

Abstract
Hypocrealean entomopathogenic fungi (HEF) produce a large variety of secondary metabolites (SMs) that are prominent virulence factors or mediate various interactions in the native niches of these organisms. Many of these SMs show insecticidal, immune system modulatory, antimicrobial, cytotoxic and other bioactivities of clinical or agricultural significance. Recent advances in whole genome sequencing technologies and bioinformatics have revealed many biosynthetic gene clusters (BGCs) potentially involved in SM production in HEF. Some of these BGCs are now well characterized, with the structures of the cognate product congeners elucidated, and the proposed biosynthetic functions of key enzymes validated. However, the vast majority of HEF BGCs are still not linked to SM products (“orphan” BGCs), including many clusters that are not expressed (silent) under routine laboratory conditions. Thus, investigations into the encoded parvome (the secondary metabolome predicted from the genome) of HEF allows the discovery of BGCs for known SMs; uncovers novel metabolites based on the BGCs; and catalogues the predicted SM biosynthetic potential of these fungi. Herein, we summarize new developments of the field from 2014 to the third quarter of 2019, and survey the polyketide, nonribosomal peptide, terpenoid and hybrid SM BGCs encoded in the currently available 39 HEF genome sequences. Studying the encoded parvome of HEF will increase our understanding of the multifaceted roles that SMs play in biotic and abiotic interactions and will also reveal biologically active SMs that can be exploited for the discovery of human and veterinary drugs or crop protection agents.
1. Introduction
Hypocrealean entomopathogenic fungi (HEF) produce a large variety of secondary metabolites (SMs) with insecticidal, immune system modulatory, antimicrobial, cytotoxic, and other bioactivities of clinical or agricultural significance.1–5 Recent advances in whole genome sequencing technologies and bioinformatics have revealed a large number of biosynthetic gene clusters (BGCs)6 potentially involved in SM biosynthesis, including those in HEF genomes.7 These BGCs are organized around genes that encode biosynthetic enzymes that yield the SM carbon skeleton (referred to as “backbone” or “core” enzymes), such as nonribosomal peptide synthetases (NRPSs), polyketide synthases (PKSs), PKS-NRPS hybrids, dimethylallyl tryptophan synthases (DMATs), geranylgeranyl diphosphate synthases (GGPSs), terpene cyclases (TCs), terpene synthases (TSs), and fatty acid synthases (FASs). Together with the genes for these core enzymes, the BGCs also feature genes encoding various enzymes that further modify the SM carbon skeleton, such as cytochrome P450 monooxygenases (CYPs), various other oxidoreductases, methyltransferases (MTs) and halogenases, together with genes for transporters, regulators, and self-resistance determinants. In addition, many BGCs also harbor genes for enzymes that synthesize specialized monomers for the pathway, such as those for α-hydroxyisocaproic acid biosynthesis for the production of destruxins in Metarhizium spp.8 BGCs are typically considered as self-contained sets of co-located genes that accomplish the coordinated and regulated biosynthesis of a single set of SM congeners. Nevertheless, exceptions include BGCs that lack genes for required modifying enzymes that are located in different parts of the genome; distributed BGCs where two or more sub-clusters located in different parts of the genome collaborate during convergent biosynthesis of a single set of SM congeners; and superclusters that contain intertwined sets of genes for the biosynthesis of more than one sets of SM scaffolds.9–11
Among the core SM biosynthetic enzymes, PKSs, NRPSs, and hybrid PKS–NRPSs are the most studied due to the outstanding diversity and varied bioactivities of their products. PKSs and NRPSs are multidomain megasynthases that assemble complex molecules by recursive condensation of various short chain carboxylic acid monomers to yield polyketides (PKs), or a large variety of aminoacyl or hydroxyacyl monomers to afford nonribosomal (depsi)peptides (NRPs). Both PKSs and NRPSs typically contain three highly conserved domains: 1) the malonyl/acyltransferase (AT) or adenylation (A) domain responsible for loading the monomer units onto the PKS or NRPS, respectively; 2) the acyl carrier protein domain (ACP) or peptidyl carrier domain (PCP, also known as thiolation domain, T) that covalently tethers the precursors and the intermediates; and 3) the ketoacyl synthase (KS) or condensation (C) domain that catalyzes the sequential condensation of the monomers with the growing polyketide or peptide chain.1 Both enzymes may also contain additional domains such as methyltransferase (MT) and epimerase (E) domains for NRPSs, and reductive domains (such as KR, ketoacyl reductase; ER, enoyl reductase; and DH, dehydratase) or starter acyltransferase (SAT) and product template (PT) domains for PKSs. These domains modify the monomers or the intermediates to provide more structural diversity for the products. Fungal PKSs are typically iterative enzymes that contain only a single set of catalytic domains that function in more than one extension cycles according to a still largely cryptic program. In contrast, fungal NRPSs may contain one or more modules (assemblies of single copies of core and modification domains) that act in a sequential (or in some cases, iterative) manner. PKS-NRPS enzymes are built from partial or complete modules of a PKS and an NRPS, and produce composite SM scaffolds that combine PK and NRP biosynthons.
Some BGCs from HEF are now well characterized, with the structures of their product congeners elucidated, and the proposed biosynthetic functions of their encoded enzymes validated by gene deletion and complementation, or by heterologous expression. Recent examples of these “voucher” HEF BGCs include those that yield the NRPs destruxins,8, 12 beauvericin,13 bassianolide14 and the serinocyclins;15 those that produce the PK oosporein;16 and those that afford the hybrid PKS-NRPS products tenellin, desmethylbassianin and NG39x.1, 17, 18 However, the vast majority of the BGCs predicted in HEF genomes are not linked to SM products (“orphan” BGCs), including many clusters that are not expressed under routine laboratory conditions. These silent, cryptic BGCs may only be transcribed under specific (and typically unknown) conditions such as those clusters that are temporarily activated during defined phases of insect infection.10, 19 The development of facile strategies to induce the expression of these silent BGCs, and to assign SM structures to orphan clusters will allow us to elucidate the parvome7, 20–26 (the encoded secondary metabolome) of microorganisms, including HEF. In the past few years, significant progress has been made towards the discovery of novel fungal SMs by engineering the activation of specific BGCs, or by remodeling global gene expression patterns that also affect BGCs.7, 20, 27 Parallel developments in metabolomics and SM structure elucidation techniques including mass spectrometry and nuclear magnetic resonance spectroscopy has played a synergistic role in expanding our understanding of the fungal parvome.28–31 Many of these methods have also been applied to HEF to discover novel, biologically active SMs, as described in the companion review in this issue.32–36 Since parvome discovery methods are still laborious, have to be applied on a case-by-case basis, and provide unpredictable results, choosing a “talented” group of microorganisms such as HEF with relatively little overlap of their predicted parvome with known SMs is important to maximize the chances of success.
1.1. Scope of this review
This review surveys the SM biosynthetic potential encoded in HEF genomes, concentrating on BGCs with PKS, NRPS, or PKS-NRPS hybrid genes at their core. It updates previous reviews that considered HEF molecular genetic SM discovery,1 or detailed PK and NRP biosynthesis in two major cosmopolitan HEF species, Beauveria bassiana and Metarhizium robertsii (M. anisopliae).2,10,37 This review focuses on the period of 2014 to the third quarter of 2019 that saw a large increase in the number of published HEF genome sequences (31 new genome sequences, compared to the eight that were published earlier), establishing a more abundant reservoir to analyze the biosynthetic potential of SMs and relationships among the BGCs of HEF. We describe recent examples of genomics-based discovery and functional characterization of BGCs for known SMs, then consider the use of genomics to predict the parvome of HEF. Finally, we provide a brief summary of the meta-parvome encoded in the recently sequenced genomes of HEF. For this review, we consider the genera Acremonium, Aschersonia, Beauveria, Cordyceps (some strains formerly known as Isaria or Paecilomyces), Gibellula, Hevansia (Akanthomyces), Hirsutella, Hypocrella, Lecanicillium, Metarhizium, Moelleriella, Ophiocordyceps, Purpureocillium (formerly Paecilomyces), Tolypocladium, Torrubiella (Verticillium), and Trichothecium. Taxonomic names of fungi featured in this review follow those in Mycobank (www.mycobank.org) wherever possible. However, SM producer strains that had been claimed to belong to Metarhizium anisopliae in the original chemical or biosynthetic papers referenced here may have now been separated from M. anisopliae (sensu stricto) and transferred to M. robertsii.38 Similarly, an entomopathogenic39 fungus isolated from an insect/Cordyceps sinensis natural complex39 remains described as Paecilomyces hepiali even after its genome sequence has been published.40 However, the genus name Paecilomyces is now restricted for fungi in the Eurotiales41–43 thus P. hepiali is classified in Mycobank (www.mycobank.org) under that order. Nevertheless, this fungus clearly belongs to the Hypocreales based on its morphological, chemical and genomic characteristics, thus we refer to this HEF in the current review as “Paecilomyces hepiali” FENG.
2. Genomics as a tool to identify BGCs for previously isolated compounds
Identification of BGCs responsible for the biosynthesis of previously isolated SMs benefited from the growing number of genome sequence assemblies in databanks and the development of more powerful bioinformatics tools. This section summarizes several recent examples where genomics was successfully used to locate target BGCs in HEF genomes and to develop and validate biosynthetic hypotheses for well-known SMs of PK, NRP, PK-NRP, and PK-meroterpenoid origin.
