OriGene (RC223406). Knockdown of SIK3 was performed using three siRNAs (Origene, SR308237) and compared to a siRNA negative control (Origene, SR30004). Electroporation was performed in a Nucleofector X system (Lonza) using Amaxa P1 primary cell kit and program DS-150 for fibroblasts and SE kit and program CM-104 for the immortal chondrocytes (Lonza).
Exome analysis
DNA was isolated, library preparation and exome sequencing was performed as previously described (36). The samples were barcoded, captured using the NimbleGen SeqCap EZ Exome Library v2.0 probe library targeting 36.5Mb of genome, and sequenced on the Illumina GAIIx platform with 50bp bidirectional reads. Novoalign was used to align the sequencing data to the human reference genome (NCBI build 37) and the Genome Analysis Toolkit (GATK) was used for post-processing and variant calling according to GATK Best Practices recommendations. For each sample, at least 90% of targeted bases were covered by at least 10 independent reads. Variants were filtered against dbSNP137, NIEHS EGP exome samples (v.0.0.8), exomes from the NHLBI Exome Sequencing Project (ESP6500), 1000 genomes (release 3.20120430), and in-house exome samples. Mutations were further compared with known disease-causing mutations in HGMD (2012.2). Variants were annotated using VAX34, and mutation pathogenicity was predicted using the programs Polyphen35, Sift36, Condel37, CADD38, and MutationTaster. The mutations reported in this work were confirmed by bidirectional Sanger sequencing of amplified DNA from the probands and the parents. Primer sequences for exon 2 were Fw: CCCAGCTGGATGAAGAAAAC and Rev: GCACACAAGCACGTAGAGGA.
Western blot and immunoprecipitations
For Western blot analyses, protein lysates were separated by electrophoresis on 10% or gradient (4–20%) SDS-polyacrylamide gels, transferred to PVDF membranes, blocked in 5% milk and probed with primary antibodies (anti-SIK3 antibody, 1:2000 Abcam 88495; anti-pSIK2/SIK3 antibody, 1:1000 Thermo Fisher Scientific PA5–64607; anti-pAKT 1:1000, Cell Signaling 4060; anti-pS6K1 1:1000 Cell Signaling 9205; anti-pS6 1:1000 Cell Signaling 2211; anti-AKT 1:1000 Cell Signaling 4685; anti-S6K1 1:1000 Cell Signaling 9202; anti-S6 1:1000 Cell Signaling 2317; anti-GAPDH 1:2000 Cell Signaling 2118S). Peroxidase-conjugated secondary antibodies (Cell Signaling 7071 and 7072) were used and immunocomplexes were identified using the ECL detection reagent (Cell Signaling 7003). FIJI was used to quantify bands following Gel Analysis recommendations from ImageJ and (37) (http://rsb.info.nih.gov/ij/docs/menus/analyze.html#gels) and the Mann-Whitney test was performed for statistical analysis using Prism software. Experiments were replicated at least three times to perform statistical analysis.
For SIK3 and SIK3-R129C immunoprecipitations HEK293T cells were transfected with the appropriate FLAG-tagged vectors. 24 h after transfections cells were harvested and protein extracts were immunopurified using anti-FLAG antibody (Sigma F1804) collected on protein A/G agarose (Santa Cruz). For DEPTOR immunoprecipitation 293T cells were transfected with HA-tagged DEPTOR and harvested 24 h after transfection. Proteins were immunoprecipitated using anti-HA magnetic beads (Thermo Fisher Scientific).
Histological analyses and immunohistochemistry
For histology and immunocytochemistry, human and mouse tissues were fixed in 4% PFA, decalcified using Immunocal decalcification solution and then paraffin embedded. Paraffin blocks were sectioned at 10 μm. For histological analyses, sections were stained with Picrosirius Red. For Picrosirius Red staining, deparaffinized and rehydrated sections were stained in a 0.1% Direct Red 80 (Sigma, 43665)/Saturated Picric Acid (Sigma, P6744) solution followed by counterstaining Hematoxylin QS.
