

Abstract
Background
Avitinib is one type of the third‐generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) for the treatment of non‐small cell lung cancer (NSCLC) with EGFR mutations. The purpose of this study was to investigate the effect of avitinib on the pharmacokinetics of osimertinib, one FDA approved third‐generation TIKI, both in vitro and in vivo.
Methods
The in vitro metabolic stability and inhibitory effect of avitinib on osimertinib were assessed with rat liver microsomes (RLM) to determine its IC50 values. For the in vivo study, 18 Sprague‐Dawley rats were randomly divided into three groups: the avitinib multiple dose group (30 mg/kg avitinib once daily for seven days), the avitinib single dose group (PEG200 once daily for six days and a dose of 30 mg/kg avitinib in PEG200 on day 7) and the control group (equal amounts of PEG200 once daily for seven days). Next, all rats were given osimertinib at a dosage of 10 mg/kg. UPLC/MS‐MS was used for the determination of the concentration of osimertinib in plasma.
Results
In vitro analysis revealed that the IC50 value of osimertinib in rat liver microsomes was 27.6 μM. When rats were pretreated with avitinib, the values of AUC and MRT of the osimertinib were increased, and its Cmax and Tmax were significantly extended, whereas the values of CLz/F were significantly decreased (P < 0.05).
Conclusions
Both in vitro and in vivo results demonstrated that a drug‐drug interaction between avitinib and osimertinib occurred and more attention should be paid when avitinib and osimertinib are synchronously administered in clinic.
Key points
Significant findings of the study
Osimertinib is the only market available third‐generation EGFR‐TKI and it has been reported that some drugs could have drug‐drug interactions with it.
What this study adds
For the first time, we systematically investigated the effect of avitinib, one newly developed third‐generation EGFR‐TKI, on the pharmacokinetics of osimertinib both in vitro and in vivo using a rat model.
Keywords: Avitinib, drug‐drug interaction, osimertinib, pharmacokinetics
We systematically investigated the effect of avitinib, one newly developed third‐generation EGFR‐TKI, on the pharmacokinetics of osimertinib both in vitro and in vivo using rat model.
Introduction
Lung cancer is the leading cause of death among all cancer types, and non‐small cell lung cancer (NSCLC) accounts for up to 85% of all lung cancer patients. 1 The epidermal growth factor receptor (EGFR) is the major therapeutic target for NSCLC, and EGFR tyrosine kinase inhibitors (TKIs) are very efficient for EGFR mutation‐positive patients. 2 To date, TKIs have been regarded as the standard first‐line treatment options for these patients. Although the first and second‐generation of EGFR‐TKIs were originally developed to target wild‐type (WT) EGFR, they are also effective for some EGFR mutations. It is well known that patients have a good initial response to the first and second‐generation EGFR‐TKIs, but the acquired resistance is very common among most patients taking them. New mutations in EGFR, especially accompanied by EGFR‐T790M mutations, are the major reason for this acquired resistance. 3 Acquired EGFR‐T790M mutations accounted for 55%–70% of first‐generation EGFR‐TKIs resistance. 4
To overcome the defects of the first/second generation of EGFR‐TKIs, a third generation of EGFR‐TKIs were developed which can specifically and selectively bind and inhibit EGFR‐T790M mutation. To date, osimertinib is the only market available third‐generation EGFR‐TKI and other similar EGFR‐TKIs, such as avitinib, olmutinib and nazartinib, are still under development. 5 Osimertinib, the FDA approved third‐generation EGFR‐TKI, can irreversibly bind to NSCLC cell lines with EGFR mutation (T790M, L858R and exon 19 deletion), with a weak inhibitory effect on the wild‐type EGFR protein. 6 Avitinib, one third‐generation TKI independently developed by China, can inhibit EGFR T790M and other sensitive mutations, with no effects on wild‐type EGFR. Now, avitinib has finished the Phase II clinical trials in China and showed similar positive results to osimertinib with reliable efficacy and safety. 1 Also, clinical trial data inferred that switching from avitinib to osimertinib as salvage therapy showed good efficacy. 4 The most common adverse drug reactions of third‐generation EGFR‐TKIs include diarrhea, rash, nausea, decreased appetite or higher toxicities, and avitinib exhibits comparable incidence of adverse drug reactions to osimertinib. Avitinib had demonstrated initial efficacy in a phase I trial, but also induced severe interstitial lung disease (ILD) in a few cases, and subsequent osimertinib therapy achieved better efficacy without the recurrence of ILD. 7
Osimertinib is metabolized by CYP3A enzymes and has a long half‐life, less clearance with slow to medium and long‐lasting absorption. 8 Thus, its exposure may be easily impacted by the CYP enzyme inducers or inhibitors. 9 It has been reported that osimertinib is a metabolic substrate of CYP3A4/5, and it is both an inhibitor and an inducer of CYP3A4 in vitro. 10 A recent study revealed that avitinib could be metabolized by rat liver microsomes (RLMs). 11 Here, we speculated that a drug‐drug interaction might occur between the two third‐generation EGFR‐TKIs osimertinib and anvitinib, and the purpose of this study was to investigate the effects of avitinib on the pharmacokinetics of osimertinib both in vitro and in vivo.