Oosporein 1 (Scheme 1) is a red 1,4-bibenzoquinone pigment isolated from the HEF Beauveria bassiana more than 50 years ago.1 Early precursor feeding experiments indicated that the simple PK orsellinic acid 2 is a precursor of oosporein.44 A genome sequence survey located 12 PKS genes in B. bassiana, two of which encoded synthases with similar domain composition to previously-validated orsellinic acid-producing PKSs from Aspergillus nidulans,45 A. terreus46 and Fusarium graminearum.47 Further bioinformatic analysis determined that one of these PKSs, encoded by BBA_08179 (opS1), is phylogenetically close to the validated orsellinic acid-producing PKS counterparts. This PKS anchors a BGC comprising of seven genes (opS1-opS7).16 Gene deletion of opS1 abrogated oosporein production in B. bassiana, while the biosynthesis of this SM resumed upon supplementation of orsellinic acid 2 to the culture of the opS1 deletion strain. Knockout of opS2, a gene encoding a putative major facilitator superfamily transporter, unexpectedly led to increased production of oosporein, identifying Ops2 as a negative regulator of oosporein biosynthesis. OpS3 encodes a Gal4-like Zyn2Cys6 domain transcription factor whose overexpression increased the expression of most genes in the BGC leading to a four-fold increase in the amount of oosporein produced. Amongst the biosynthetic enzymes, the salicylate hydroxylase OpS4 converted orsellinic acid to 6-methyl-1,2,4-benzenetriol 3 and a new product, 5,5’-dideoxy-oosporein 5 when heterologously produced in yeast (Scheme 1). Spontaneous oxidation of benzenetriol 3 afforded the benzoquinone 4. However, on-pathway oxidation of benzenetriol 3 by the cupin-family oxygenase Ops7 yielded the benzenetetrol 7 that tautomerizes to quinone 6 or dimerizes to oosporein 1 due to the catalytic activity of the laccase Ops5. Meanwhile, the glutathione S-transferase Ops6 protects the fungus from the harmful effects of the free radical intermediate 8 formed by Ops5 (Scheme 1). A syntenic BGC with orthologous genes to those of the B. bassiana oosporein BGC has recently been identified in the genome sequence assembly of Cordyceps cycadae, and production of oosporein was confirmed by HPLC both in the liquid culture and the fruiting bodies of this HEF.48
Scheme 1. Biosynthesis of oosporein 1 in Beauveria bassiana.
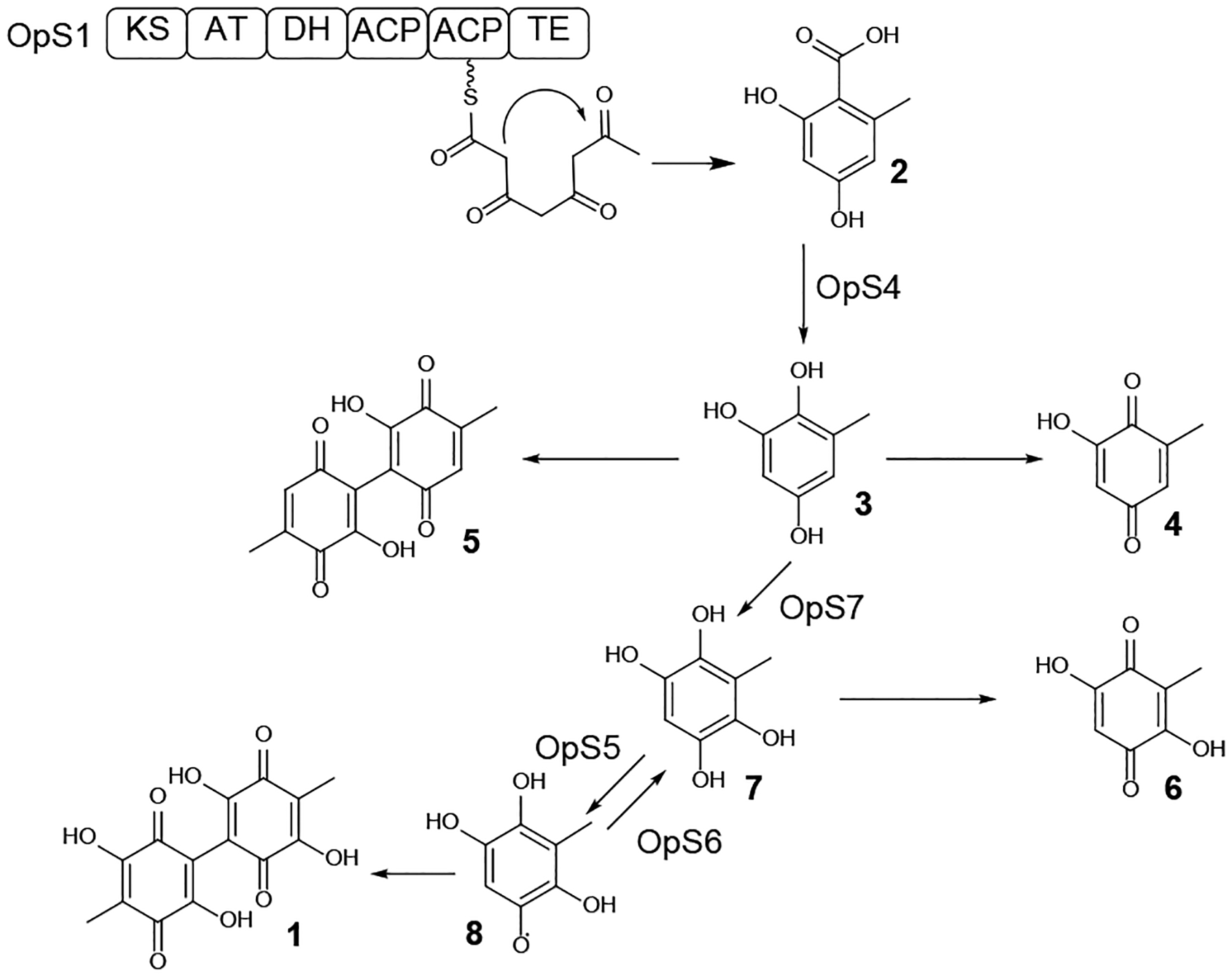
Reactions catalyzed by enzymes encoded in the oosporein BGC are indicated with the name of the enzyme. Adapted from Feng et al.16 with permission (Copyright 2015, National Academy of Sciences).
Leucinostatins such as leucinostatin A 9 are linear, microheterogenous peptaibiotic NRPs with nine amino acid residues where the N-terminal methylproline is acylated with 4-methylhex-2-enoic acid, while the C-terminal β-alanine is amidated by N,N-dimethylpropane-1,2-diamine (DPD).49 Leucinostatins were isolated from Purpureocillium lilacinum (formerly Paecilomyces lilacinus), an ophiocordycipitalean fungus that infects insects, nematodes and even mammals, but is also often isolated from soil or from plant tissues. Leucinostatins show potent cytotoxic,50–52 anti-trypanosomal,53 antifungal and antibiotic49, 54 activities due to their inhibition of the mitochondrial oxidative phosphorylation. The leucinostatin BGC was identified in the Pu. lilacinum genome assembly based on the clustering of genes encoding a 10-module NRPS and two PKSs.49 The BGC was validated by knocking out several key biosynthetic genes, while cluster boundaries were delimited by differential transcriptomics and qRT-PCR analysis under leucinostatin producing vs. nonproducing conditions. Overexpression of the LcsF cluster-specific regulator increased leucinostatin production and induced the expression of genes in the BGC. The broad outlines of the biosynthetic steps were proposed based on the genes present in the BGC (Scheme 2).49 Thus, one of the two hrPKSs would produce 4-methylhex-2-enoic acid that is transferred to the N-terminal T domain of the LcsA NRPS by the LcsE thioesterase and the LcsD acyl CoA-ligase. The other hrPKS from the BGC was proposed to be involved in the assembly of the nonproteinogenic amino acid 2-amino-6-hydroxy-4-methyl-8-oxodecanoic acid. Peptide chain assembly is terminated by the R (reductive release) domain at the C-terminus of LcsA, and the released aldehyde may undergo transamination catalyzed by the LcsP aminotransferase to yield the 1,2-diaminopropane-capped peptide. Oxidation by one of the LcsI, LcsK and LcsN cytochrome P450 enzymes, and N-dimethylation of the C-terminal cap residue are expected to complete the biosynthetic process. However, these reactions, as well as the formation of the β-alanine, α-aminoisobutyric acid and methylproline residues remain uncharacterized.49
Scheme 2. Proposed biosynthesis of leucinostatin A 9 and leucinostatin B 10 in Purpureocillium lilacinum.
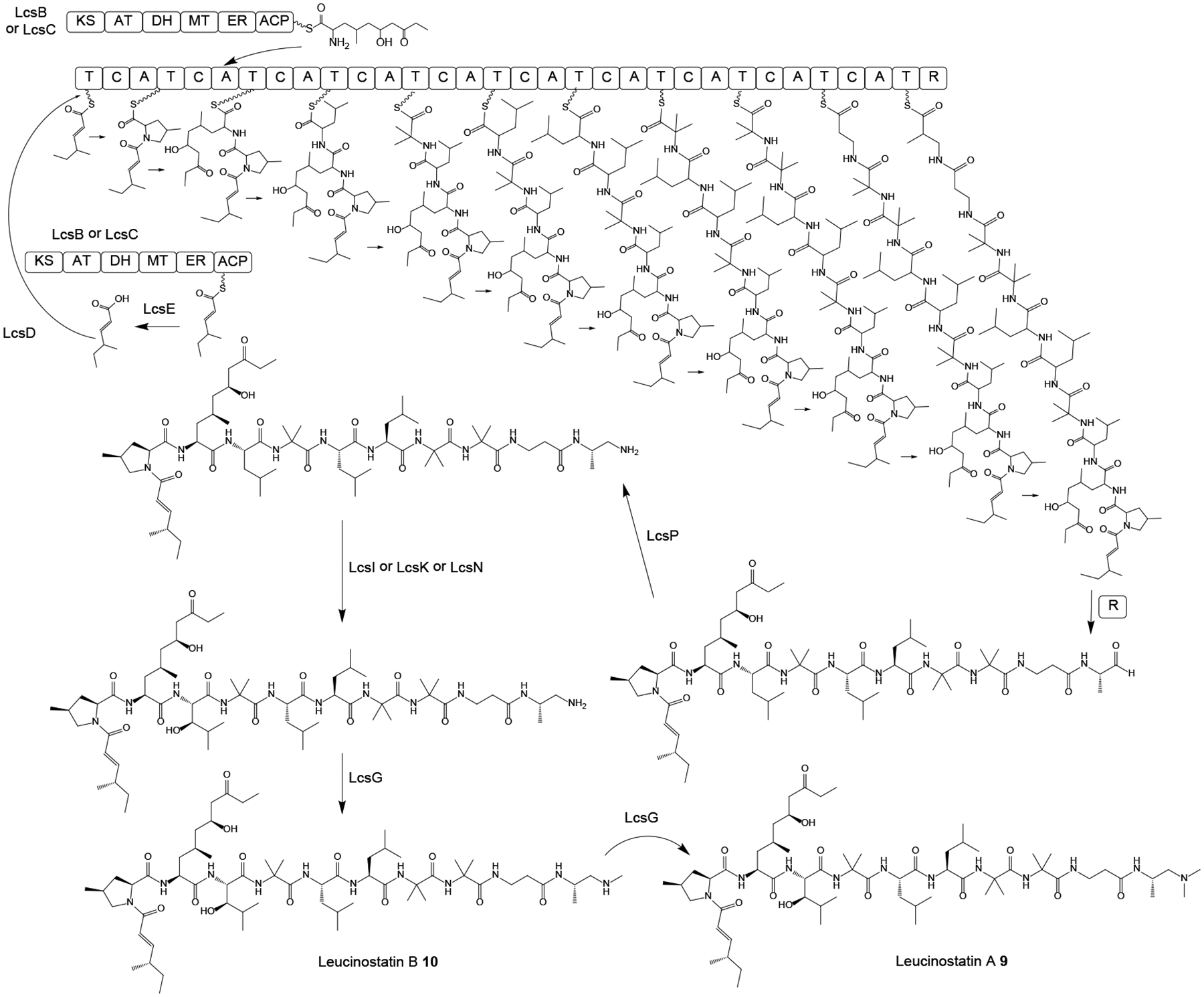
Adapted from Wang et al.49 with permission (Copyright 2016, Public Library of Science).