For immunohistochemistry, paraffin sections were boiled for 20 minutes in Antigen Unmasking Solution (Vector) and subsequently stained using a Rabbit Specific HRP/DAB (ABC) Detection IHC Kit (Abcam). Primary antibodies used were: anti-SIK3 antibody, 1:50 Abcam 88495; anti-DEPTOR 1:500, Proteintech 20985–1-AP; anti-pS6 1:400 Cell Signaling 2211.
Site Directed Mutagenesis
Site-directed mutagenesis was performed to generate SIK3R129C plasmid using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent 200521). Mutagenesis primers were designed using the QuikChange Pimer Design Program (http://www.genomics.agilent.com/primerDesignProgram.jsp). Primers used were Fw: TTTTGTCACTGTCGGAACATTGTTCATTGTGATTTAAAAGCTGAAA and Rev: TTTCAGCTTTTAAATCACAATGAACAATGTTCCGACAGTGACAAAA.
RNA extraction and qPCR
RNA was extracted from fibroblasts using TRIzol reagent (Life Technologies). cDNA was prepared from 1 ug of RNA using RevertAid First strand cDNA synthesis kit (Thermo Scientific) and amplified using Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific). Gene expression was calculated using the 2^deltaCT-method of analysis against the stable housekeeping gene beta-2-microglubulin (B2M). Significance was determined via Student’ s t-test. Three biological replicates were performed with each two technical replicates each. qPCR primers were 1) SIK3 Fw: TCGGCTACTACGAGATCGAC and Rev: TCCCGGAAAATCTTCTTCAA; 2) B2M Fw: TGACTTTGTCACAGCCCAAG and Rev: AGCAAGCAAGCAGAATTTGG
3D in silico modeling and kinase assay
For SIK3 structural modeling, the three-dimensional (3D) model for wildtype human SIK3 was obtained via template-based homology modeling using the PHYRE software (38). The SIK3-specific functional elements, predicted using the NCBI Conserved Domain Database (39), were mapped onto a three-dimensional model of the SIK3 using CHIMERA software (40).
For kinases assays, the C-terminally FLAG tagged SIK3 was expressed in 293T cells and purified by FLAG immunoprecipitation 24 hours later. Kinase assays were performed using immunopurified SIK3 or 200 ng of recombinant active SIK3 (SignalChem) together with 4.6 μg of RBER-CHKtide peptide (ProQuinase) or 10 μg of AMARA peptide (obtained from SignalChem or Abcam) as a substrate, respectively. Kinase assays were carried-out in Kinase Assay Buffer (SignalChem) according to manufacturer’s instructions, supplemented with 1 μCi 32P-ATP (Hartmann Analytics) for 30 minutes at 30°C. The reactions were terminated by spotting 4 μl from each reaction onto nitrocellulose membrane (for autoradigraphy), or spotting 10 μl onto 1 cm2 rectangular piece of nitrocellulose membrane (for scintillation). Nitrocellulose membranes were allowed to dry for 10 minutes at room temperature and washed three times for 10 minutes in 1% phosphoric acid, 0.5M NaCl. The SIK3 kinase activity was determined using 1214 RackBeta Liquid Scintillation Counter (LKB Instruments).
Statistical analysis
GraphPad Prism was used for statistical analysis. All values are means ± SE as indicated in figure legends. All comparisons in the study were performed using Student’s t-test.
Supplementary Material
Figure S1. Patients show a missense mutation in the catalytic domain of SIK3.
Figure S2. Quantification of protein amounts of western blots in Figure 3.
Figure S3. Serum and insulin treatment in control and patient cells.
Figure S4. Quantification of protein amounts of western blots in Figure 4.
Table S1. Clinical findings.
Acknowledgements:
We thank the families for their participation in this study.
Funding: D.K. and D.H.C are supported by NIH RO1 AR066124, R01 AR062651, and RO1 DE019567. I.D. is supported by a Geisman Fellowship award from the OIF. P.K is supported by Agency for Healthcare Research of the Czech Republic (15-33232A, 15-34405A, NV18-08-00567); Czech Science Foundation (GA17-09525S); Ministry of Education, Youth and Sports of the Czech Republic(LQ1605,NPU II).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All materials will be made available to the scientific community. All data are in the paper and supplementary materials.