Methods
Chemicals and reagents
Avitinib (purity >98%) was purchased from Send Pharm (Beijing, China). Osimertinib (purity >98%) were purchased from Beijing Sunflower and Technology Development Co. Ltd. (Beijing, China). Diazepam (Internal standard, IS) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Reduced nicotinamide adenine dinucleotide phosphate (NADPH) was purchased from Roche Pharmaceuticals Ltd. (Basel, Switzerland). Methanol and acetonitrile (gradient grade) were purchased from Merck (Billerica, MA, USA). Formic acid were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Ultrapure water was obtained from the Milli‐Q plus filtration system (Millipore, Billerica, MA, USA). All chemicals and solvents were analytical grade.
Instrumentation and analytical conditions
Waters Acquity UPLC I‐Class (Waters, USA) was used for the separation of osimertinib and diazepam. Binary solvent manager (BSM) and a sample manager with flow‐through needle (SM‐FTN) were used in the UPLC system. The effective separation of metabolites was achieved on Acquity UPLC BEH C18 (50 × 2.1 mm, 1.7 μm) (Waters Corporation, Milford, MA, USA) using mobile phase acetonitrile (solvent A) and 0.1% formic acid water containing 5 mmol Ammonium formate (solvent B). The gradient program was set as follows: 0 minutes 10% A; 0.5 minutes 30% A; 1 minute 95% A; 2 minutes 95% A; 2.6 minutes 10% A. The flow rate of mobile phase was 0.4 mL/minute, the column temperature was maintained at 40°C and injection volume was 2 μL.
XEVO TQD triple quadrupole mass spectrometer was equipped with electrospray ionization (ESI), and multiple reaction monitoring (MRM) mode was selected for quantitation. Osimertinib was quantified using the multiple reaction monitoring (MRM) scanning mode, for the mass reactions (parent to fragment ions) from 500.57 → 385.13 and 500.57 → 427.78 for osimertinib and 285.10 → 193.10 for diazepam (IS). The cone voltage was set at 30, 25 and 30 V for osimertinib and IS with collision energy of 33, 35 and 30 V. MassLynx 4.1 software (Waters Corp., Milford, MA, USA) was used for data acquisition.
Preparation of RLMs
Rats were anesthetized with 10% chloral hydrate after being starved for 12 hours prior, then sterilized above the chest with 75% alcohol. The liver was exposed by opened the abdominal cavity, perfused using 0.15 mol/L KCl buffer (PH = 7.4) through the superior vena cava until the blood had flushed away, and 3 g liver was precisely removed and transferred to a petri dish (prekept at 4°C), 0.15 mol/L KCl‐PBS buffer (stored at 4°C) was added and homogenized. The homogenate was centrifuged at 9000 g for 30 minutes at 4°C, the supernatant was precipitated at 105000 g for 60 minutes at 4°C after removal of the precipitate (the precipitate was the liver microsomes). It was then transferred into 0.15 mol/L KCL‐PBS buffer and homogenized for resuspension. Centrifugation (105 000 g for 60 minutes at 4°C) and resuspension (0.15 mol/L KCl‐PBS buffer containing 0.25 mol/L sucrose) procedures were repeated for RLMs homogenate and it was then stored at −80°C until use. The protein concentration of RLMs was 8.838 mg/mL determined by the Bradford Protein Assay Kit (Thermo Scientific, Waltham, MA, USA).