The BGC for the cyclic lipohexadepsipeptides verlamelin A 10 and verlamelin B 11 (Scheme 3) was identified from Lecanicillium sp. HF627, a pathogen of Thrips spp. (Thysanoptera).55 RT-PCR with NRPS consensus primers was used to collect A domain ESTs under verlamelin production conditions, and gene disruption validated one of these A domains as part of the NRPS responsible for the production of compounds 10 and 11. The corresponding NRPS, encoded by the vlmS gene was discovered in the draft genome sequence of Lecanicillium sp. as part of a BGC that also features the fatty acid hydroxylase vlmA, the thioesterase vlmB and the AMP-dependent ligase vlmC. The VlmS NRPS consists of seven modules with epimerization domains in the 2nd, 3rd, and 6th modules, matching the configurations of the peptidyl moieties of verlamelins A and B (Scheme 3). The starter module at the N-terminus contains an inactive epimerization domain (E0) where the catalytic histidine residue is substituted by an alanine. This starter module lacks the A domain whose function may be substituted by the AMP-dependent ligase VlmC. VlmC may activate 5-hydroxytetradecanoic acid, which is derived by the thioesterase VlmB and the hydroxylase VlmA from myristic acid provided by the primary metabolic fatty acid synthase (FAS). After six rounds of elongation with the appropriate amino acids, the depsipeptide is cyclized by the terminal CT domain of VlmS through the C5 alcohol of the fatty acyl moiety (Scheme 3).55
Scheme 3. Proposed55 biosynthesis of verlamelin A 10 and verlamelin B 11 in Lecanicillium sp. HF627.
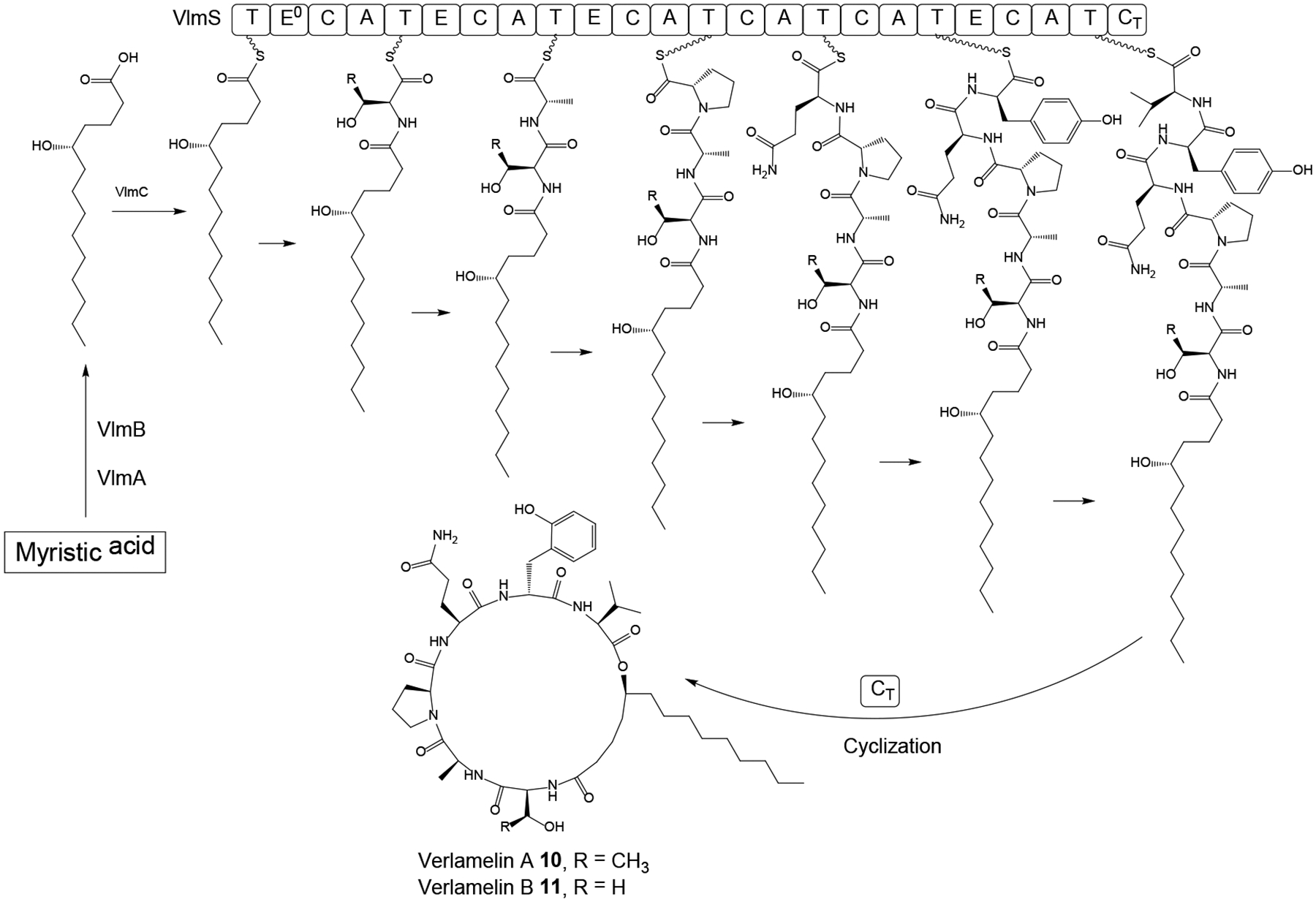
E0, inactive epimerization domain; CT, terminal C domain for intramolecular cyclization.
Cyclosporine A 12 (Scheme 4) is a cyclic undecapeptide that was discovered from the HEF Tolypocladium inflatum in the early 1970s on the basis of its antifungal activity.56 It was later developed as an immunosuppressant drug that revolutionized organ transplantation since its clinical introduction in 1983.57 Although SimA, the NRPS responsible for the assembly of 12,58, 59 and the alanine racemase that supplies D-Ala for this NRPS have both been identified,60, 61 and a proposed PKS that generates another precursor, (4R)-4-[(E)-2-butenyl]-4-methyl-l-threonine (Bmt) has been biochemically characterized,62, 63 the whole BGC for cyclosporine A production was only uncovered after whole genome sequencing of T. inflatum.56 This allowed the functional characterization of the individual BGC genes by gene deletions, the isolation of intermediates from the knockout strains, and substrate feeding experiments.57 These studies confirmed the role of the NRPS SimA in assembling cyclosporine A from 11 amino acid monomers (Scheme 4), including N-methylation of the growing peptide at the appropriate extension cycles by integrated N-methyltransferase domains in the corresponding modules of SimA, and cyclization of the final product by a terminal CT domain. These studies also clarified that Bmt is biosynthesized by the PKS SimG, the cytochrome P450 monooxygenase SimI, and the aminotransferase SimJ; and that d-alanine is produced from l-alanine by the alanine racemase SimB as a free substrate, instead of being generated by an on-board NRPS epimerase domain from a thioester-bound intermediate. The transcription of the BGC was shown to be regulated by SimL, a basic leucine zipper (bZIP)-type protein. Meanwhile, the cyclophilin SimC and the ABC (ATP-binding cassette) transporter SimD both contribute to cyclosporine A resistance in T. inflatum. Finally, these genomic and functional studies also validated a dual function for cyclosporine A during the lifecycle of T. inflatum: this SM enables T. inflatum to outcompete other fungi during co-cultivation, while the compound also acts as an immunosuppressive virulence factor against the insect hosts.57
Scheme 4. Proposed biosynthesis of cyclosporine A 12 in Tolypocladium inflatum.