REFERENCES
- 1.Kozhemyakina E, Lassar AB, Zelzer E, A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation, Development 142, 817–831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, Mulligan RC, Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene, Genes Dev. 8, 277–289 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Lee K, Lanske B, Karaplis AC, Deeds JD, Kohno H, Nissenson RA, Kronenberg HM, Segre GV, Parathyroid hormone-related peptide delays terminal differentiation of chondrocytes during endochondral bone development, Endocrinology 137, 5109–5118 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LH, Ho C, Mulligan RC, Abou-Samra AB, Jüppner H, Segre GV, Kronenberg HM, PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth, Science 273, 663–666 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Kozhemyakina E, Cohen T, Yao T-P, Lassar AB, Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway, Mol. Cell. Biol 29, 5751–5762 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Correa D, Hesse E, Seriwatanachai D, Kiviranta R, Saito H, Yamana K, Neff L, Atfi A, Coillard L, Sitara D, Maeda Y, Warming S, Jenkins NA, Copeland NG, Horne WC, Lanske B, Baron R, Zfp521 is a target gene and key effector of parathyroid hormone-related peptide signaling in growth plate chondrocytes, Dev. Cell 19, 533–546 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schipani E, Kruse K, Jüppner H, A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia, Science 268, 98–100 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Cohen MM, Some chondrodysplasias with short limbs: Molecular perspectives, Am. J. Med. Genet 112, 304–313 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Sarbassov DD, Ali SM, Sabatini DM, Growing roles for the mTOR pathway, Curr. Opin. Cell Biol 17, 596–603 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Guertin DA, Sabatini DM, Defining the role of mTOR in cancer, Cancer Cell 12, 9–22 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Long F, mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis, Development 141, 2848–2854 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Holguin N, Shi Y, Silva MJ, Long F, mTORC2 signaling promotes skeletal growth and bone formation in mice, J. Bone Miner. Res 30, 369–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan B, Zhang Z, Jin D, Cai C, Jia C, Liu W, Wang T, Li S, Zhang H, Huang B, Lai P, Wang H, Liu A, Zeng C, Cai D, Jiang Y, Bai X, mTORC1 regulates PTHrP to coordinate chondrocyte growth, proliferation and differentiation, Nat Commun 7, 11151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mihaylova MM, Shaw RJ, The AMPK signalling pathway coordinates cell growth, autophagy and metabolism, Nature Publishing Group 13, 1016–1023 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walkinshaw DR, Weist R, Kim G-W, You L, Xiao L, Nie J, Li CS, Zhao S, Xu M, Yang X-J, The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases, J. Biol. Chem 288, 9345–9362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, Shaw RJ, Yates JR, Fischer WH, Thomas JB, Montminy M, A hormone-dependent module regulating energy balance, Cell 145, 596–606 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi S, Lim D-S, Chung J, Taghert PH, Ed. Feeding and Fasting Signals Converge on the LKB1-SIK3 Pathway to Regulate Lipid Metabolism in Drosophila, PLoS Genet. 11, e1005263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakamaki J-I, Fu A, Reeks C, Baird S, Depatie C, Al Azzabi M, Bardeesy N, Gingras A-C, Yee S-P, Screaton RA, Role of the SIK2-p35-PJA2 complex in pancreatic β-cell functional compensation, Nat. Cell Biol 16, 234–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasagawa S, Takemori H, Uebi T, Ikegami D, Hiramatsu K, Ikegawa S, Yoshikawa H, Tsumaki N, SIK3 is essential for chondrocyte hypertrophy during skeletal development in mice, Development 139, 1153–1163 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, O’Meara MJ, Govea N, Beqo B, Nishimori S, Nagano K, Brooks DJ, Martins JS, Corbin B, Anselmo A, Sadreyev R, Wu JY, Sakamoto K, Foretz M, Xavier RJ, Baron R, Bouxsein ML, Gardella TJ, Divieti-Pajevic P, Gray NS, Kronenberg HM, SIKs control osteocyte responses to parathyroid hormone, Nat Commun 7, 13176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen J, Snow