Effect of avitinib on the metabolism of osimertinib in vitro
The metabolic stability study for osimertinib was performed by calculating the concentration of osimertinib in the RLMs matrix from the displacement of the peak area ratios in the regression equation of the calibration curve and determining the half maximal inhibitory concentration (IC50) of avitinib. The metabolic stability incubation system of RLMs was a total volume of 200 μL, and the components of the system were as follows: osimertinib with final concentration of 0.01 μM which was used as metabolic substrate, avitinib with final concentration of 1 μM used as the inhibitor, 20 μL 100 mM potassium phosphate buffer (pH 7.4) and 10 μL RLMs were added and vortexed. The mixture was preincubated for 5 minutes in a 37°C water bath, followed by adding 1 mM NADPH to initiate the metabolic reaction. The metabolic reaction was terminated by adding 200 μL iced ACN at the series time of 0, 10, 20, 30, 40, and 50 minutes. All incubations were centrifuged at 13000 rpm (10 minutes at 4°C) after mixing with 20 μL internal standard (50 ng/mL diazepam). The supernatant mixture (2 μL) was injected into the UPLC–MS/MS system for analysis.
Animal experiments
Male SPF SD rats with bodyweights of 180–220 g were obtained from the Laboratory Animal Center of the Wenzhou Medicine University (Wenzhou, Zhejiang, China). The animals were kept in a controlled environment at 20°C–26°C and 55 ± 15% relative humidity under a 12 hour light‐dark cycle. Diet was prohibited for 12 hours before the experiment, but water was freely available. All experimental procedures and protocols were reviewed and approved by the Animal Ethics Committee of Wenzhou Medical University, in line with the Guidelines for the Care and Use of Laboratory Animals.
Effects of avitinib on the pharmacokinetics of osimertinib in vivo
A total of 18 rats were randomly divided into three groups: the avitinib multiple dose group (pretreated with 30 mg/kg avitinib once daily for seven days), the avitinib single dose group (treatment with PEG200 once daily for six days and a dose of 30 mg/kg avitinib in PEG200 on day 7) and the control group (pretreated with the equal amounts of PEG200 once daily for seven days). All rats were given osimertinib at a dosage of 10 mg/kg after administration of avitinib or PEG200, then 300 μL of blood samples were collected into 1.5 mL heparinized tubes via the caudal vein at 0.083, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24 and 36 hours. Blood samples was centrifuged at 4000 rpm/min for 10 minutes at 4°C to obtain the plasma, separated the supernatant and stored at −80°C until analysis.
Sample preparation
The frozen plasma sample was thawed at room temperature and uniformly mixed, and 100 μL plasma was taken out and mixed with 20 μL internal standard (50 ng/mL diazepam), then 200 μL acetonitrile was added to precipitate protein followed by vortexing for 30 seconds and centrifugation at 13000 rpm for five minutes. The supernatant (2 μL) was injected into the UPLC‐MS/MS system for analysis.
Statistical analysis
The pharmacokinetic parameters, including maximal plasma concentration (Cmax), the time to peak plasma concentration (Tmax), the area under the plasma concentration‐time curve (AUC), the elimination half‐life (t1/2) and the mean residence time (MRT), were analyzed using DAS (Drug and Statistics) software (Version 3.2.8, The People's Hospital of Lishui, China). 12 , 13 , 14
All the pharmacokinetic parameters are expressed as the mean ± SD. The GraphPad (version 7.0; GraphPad Software Inc., San Diego, CA, USA) was applied to calculate IC50 values and plot plasma concentration‐time curves. Statistical comparisons within groups were conducted by SPSS (version 24.0; SPSS Inc., Chicago, IL, USA), using One‐way ANOVA. A P‐value <0.05 was considered statistically significant.
Results
Effect of avitinib on the metabolism of osimertinib in vitro
A metabolic stability curve was drawn by plotting the natural log (Ln) of the remaining percent of osimertinib on the y‐axis against the incubation time on the x‐axis (Fig 1a). The linear portion of the plotted curve was used to calculate in vitro t1/2 value. 15 By using the following equation, t1/2 = , where the slope was 0.02062, the regression equation of osimertinib was calculated and shown in Table 1.
Figure 1.