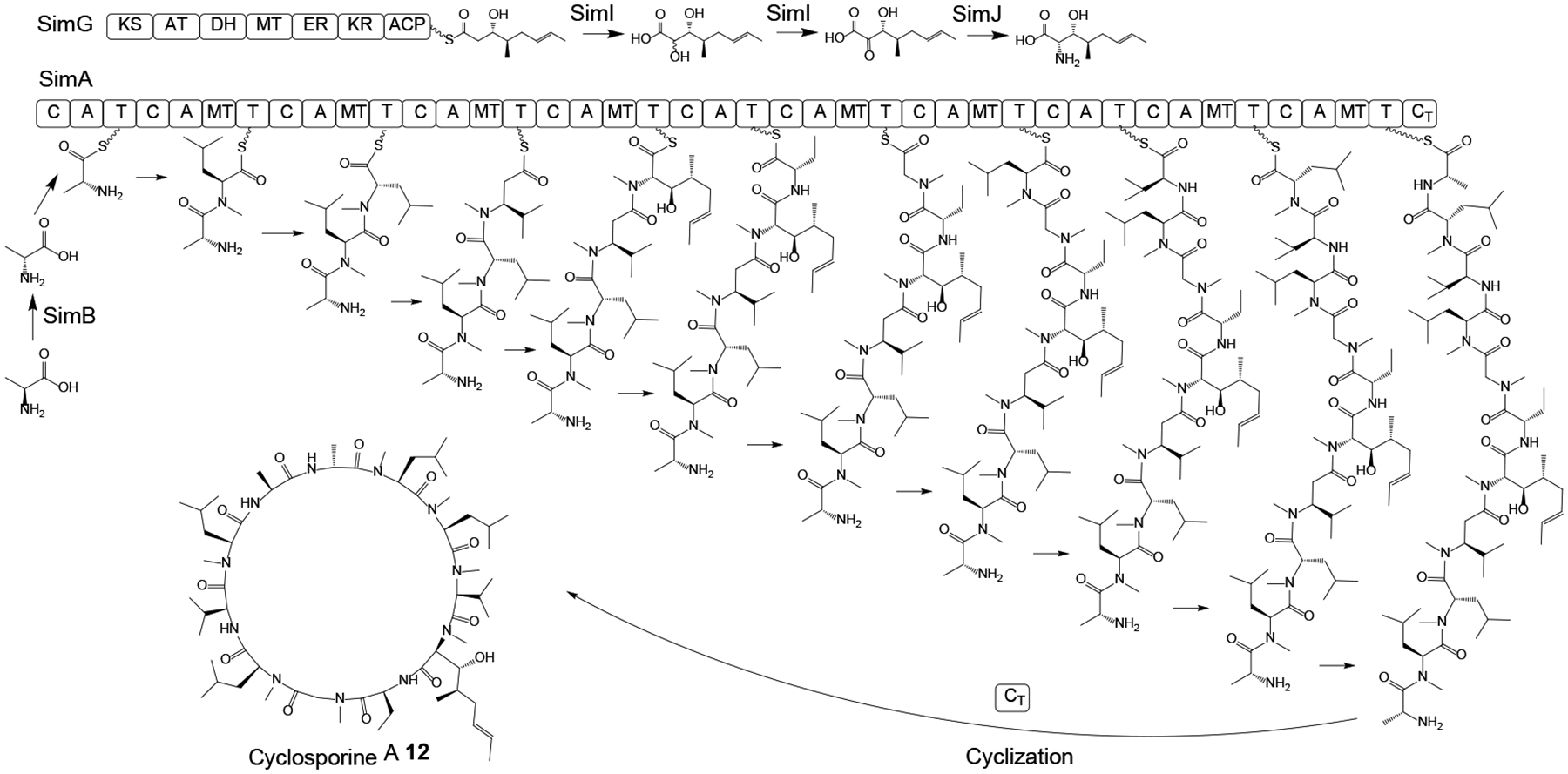
Adapted from Yang et al.57 with permission (Copyright 2018, American Society for Microbiology).
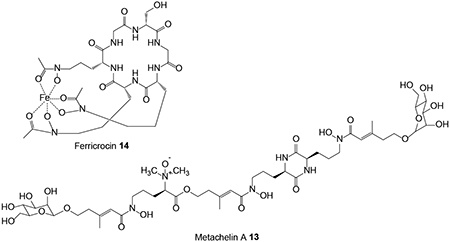
BGCs for NRP-based siderophores have been identified in the genome sequence assembly of the HEF M. robertsii based on the orthologous relationships of the NRPSs anchoring these clusters to those of functionally validated siderophore synthetases in other fungi.64 MrSidD is a bimodular NRPS with an ATC-TC domain architecture that produces the coprogen-type extracellular siderophore metachelin A 13. MrSidC is a type IV ferrichrome synthetase NRPS with a domain structure of ATC-ATC-TC-ATC-TC-TC. These NRPSs are clustered with conserved genes for siderophore biosynthesis, excretion and uptake such as the MrAbcB membrane transporter, the MrSidF triacetylfusarinine transacylase and the MrSidI triacetylfusarinine enoyl-CoA hydratase for MrSidD; and the MrSidJ triacetylfusarinine esterase and the MrSidA l-ornithine-N5-monooxygenase for MrSidC. However, the glycosyltransferase necessary for the O-mannosylation of metachelin A has not been found clustered with the MrSidD NRPS. Gene knockouts of the NRPSs validated their involvement in the production of the appropriate siderophores. Deletion of MrSidD led to only mild physiological effects and did not reduce virulence, indicating that other iron acquisition systems can efficiently compensate for the lack of metachelin production. Deletion of MrSidC led to more pronounced delays in growth and germination and reduced but did not eliminate virulence during insect infection, emphasizing the importance of the siderophore ferricrocin 14 for intracellular iron homeostasis.
Fumosorinone 15 (Scheme 5) is a 2-pyridone alkaloid isolated from the HEF Cordyceps (formerly Isaria) fumosorosea. Compound 15 is a potent noncompetitive inhibitor of protein tyrosine phosphatase 1B, a promising target for the treatment of type II diabetes.65 Its BGC, anchored by the hybrid PKS-NRPS FUMOS, was identified in the genome sequence of C. fumosorosea ARSEF 2679, and validated via the knockout of FUMOS using Agrobacterium-mediated transformation. FUMOS consists of one PKS and one NRPS module with a domain composition of KS-AT-DH-MT-KR-ACP – C-A-T-DKC, where the PKS module assembles a reduced and C-methylated octaketide that is fused with tyrosine by the NRPS module, and released by the terminal Dieckmann cyclase (DKC) domain as an acyltetramic acid (Scheme 5).65 The BGC also encodes a trans enoyl reductase that partners with FUMOS to reduce the PK chain during the first two extension cycles; and two cytochrome P450 monooxygenases that are involved in the oxidative ring expansion of the acyltetramic acid intermediate and the subsequent N-hydroxylation that affords the 1,4-dihydroxy-2-pyridone core of fumosorinone 15. The BGC is completed by two genes encoding deduced transcriptional regulators. BGCs with orthologous genes for the production of desmethylbassianin and tenellin, 1,4-dihydroxy-2-pyridones from two B. bassiana strains, and for an unknown 2-pyridone from the genome sequence of C. militaris have also been described.66–69
Scheme 5. Proposed biosynthesis of fumosorinone 15 in Cordyceps fumosorosea.
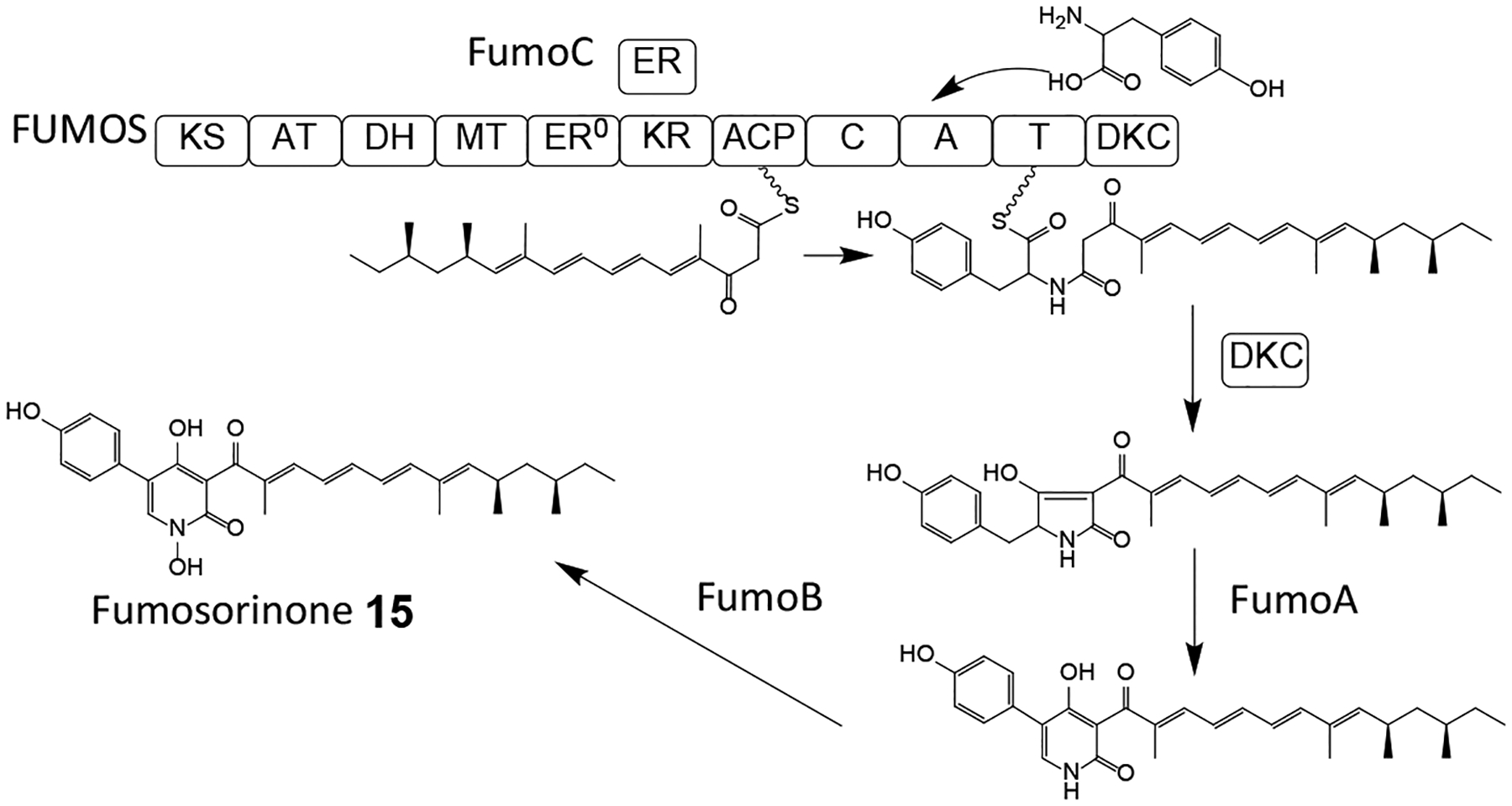
Adapted from Liu et al.65 with permission (Copyright 2015, Elsevier).