C, Tuttle E, Ghoneim DH, Yang C-S, Spencer A, Gunter SA, Smyser CD, Gurnett CA, Shinawi M, Dobyns WB, Wheless J, Halterman MW, Jansen LA, Paschal BM, Paciorkowski AR, De novo mutations in SIK1 cause a spectrum of developmental epilepsies, Am. J. Hum. Genet 96, 682–690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoki K, Zhu T, Guan K-L, TSC2 mediates cellular energy response to control cell growth and survival, Cell 115, 577–590 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ, AMPK phosphorylation of raptor mediates a metabolic checkpoint, Molecular Cell 30, 214–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM, DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival, Cell 137, 873–886 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao D, Inuzuka H, Tan M-KM, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, Lyssiotis CA, Gygi SP, Toker A, Cantley LC, Asara JM, Harper JW, Wei W, mTOR Drives Its Own Activation via SCFβTrCP-Dependent Degradation of the mTOR Inhibitor DEPTOR, Molecular Cell 44, 290–303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y, Xiong X, Sun Y, DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy, Molecular Cell 44, 304–316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, Pagano M, mTOR Generates an Auto-Amplification Loop by Triggering the βTrCP- and CK1α-Dependent Degradation of DEPTOR, Molecular Cell 44, 317–324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.González-Terán B, López JA, Rodríguez E, Leiva L, Martínez-Martínez S, Bernal JA, Jiménez-Borreguero LJ, Redondo JM, Vazquez J, Sabio G, p38γ and δ promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation, Nat Commun 7, 10477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berggreen C, Henriksson E, Jones HA, Morrice N, Goransson O, cAMP-elevation mediated by β-adrenergic stimulation inhibits salt-inducible kinase (SIK) 3 activity in adipocytes, Cell. Signal 24, 1863–1871 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, Alessi DR, LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1, EMBO J. 23, 833–843 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Al Kaissi A, Ghachem MB, Nessib N, Chehida FB, Hammou A, Kozlowski K, Distinctive new form of spondyloepimetaphyseal dysplasia with severe metaphyseal changes similar to Jansen metaphyseal chondrodysplasia, Australas Radiol 49, 57–62 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Laplante M, Sabatini DM, mTOR signaling in growth control and disease, Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoncu R, Efeyan A, Sabatini DM, mTOR: from growth signal integration to cancer, diabetes and ageing, Nat. Rev. Mol. Cell Biol 12, 21–35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikegami D, Akiyama H, Suzuki A, Nakamura T, Nakano T, Yoshikawa H, Tsumaki N, Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways, Development 138, 1507–1519 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Kita K, Kimura T, Nakamura N, Yoshikawa H, Nakano T, PI3K/Akt signaling as a key regulatory pathway for chondrocyte terminal differentiation, Genes to Cells 13, 839–850 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Graham JM, Rimoin DL, Lachman RS, Krejci P, Tompson SW, Nelson SF, Krakow D, Cohn DH, Exome sequencing identifies PDE4D mutations in acrodysostosis, Am. J. Hum. Genet 90, 746–751 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gassmann M, Grenacher B, Rohde B, Vogel J, Quantifying Western blots: Pitfalls of densitometry, Electrophoresis 30, 1845–1855 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Kelley LA, Sternberg MJE, Protein structure prediction on the Web: a case study using the Phyre server, Nat Protoc 4, 363–371 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH, CDD: conserved domains and protein three-dimensional structure, Nucleic Acids Research 41, D348–D352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera?A visualization system for exploratory research and analysis, J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Patients show a missense mutation in the catalytic domain of SIK3.
Figure S2. Quantification of protein amounts of western blots in Figure 3.
Figure S3. Serum and insulin treatment in control and patient cells.
Figure S4. Quantification of protein amounts of western blots in Figure 4.
Table S1. Clinical findings.