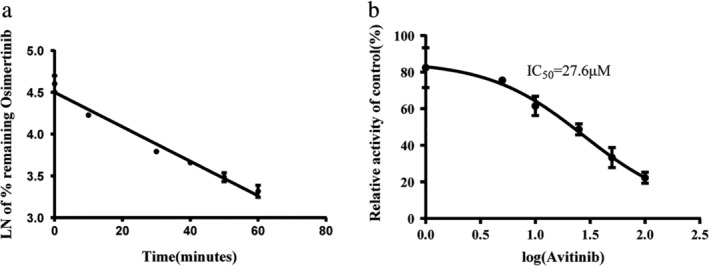
(a) Curve of the metabolic stability of osimertinib in RLMs (n = 3) and (b) Curve of the inhibition ratio of avitinib in RLMs (n = 3).
Table 1.
Metabolic stability parameters for osimertinib incubation with RLMs
| Parameter | Value |
|---|---|
| Regression equation | y = −0.02062x + 4.5 |
| r2 | 0.9775 |
| Slope | 0.02062 |
| t1/2 | 33.61 minutes |
A metabolic stability curve was drawn by plotting the percentage of mean inhibition rate of avitinib on the y‐axis against the Log value of determined concentration of avitinib on the x‐axis. As illustrated in Fig 1b, the IC50 value of avitinib to osimertinib was 27.6 μM with r2 = 0.9608. These data indicated that avitinib could significantly inhibit the metabolism of osimertinib in vitro.
Effect of avitinib on the pharmacokinetics of osimertinib in vivo
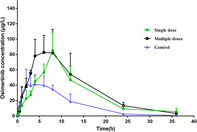
Typical MRM chromatograms of osimertinib and internal standard in plasma samples at 6 hours after the oral administration of osimertinib in rats are shown in Fig 2. The mean plasma concentration‐versus‐time curves of osimertinib after administration by gavage at a dose of 10 mg/kg, with different pretreatments, are shown in Fig 3. Detailed pharmacokinetic parameters of osimertinib with different pretreatments in rats are shown in Table 2. These data indicated that the pharmacokinetic parameters of rats administered osimertinib in the avitinib treatment groups, including multiple doses and single dose, exhibited significant differences (P < 0.05) compared with those of the control group.
Figure 2.
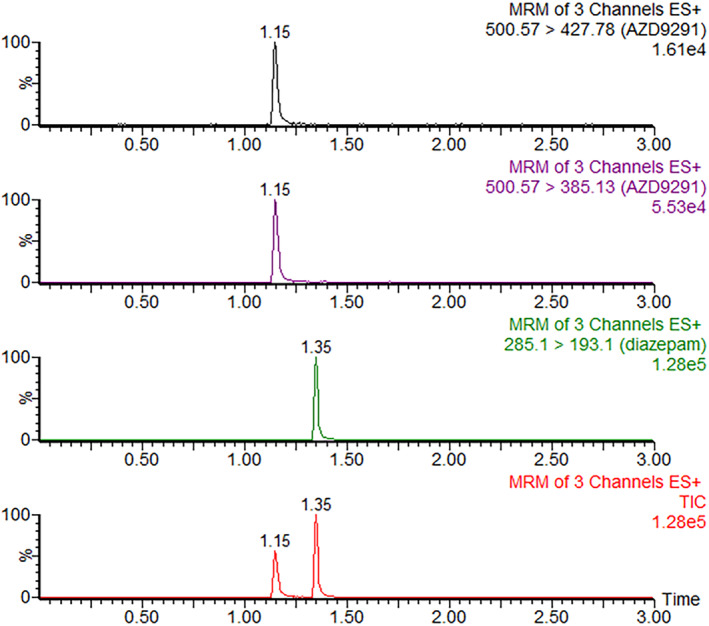
Typical MRM chromatograms of osimertinib and internal standard in plasma samples at 6 hours after oral administration of osimertinib. TIC, total ion chromatograph.
Figure 3.
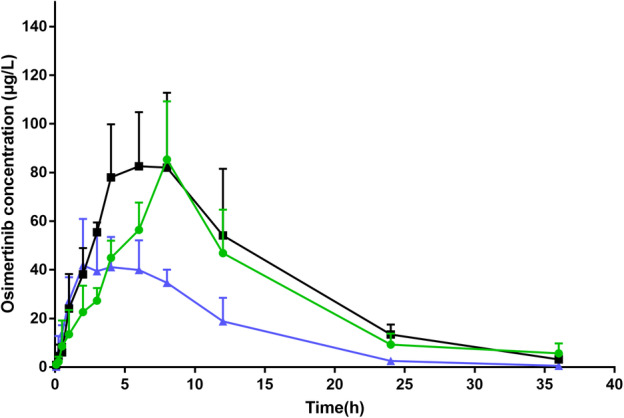
Mean plasma concentration time profile of orally administrated osimertinib (10 mg/kg) after pretreatment with avitinib in rats (Mean ± SD) ( ) Single dose (
) Single dose ( ) Multiple dose (
) Multiple dose ( ) Control.