The indolizidine alkaloid swainsonine 16 (Scheme 6) is a potential drug for cancer chemotherapy70 that disrupts the endomembrane system of eukaryotic cells by specifically inhibiting the enzyme α-mannosidase II in the Golgi apparatus.71 Swainsonine 16 is produced by various endophytic and plant pathogenic fungi as well as certain HEF such as the plant symbiotic HEF M. robertsii.71, 72 Orthologous BGCs responsible for the biosynthesis of this SM were located by comparing PKS-NRPS-related genes from the genome sequence assemblies of the swainsonine producers M. robertsii and ICE, the fungal endophyte of Ipomoea carnea. These BGCs (termed the SWN clusters) were found organized around the swnK gene that codes for a multidomain synthase with an A, an AT, a KS, two carrier protein and two reductase domains. Knockout of the swnK gene in M. robertsii eliminated swainsonine biosynthesis, while complementation of the mutant with an intact copy of the swnK gene restored the production of this SM. Both SWN clusters also encode two putative hydroxylases (SwnN and SwnR) and two dioxygenases (SwnH1 and SwnH2) each. Orthologous SWN clusters were subsequently identified in the genome sequences of multiple Metarhizium spp., the plant pathogen Slafractonia leguminicola, the endophyte Alternaria oxytropis, and the saprobiont Pseudogymnoascus sp. Unexpectedly, orthologous SWN clusters were also found in the genome sequences of multiple Anthrodermataceae fungi: these skin pathogens had not previously been known to produce swainsonine-like alkaloids. The wide phylogenetic distribution of the SWN cluster suggests that swainsonine has a role in mutualistic or pathogenic interactions of the producer fungi with plants or animal hosts.71 Swainsonine biosynthesis was proposed to use pipecolic acid and malonyl-CoA as the precursors (Scheme 6), and proceed through aldehyde intermediate 17 that spontaneously cyclizes to iminium 18. Reduction of compound 18 by SwnR or SwnN yields the 1-hydroxyindolizidine 19, which is oxidized by SwnH1 and SwnH2 and epimerizes to a second iminium 20 whose reduction by SwnR or SwnN finally yields swainsonine 16 (Scheme 6).
Scheme 6. Proposed biosynthesis of swainsonine 16 in Metarhizium robertsii.
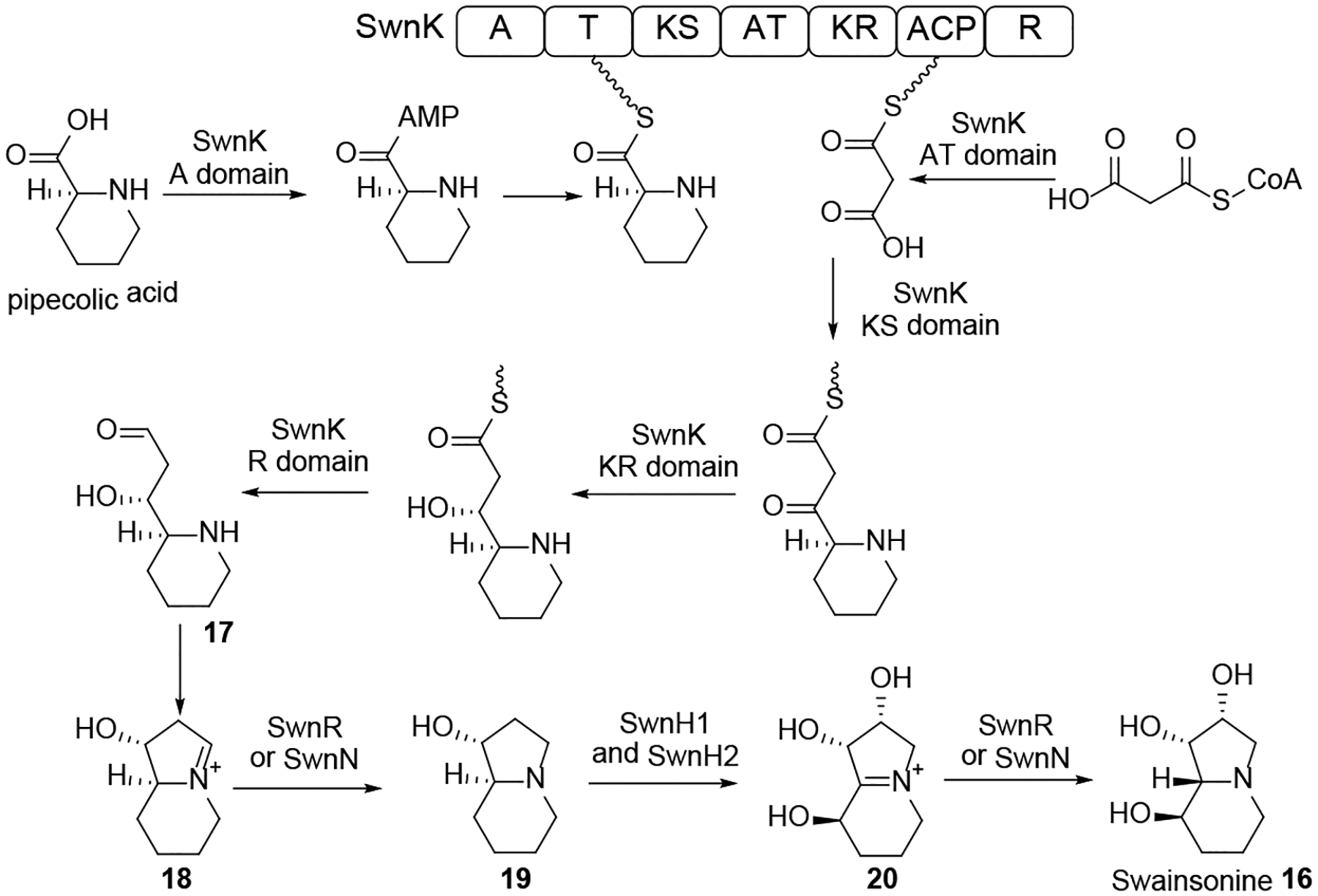
Adapted from Cook et al.71 with permission (Copyright 2017, G3: Genes, Genomes, Genetics).
Tolypocladium ophioglossoides is a close relative of the HEF T. inflatum whose lineage switched to a mycoparasitic lifestyle. Genome sequence analysis of T. ophioglossoides led to the identification of the BGC for balanol 24 (Scheme 7), a composite PK-NRP metabolite that is a potent ATP-competitive inhibitor of protein kinases C and A.73 Overexpression of the putative cluster-specific regulator BlnR allowed the expression of the otherwise silent 18-gene BGC, and subsequent deletions of key biosynthetic genes in the BlnR-overexpressing background helped to clarify the biosynthesis of balanol 24. The BGC contains a subcluster organized around the BlnA nrPKS with orthologous genes to that for monodictyphenone biosynthesis from A. nidulans.74 This subcluster yields monodictyphenone analogues such as 21 (Scheme 7) as well as xanthones and endocrocin. The BGC also encodes the bimodular NRPS BlnO that produces a p-hydroxybenzoic acid – L-lysine amide that is released through intramolecular cyclization by amide formation between the terminal amino group of the lysine side chain and its main chain carboxylate, catalyzed by the terminal TE domain. After carbonyl reduction, hydroxylation of the azepane 22 by the dioxygenase BlnF ensues to yield compound 23. Finally, the single-module NRPS BlnN catalyzes intermolecular ester bond formation between the hydroxyazepane 23 and the product of the PKS subcluster, the oxidized monodictyphenone analogue 21 activated as the BlnN T-domain-bound thioester. Thus, the balanol BGC is the composite of two semi-independent subclusters that produce a PK and an NRP moiety in a parallel manner, with biosynthesis converging on the BlnN NRPS that fuses the two biosynthons.1, 73
Scheme 7. Proposed biosynthesis of balanol 24 in Tolypocladium ophioglossoides.
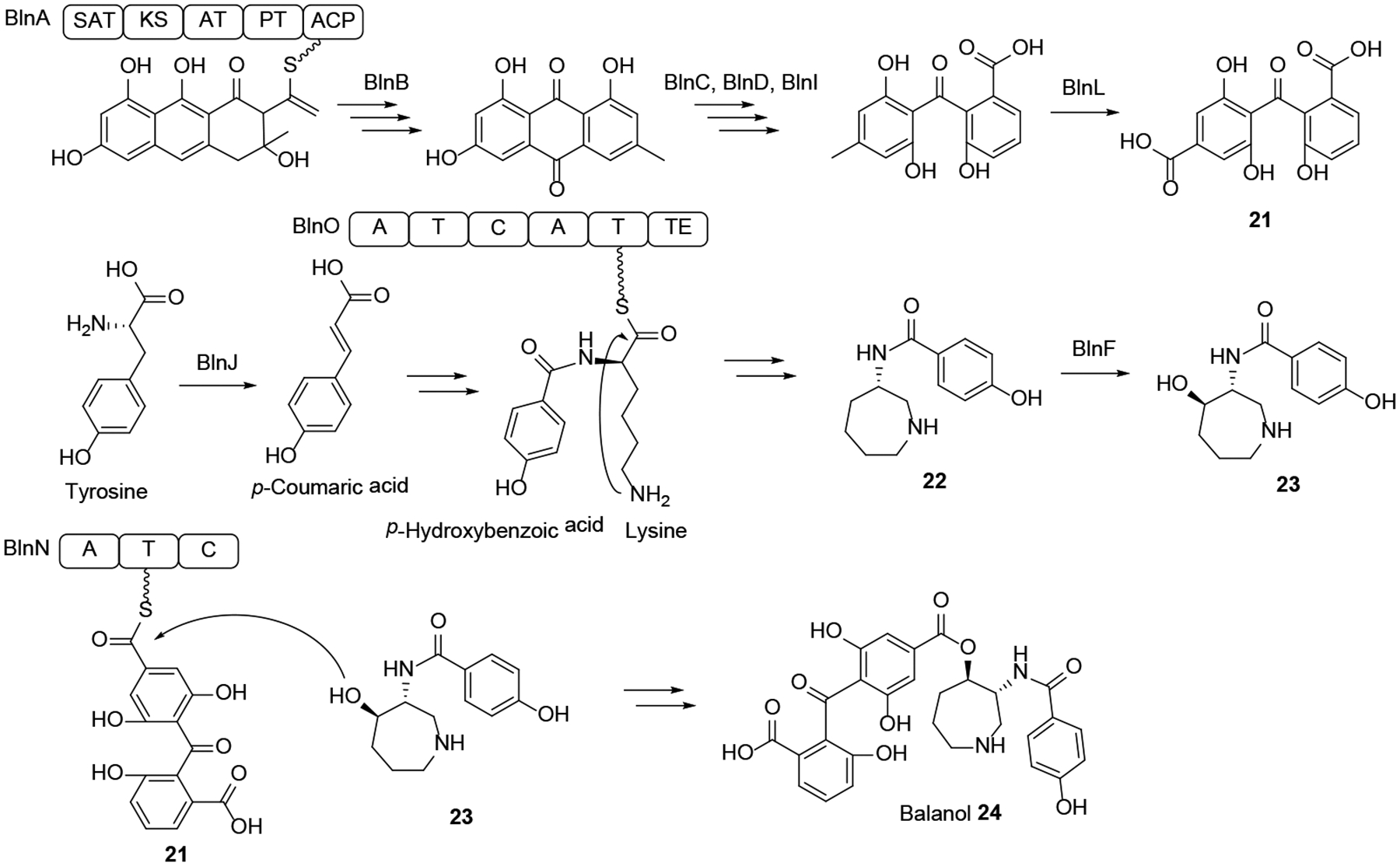
Adapted from He et al.73 with permission (Copyright 2018, American Chemical Society).