) Control.
Table 2.
The pharmacokinetic parameters of osimertinib after pretreated with avitinib in rats (n = 6)
| Parameter | Unit | Multiple dose | CV% | Single dose | CV% | Control | CV% |
|---|---|---|---|---|---|---|---|
| AUC(0‐t) | ug/L*hour | 1019.49 ± 240.07* | 23.5 | 1255.82 ± 419.43* | 33.4 | 534.50 ± 142.84 | 26.7 |
| AUC(0‐∞) | ug/L*hour | 1035.47 ± 247.53* | 23.9 | 1283.21 ± 429.78* | 33.5 | 539.32 ± 144.35 | 26.8 |
| MRT(0‐t) | hour | 11.17 ± 0.93* | 8.3 | 10.57 ± 0.71* | 6.8 | 8.04 ± 1.10 | 13.6 |
| MRT(0‐∞) | hour | 11.65 ± 1.13* | 9.7 | 11.28 ± 0.88* | 7.8 | 8.28 ± 1.13 | 13.6 |
| t1/2 | hour | 5.12 ± 0.92 | 17.9 | 5.74 ± 0.67* | 11.7 | 4.08 ± 0.98 | 24.1 |
| Tmax | hour | 8.00 ± 0.00* | 0 | 6.80 ± 1.79 | 26.3 | 4.60 ± 2.41 | 52.3 |
| Vz/F | L/kg | 75.55 ± 27.50 | 36.4 | 71.70 ± 28.01 | 39.1 | 112.45 ± 24.31 | 21.6 |
| CLz/F | L/h/kg | 10.27 ± 3.25* | 31.7 | 8.65 ± 3.20* | 37 | 19.76 ± 5.84 | 29.6 |
| Cmax | ug/L | 85.35 ± 23.93* | 28 | 88.30 ± 23.65* | 26.8 | 51.47 ± 10.74 | 20.9 |
P < 0.05 indicates significant differences from the control group.
As compared with those of the control group, the parameters of rats pretreated with avitinib in the multiple dose group and the single dose group exhibited little difference. In detail, the AUC(0‐t) values of multiple doses group and single dose group were both increased greatly, by 1.91‐fold and 2.35‐fold, respectively. The AUC(0‐∞) values of multiple doses group and single dose group were elevated by 1.92‐fold and 2.38‐fold, respectively. The MRT(0‐t) values of multiple doses group and single dose group were increased by 1.39‐fold and 1.31‐fold, respectively. The MTR(0‐∞) values of multiple doses group and single dose group were elevated by 1.41‐fold and 1.36‐fold, respectively. Specifically, multiple doses avitinib treatment group and single dose avitinib treatment group could significantly increase the Tmax values of osimertinib by 1.74‐fold and 1.48‐fold, respectively. A similar pattern could also be found in the Cmax values of osimertinib, increased by 1.66‐fold and 1.72‐fold, respectively. However, pretreatment with multiple or single doses of avitinib could both significantly decrease the CLz/F of osimertinib by 52% and 44%, respectively. These results indicated that avitinib had an inhibitory effect on the metabolism of osimertinib in rats, which was in good agreement with the in vitro results obtained with rat liver microsomes.
Discussion
Lung cancer can be divided into non‐small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), and NSCLC accounts for 85% of the incidence of all lung cancers. It is reported that approximately 10% of patients with epidermal growth factor receptor (EGFR) mutation‐positive NSCLC harbor uncommon mutations, and EGFR tyrosine‐kinase inhibitor (KTI) are currently used as the standard first‐line treatment options for NSCLC patients harboring sensitizing EGFR mutations. 2 Osimertinib is the first FDA approved third‐generation EGFR‐TKI that selectively targets the T790M mutated EGFR and is now market‐available in China. Osimertinib can irreversibly bind to NSCLC cell lines with EGFR mutation (T790M, L858R and exon 19 deletion), with a weak inhibitory effect on wild‐type EGFR protein. 6 The safety profile of osimertinib is quite acceptable. However, about 1.2% of patients do not pursue curative treatment because of adverse reactions or abnormal laboratory tests. According to the pharmacokinetic parameters, osimertinib has a long half‐life and less clearance with long‐lasting absorption, 8 thus can accumulate easily after multiple administration. Avitinib is one third‐generation TKI independently developed by China and its phase II clinical trial tests are now complete. Similar to osimertinib, avitinib can also inhibit EGFR T790M and other sensitive mutations, but it has no inhibitory effects on wild‐type EGFR. 1 Avitinib can overcome the shortcomings of first‐generation EGFR‐TKIs that T790M‐induced resistance in NSCLC patients and shows a good prospect for clinical application. It is reported that switching from avitinib to osimertinib as salvage therapy showed a good efficacy. 4 A previous study revealed that osimertinib is metabolized by CYP3A enzymes, and its exposure might be influenced by CYP enzyme inducers or inhibitors. 16 Because of the similar metabolism pathways of osimertinib and avitinib, it is estimated that a drug‐drug interaction might occur between these two third‐generation EGFR‐TKIs.
In this study, we evaluated the effects of avitinib on osimertinib both in vitro and in vivo. The in vitro metabolic stability results showed that avitinib had direct inhibitory effects on osimertinib with IC50 = 27.6 μM. In addition, the metabolic stability of osimertinib was characterized by a very long in vitro T1/2 value (33.61 minute), which indicated that a very slow clearance of osimertinib from the blood might occur. To further consolidate these results, pharmacokinetic interaction studies in rats were then implemented in vivo. As a result, the main pharmacokinetic parameters (AUC(0‐t), AUC(0‐∞), MRT(0‐t) and MRT(0‐∞)) of osimertinib all increased significantly in the avitinib treatment groups, compared with those of the control group, indicating that avitinib truly could inhibit the metabolism of osimertinib in vivo. Specifically, the oral clearance (CLz/F) of osimertinib was decreased with extended Tmax value, indicating that avitinib might affect osimertinib by prolonging its exposure time and reducing its clearance rate.
As one novel third‐generation EGFR TKI target of T790M mutations, avitinib has demonstrated initial efficacy in a phase I trial, but it has also been reported to induce severe ILD in a few cases. Subsequent treatment with osimertinib achieved better efficacy and prolonged PFS without the recurrence of ILD. 7 Osimertinib is a metabolic substrate of CYP3A4/5, its pharmacokinetics are influenced by other substances metabolized by CYP3A. In addition, osimertinib is both an inhibitor and an inducer of CYP3A4 as reported in one in vitro study, 10 and it can also affect the in vivo process of certain drugs. Osimertinib has a long half‐life and a less clearance with slow to medium and long‐lasting absorption, and inhibition of osimertinib can lead to accumulation in vivo. Excessive exposure of osimertinib could induce some adverse drug reactions such as diarrhea, rash, nausea, decreased appetite or higher toxicities, and may lead to a dose reduction or drug discontinuation that would accelerate the progression of the disease. 5 Our data revealed that pretreatment with avitinib and then administration of osimertinib could lead to the extended exposure time and reduced clearance rate of osimertinib in rats. Based on these data, we speculated that treatment with avitinib and subsequent osimertinib therapy would affect the in vivo process of osimertinib in humans. Thus, performing therapeutic drug monitoring (TDM) and adjusting the drug dosage in time in a clinical environment are very important before and after switching drugs from avitinib to osimertinib.
In conclusion, our data clearly illustrated that the pharmacokinetics of osimertinib could be significantly influenced by avitinib both in vitro and in vivo. In spite of this, considering for the differences in metabolic enzymes and physiological conditions between rats and human beings, further pharmacokinetic studies in humans are still required in the future.
Disclosure
The authors had no conflicts of interest and no disclosures to declare.
Acknowledgments
This work was supported by grants funded by High‐Level Talent Training Project of Lishui (2018RC18), Natural Science Foundation of Zhejiang and Zhejiang Pharmaceutical Association Joint Foundation (LYY18H280003), City‐level public welfare technology application research project of Lishui (2017GYX15) and CAMS Innovation Fund for Medical Sciences (2018‐I2M‐1‐002).
Contributor Information
Qingjun Wu, Email: wooqj_bj@126.com.
Debiao Pan, Email: pandebiao@163.com.