The meroterpenoid immunosuppressants subglutinol A (25, Scheme 8) and subglutinol B were first isolated from Fusarium subglutinans,75 but these and additional congeners such as compounds 26 and 27 were also obtained from the HEF M. robertsii.76 Subglutinols feature a PK-derived α-pyrone moiety attached to a terpenoid-derived decalin core fused with a tetrahydrofuran with a prenyl side chain. The subglutinol BGC was identified in the genome sequence assembly of M. robertsii based on the predicted presence of clustered genes for a nonreducing PKS, a prenyltransferase and a terpene cyclase. Heterologous expression of the PKS SubA in A. nidulans yielded the expected α-pyrone, while co-expression of SubA with the geranylgeranyl diphosphate synthase SubD and the prenyltransferase SubC afforded the linear geranylgeranyl α-pyrone intermediate of subglutinols (Scheme 8). Formation of the fused decalin - tetrahydrofuran ring system was predicted to be initiated by epoxidation of the prenyl chain by the FAD-dependent oxidase SubE, followed by cyclization catalyzed by the terpene cyclase SubB, and cyclic ether formation by the FAD-dependent dehydrogenase SubF.76
Scheme 8.
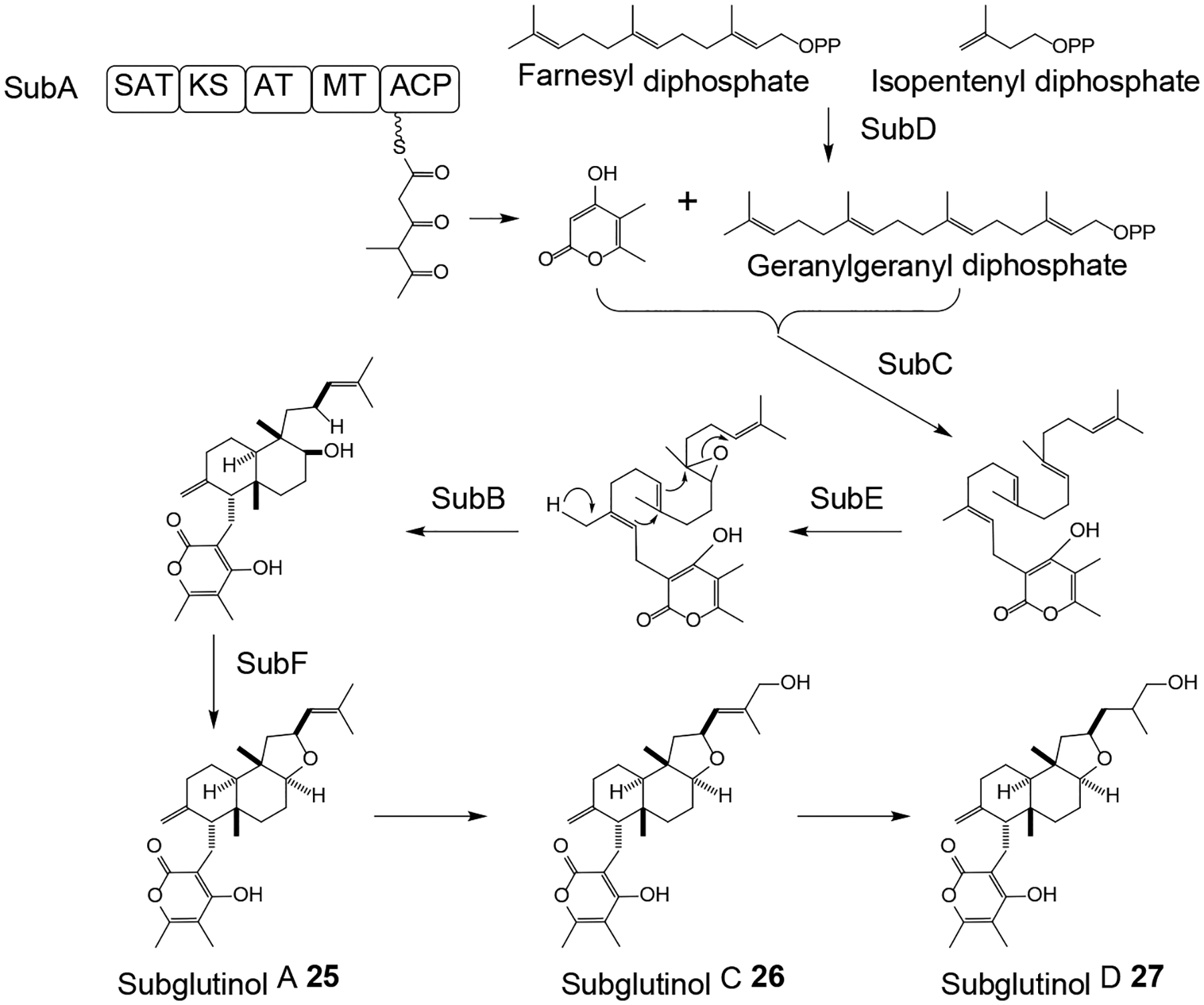
Proposed76 biosynthesis of subglutinol A 25 and its congeners in Metarhizium robertsii.
The meroterpenoid ascofuranone 28 and its congener ascochlorin 29 (Scheme 9) were obtained from the HEF Acremonium egyptiacum.77 Ascofuranone 28 shows therapeutic potential in a rodent infection model of African trypanosomiasis (sleeping sickness) due to its potent inhibition of cyanide-insensitive alternative oxidases from trypanosomes. The biosynthetic pathway that yields both compounds 28 and 29 were elucidated by comparing the transcriptomes of Ac. egyptiacum under ascofuranone-competent vs. ascofuranone non-producing culture conditions and mapping differentially expressed genes on the draft genome sequence of the strain. This led to the identification of two disjointed gene clusters that are collectively responsible for the biosynthesis of compounds 28 and 29 and form a dispersed BGC regulated by the common cluster-specific Zn2Cys6-type regulator AscR. Heterologous expression of selected genes from this disjointed BGC in A. oryzae led to the reconstruction of the initial steps of the biosynthesis (Scheme 9), including the assembly of orsellinic acid 30 on the PKS AscC; the prenylation of this intermediate with farnesyl diphosphate by AscA to yield ilicicolinic acid B 31; the reduction of compound 31 by the NRPS-like reductase AscB to afford ilicicolin B 32; and the chlorination of 32 by the halogenase AscD to produce ilicicolin A 33. In vitro reconstitution of the subsequent steps catalyzed by the cytochrome P450 epoxidase AscE, the terpene cyclase AscF, and another cytochrome P450 monooxygenase AscG yielded ascochlorin 29 (Scheme 9). Ascofuranone biosynthesis branches from ilicicolin A epoxide 34, the product of AscE, and requires the cytochrome P450 monooxygenase AscH, the terpene cyclase AscI, and the dehydrogenase AscJ, all encoded by genes in the second, distant part of the disjointed BGC. Phylogenomic considerations support a scenario whereby this AscJIH subcluster evolved late to channel an advanced ascochlorin intermediate towards a novel metabolite, ascofuranone 28. Importantly, the differential transcriptomics approach used in this study was essential for the BGC discovery process since AscI, the core enzyme anchoring the second part of the BGC shows very little similarity to known terpene cyclases, and AscH, the enzyme that is responsible for diverting compound 34 towards ascofuranone production is a cytochrome P450 from which there are more than a hundred encoded in the Ac. egyptiacum genome.77
Scheme 9. Proposed biosynthesis of ascofuranone 28 and ascochlorin 29 in Acremonium egyptiacum.