Quan Zhou, Email: zhouquan1991@lsu.edu.cn.
Chunjie Wang, Email: 1024172645@qq.com.
References
- 1. Xu X, Mao L, Xu W et al AC0010, an irreversible EGFR inhibitor selectively targeting mutated EGFR and overcoming T790M‐induced resistance in animal models and lung cancer patients. Mol Cancer Ther 2016; 15: 2586–97. [DOI] [PubMed] [Google Scholar]
- 2. Cho JH, Lim SH, An HJ et al Osimertinib for patients with non‐small‐cell lung cancer harboring uncommon EGFR mutations: A multicenter, open‐label, phase II trial (KCSG‐LU15‐09). J Clin Oncol 2020; 38: 488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Costa DB, Kobayashi SS. Whacking a mole‐cule: Clinical activity and mechanisms of resistance to third generation EGFR inhibitors in EGFR mutated lung cancers with EGFR‐T790M. Transl Lung Cancer Res 2015; 4: 809–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang H, Pan R, Zhang X, Si X, Wang M, Zhang L. Abivertinib in patients with T790M‐positive advanced NSCLC and its subsequent treatment with osimertinib. Thorac Cancer 2020; 11: 594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan CS, Kumarakulasinghe NB, Huang YQ et al Third generation EGFR TKIs: Current data and future directions. Mol Cancer 2018; 17: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cross DA, Ashton SE, Ghiorghiu S et al AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014; 4: 1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang H, Zhang L, Shi X, Zhang X, Si X. Successful treatment with osimertinib and its subsequent resistance mechanism in a patient with non‐small‐cell lung cancer harboring acquired EGFR T790M mutation after recovery from AC0010‐induced interstitial lung disease. Onco Targets Ther 2019; 12: 5545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Janne PA, Yang JC, Kim DW et al AZD9291 in EGFR inhibitor‐resistant non‐small‐cell lung cancer. N Engl J Med 2015; 372: 1689–99. [DOI] [PubMed] [Google Scholar]
- 9. Kucharczuk CR, Ganetsky A, Vozniak JM. Drug‐drug interactions, safety, and pharmacokinetics of EGFR tyrosine kinase inhibitors for the treatment of non‐small cell lung cancer. J Adv Pract Oncol 2018; 9: 189–200. [PMC free article] [PubMed] [Google Scholar]
- 10. Calvo E, Lee JS, Kim SW et al Modulation of exofenadine pharmacokinetics by Osimertinib in patients with advanced EGFR‐mutated non‐small cell lung cancer. J Clin Pharmacol 2019; 59: 1099–109. [DOI] [PubMed] [Google Scholar]
- 11. Attwa MW, Kadi AA, Abdelhameed AS. Reactive intermediates and bioactivation pathways characterization of avitinib by LC‐MS/MS: in vitro metabolic investigation. J Pharm Biomed Anal 2019; 164: 659–67. [DOI] [PubMed] [Google Scholar]
- 12. Ma JS, Wang SH, Zhang ML et al Simultaneous determination of bupropion, metroprolol, midazolam, phenacetin, omeprazole and tolbutamide in rat plasma by UPLC‐MS/MS and its application to cytochrome P450 activity study in rats. Biomed Chromatogr 2015; 29: 1203–12. [DOI] [PubMed] [Google Scholar]
- 13. Wang S, Wu H, Huang X et al Determination of N‐methylcytisine in rat plasma by UPLC‐MS/MS and its application to pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 2015; 990: 118–24. [DOI] [PubMed] [Google Scholar]
- 14. Wang X, Wang S, Ma J et al Pharmacokinetics in rats and tissue distribution in mouse of berberrubine by UPLC‐MS/MS. J Pharm Biomed Anal 2015; 115: 368–74. [DOI] [PubMed] [Google Scholar]
- 15. Abdelhameed AS, Kadi AA, Attwa MW, AlRabiah H. Validated LC‐MS/MS assay for quantification of the newly approved tyrosine kinase inhibitor, dacomitinib, and application to investigating its metabolic stability. PLOS One 2019; 14: e0214598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kucharczuk CR, Ganetsky A, Vozniak MJ. Drug‐drug interactions, safety, and pharmacokinetics of EGFR T yrosine kinase inhibitors for the treatment of non–small cell lung cancer. J Adv Pract Oncol 2018; 9 (2): 189–200. [PMC free article] [PubMed] [Google Scholar]