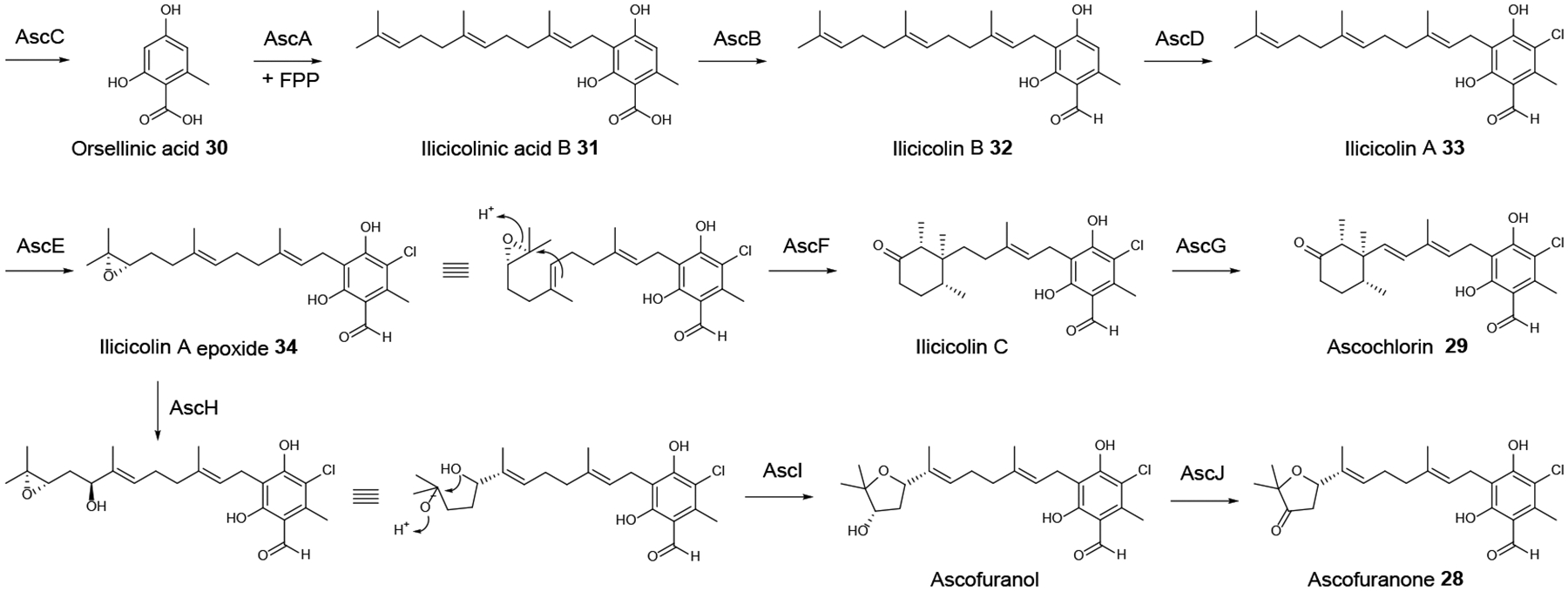
FPP, farnesyl diphosphate. Adapted from Araki et al.77 with permission (Copyright 2019, National Academy of Sciences).
Peramine 35 (Scheme 10) is an NRP-derived pyrrolopyrazine-containing SM with insecticidal activities.78 Peramine was first identified from Epichloë spp. that form mutualistic endophytic associations with cool-season grasses, whereby this compound contributes to the protection of the fungus-grass association against herbivory. Compound 35 has a potential use in agriculture due to its potent insect anti-feeding activities that can provide deterrence against insect pests such as the Argentine stem weevil (Listronotus bonariensis).79 Peramine biosynthesis in Epichloë spp. from pyrroline-5-carboxylic acid and arginine was proposed to require only the bimodular NRPS PerA with the domain composition of A-T-C-A-M-T-R;80 however this has not been conclusively proven. PerA orthologues have recently been found to be encoded in the genome sequences of the insect-pathogenic fungi M. rileyi and M. majus, and the stalked-cup lichen fungus Cladonia grayi.78 The M. rileyi perA gene was expressed in the heterologous host Penicillium paxilli and shown to be sufficient for peramine production. The Metarhizium and Cladonia perA orthologues are located in a predicted seven-gene BGC each that encodes an MFS transporter, two ketoglutarate-dependent dioxygenases, two cytochrome P450 monooxygenases, and one p-loop NTPase. These deduced enzymes may be involved in the biosynthesis of the pyrroline-5-carboxylic acid precursor and/or yield putative pyrrolopyrazine SMs by further elaborating peramine 35. Orthologues of these genes (apart from perA) are absent in Epichloë spp., thus the orphaned perA gene in those endophytes may represent an example of reductive evolution.78
Scheme 10. Proposed biosynthesis of peramine 35 in Metarhizium rileyi.
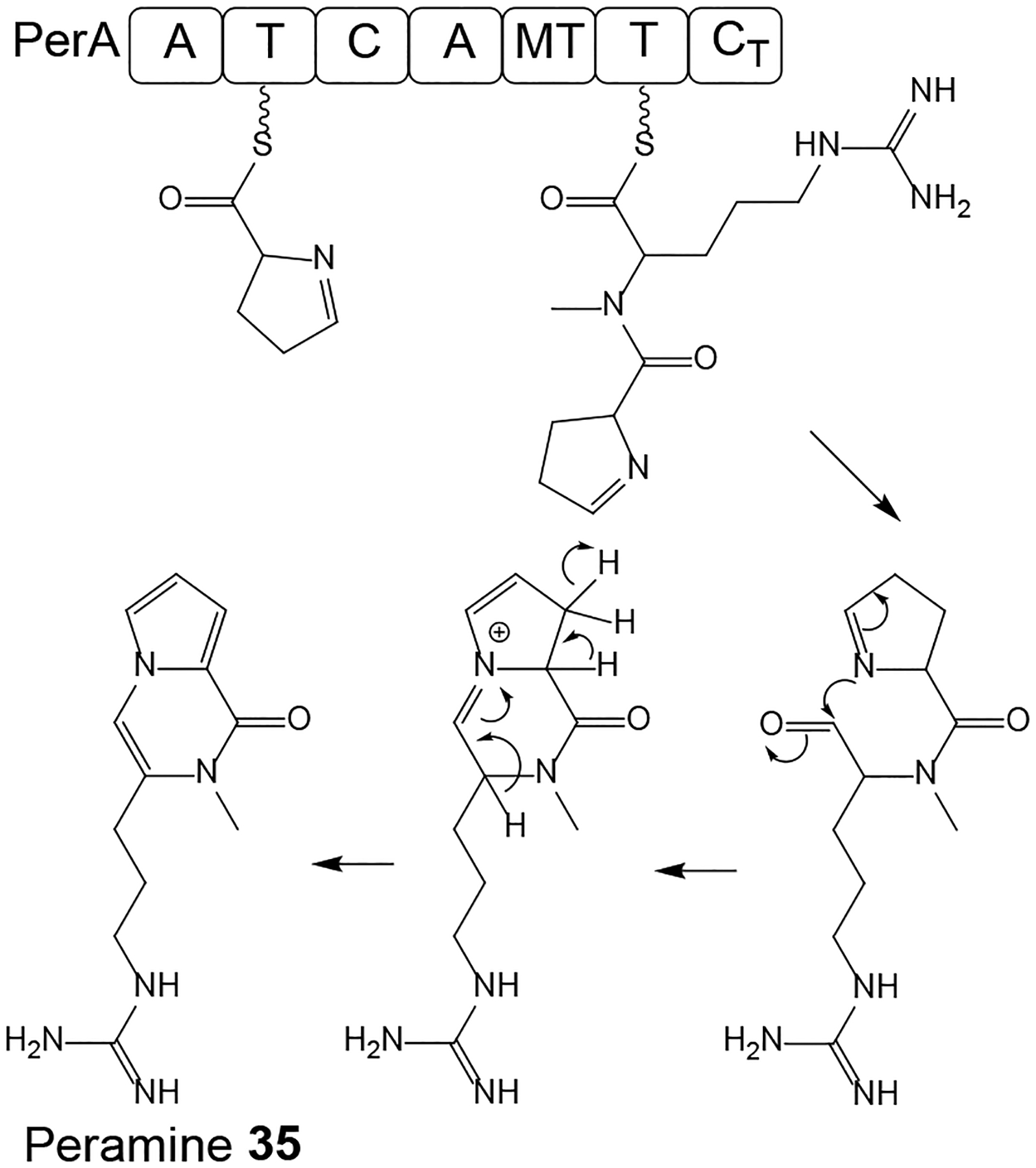
Adapted from Berry et al.78 with permission (Copyright 2019, John Wiley and Sons).
The nucleoside antibiotic and RNA synthesis inhibitor cordycepin (3’-deoxyadenosine 36, Scheme 11) is the best known biologically active ingredient of the caterpillar fungus Ophiocordyceps sinensis and similar Cordyceps spp. such as C. militaris and C. cicadae.1, 5 The fruiting bodies of these fungi are high-value traditional medicines that have been used since ancient times as tonics and roborants in East Asia. Contemporary studies indicate cancer cell antiproliferative, pro-apoptotic, anti-inflammatory and intracellular signal transduction modulatory activities for this compound. Comparative genomics of C. militaris and A. nidulans (a strain that had been reported to also produce cordycepin 36) revealed a syntenic BGC with four orthologous genes each in these fungi.81 Gene deletions in C. militaris and A. nidulans, and heterologous expression of the genes in S. cerevisiae proved that Cns1 and Cns2 (and their A. nidulans orthologues) are necessary and sufficient for 3’-deoxyadenosine 36 production. Deletion of cns3 eliminated the production of another adenosine analogue, pentostatin 37, a SM that had first been isolated from the bacterium Streptomyces antibioticus. Heterologous expression of cns3 in B. bassiana and M. robertsii validated that Cns3 alone is sufficient for pentostatin 37 biosynthesis. Deletion of the cns3 gene also allowed the production of yet another nucleoside analogue, 3’-deoxyinosine 38 that is the deaminated derivative of 3’-deoxyadenosine 36. Compound 38 was produced only when C. militaris was cultivated on insects, but not when it was cultured in artificial media. Considering that pentostatin 37 inhibits adenosine deaminase in an irreversible manner, this composite BGC produces synergistic SMs where pentostatin 37 protects cordycepin 36 from adenosine deaminases from the host or the producer fungus. Taken together (Scheme 11), the 3’-OH of adenosine is phosphorylated by the kinase activity of Cns3 to yield 3’-adenosine monophosphate 39. Compound 39 is dephosphorylated by Cns2 to 2’-carbonyl-3’-deoxyadenosine 40 that is then reduced to 3’-deoxyadenosine 36 by Cns1. In Δcns3 mutants or heterologous production systems lacking Cns3, 3’-AMP may be supplied by phosphoesterases from 2’,3’-cyclic adenosine monophosphate 41, a product of RNA degradation. Meanwhile, pentostatin 37 is produced by the C-terminal HisG ATP phosphoribosyltransferase domain of Cns3. The fourth conserved gene of the BGC encodes an ATP-binding cassette transporter for pentostatin 37 export from the mycelia. Syntenic BGCs were also found in the genome of Ac. chrysogenum and C. kyushuensis, but only C. kyushuensis was shown to produce cordycepin 36 and pentostatin 37. Surprisingly, the O. sinensis genome assembly was missing orthologous genes for cns1–3. Although transcriptomic studies from O. sinensis,82 C. cicadae,83 and “Paecilomyces hepiali” FENG84 (a cordycepin-producing HEF that is often encountered as an endoparasite of O. sinensis) catalogued ESTs for hypothetical steps of deoxyadenosine analogue conversions, elucidation of cordycepin 36 biosynthesis in these fungi has still not been completed.
Scheme 11. Proposed biosynthesis of cordycepin 36 and pentostatin 37 in Cordyceps militaris.
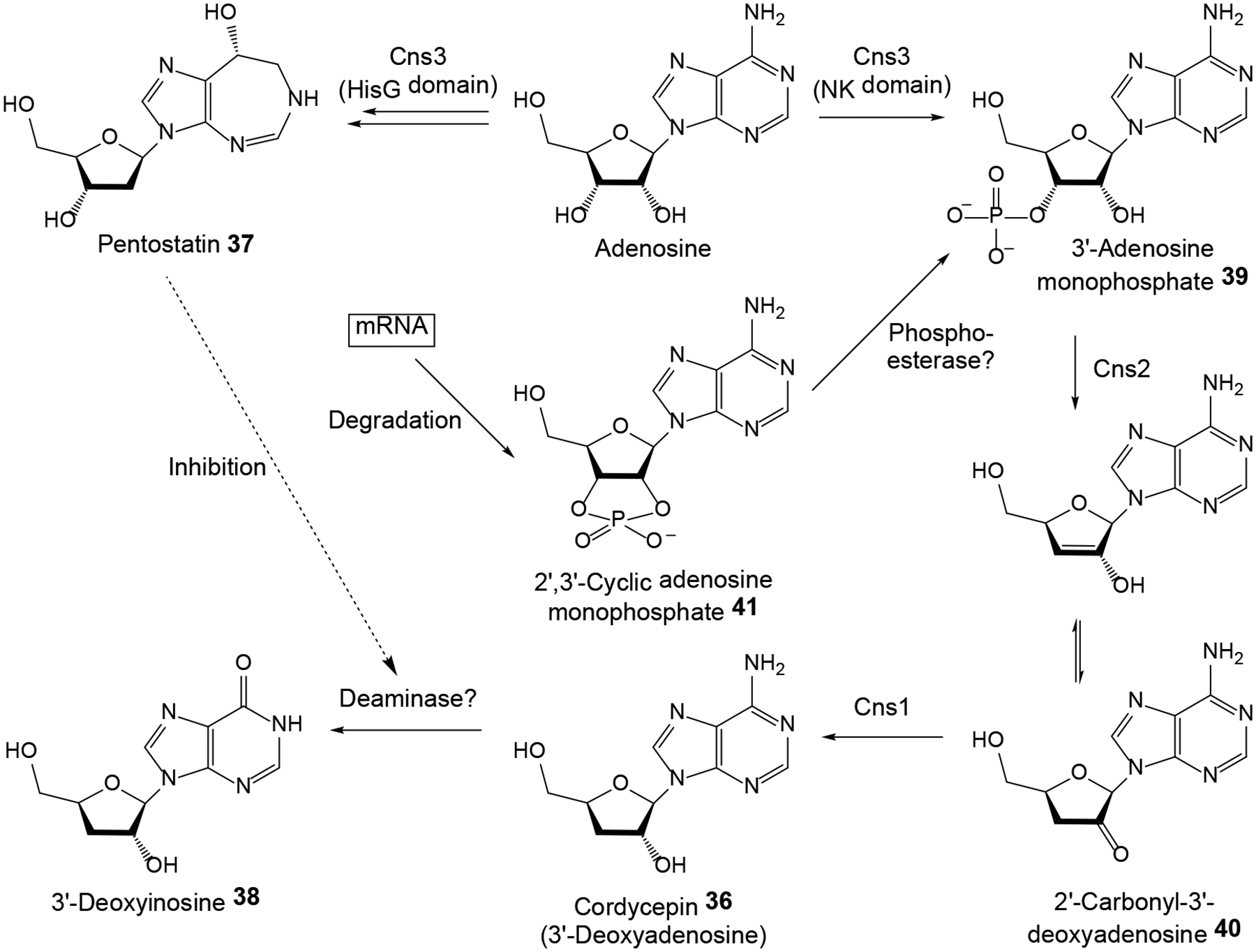
Adapted from Xia et al.81 with permission (Copyright 2017, Elsevier).
3. Comparative genomics as a tool to predict the products of orphan BGCs
As described in the previous section, the availability of HEF genome sequences allows researchers to functionally validate BGCs that were identified using retrobiosynthetic hypotheses formulated on the basis of the structures of known SMs. However, comparative genome analysis can also be used to reverse this workflow, and to derive hypotheses for the structures (of at least the scaffolds) of the products of orphan HEF BGCs. Thus, de novo sequenced BGCs may be predicted to afford metabolites belonging to known SM structural families if the core genes are orthologous, and the constituent tailoring gene assemblages are similar (or in some cases even syntenic) to those of BGCs that had been functionally characterized in other fungi. Isolation and structure elucidation of these predicted products from the genome-sequenced strain (or from heterologous hosts expressing the new BGC) are then used to validate such hypotheses. The growing number of HEF genomes in public databanks made these predictions even more informative by revealing lineage-specific conservation of certain orthologous BGCs, or by pointing out the absence of certain widely present BGCs in some species, thereby allowing the generation of evolutionary hypotheses correlating the production of a given SM with the lifestyle and the evolutionary history of the producer fungus.
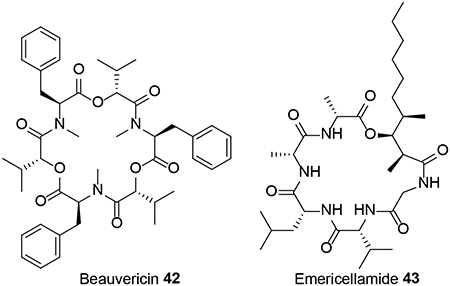
Sequencing and bioinformatic analysis of the whole genome of C. cicadae identified highly syntenic BGCs to those of the PK oosporein 1 and the cyclic oligodepsipeptide NRP beauvericin 42, both characterized earlier in B. bassiana.48 An orthologous and syntenic beauvericin BGC was also found in the genome sequence assembly of the HEF Cordyceps (formerly Isaria) fumosorosea.48 Production of oosporein in C. cicadae, and that of beauvericin in C. cicadae and C. fumosorosea was ascertained by HPLC analysis of fermentation extracts of these fungi.48 The genome sequence assembly of the HEF C. militaris ATCC 34164 revealed a BGC anchored by a PKS-NRPS that contains orthologous genes to that of fumosorinone 15 from C. fumosorosea and thus should yield a 1,4-dihydroxy-2-pyridone alkaloid.68 This C. militaris strain was also predicted to produce an acyl-cyclohexadepsipeptide as suggested by the presence of a BGC with genes for a PKS, a NRPS, an acyl-CoA ligase, and an acyltransferase that are all similar to those involved in the biosynthesis of emericellamide 43 in A. nidulans. Another C. militaris BGC was speculated to yield an equisetin-related SM based on partial similarities with the corresponding BGC from Fusarium heterosporum.68 None of these three predicted products have yet been isolated from the fungus, in spite of its widespread use as a nutraceutical TCM product.
Sbaraini et al. conducted an exhaustive survey of the M. anisopliae E6 genome sequence assembly to identify BGCs for SM production, and analyzed these clusters by comparing them to other Metarhizium spp. genomes and to those of other fungi.10 They also conducted a comparative transcriptomic study under standard growth conditions vs. growth on insect cuticle mimicking the infection process. The analysis indicated that M. anisopliae BGCs are well conserved in other host-generalist Metarhizium spp., but only a minority of the M. anisopliae BGCs are also present in host-specialist Metarhizium strains. Their analysis also confirmed or suggested potential SM products for several of the orphan M. anisopliae BGCs. In a similar study with a somewhat broader focus, Donzelli and Krasnoff concentrated on the available nine Metarhizium genome sequence assemblies and grouped the core genes of Metarhizium BGCs into homogenous sets that could be compared to other Ascomycota genomes and to a curated set of fungal BGCs with known products.37 This analysis established 27 NRPS and 14 NRPS-like Metarhizium families/pathways (based on A domain orthology); 32 PKS groups (based on KS phylogeny); 8 PKS-NRPS and 2 NRPS-PKS hybrids (either KS or A domain similarity, as the two constituent parts of these proteins are monophyletic); and an assortment of more than 40 terpene biosynthetic gene families. Some of these groups are widely present within Metarhizium, others appear only in host generalist or host specialist Metarhizium genomes; some are unique to the Metarhizium lineage while others have orthologues in distantly related fungal groups. This analysis37 which is congruent with that of Sbaraini et al.10 also provided interesting hints towards the nature of the potential products of some of these core gene families and their respective BGCs (See Supplementary Information Table S1 and Figure S1 for a detailed summary of the combination of these two analyses and some newer work10,37,85,78,76).
4. HEF genomes as a growing resource for SM discovery
The rapidly increasing number of HEF genomes in publicly available sequence databases provides an opportunity to use genome sequence data to guide the discovery of new chemical entities through the identification of BGCs of interest. To map out the extent of the encoded parvome of HEF, we surveyed the BGCs encoded in the 40 HEF genomes sequenced and deposited in GenBank until December 2019. 31 of these genome sequences were published after 2014, including eight species from Cordyceps, seven from Ophiocordyceps, five from Metarhizium, two each from Beauveria, Trichothecium and Lecanicillium, and one each from Moelleriella, “Paecilomyces”, Tolypocladium, Torrubiella, and Hypocrella, respectively (Supplementary Information).10, 19, 48, 68, 86–93 The typical genome of a sequenced HEF ranges from 20 to 50 Mb in size and encodes 8,000–12,000 genes. Exceptions are found among the Ophiocordyceps spp. that feature large genomes but a relatively small number of genes. For example, O. sinensis has a genome of 112.1 Mb in size but encodes only 6,972 predicted proteins (Fig. 1).94 The GC content of these genomes ranges from 40 to 60%, with Ophiocordyceps sp. ‘camponoti-saundersi’ possessing the lowest GC content (40%) and Cordyceps sp. RAO-2017 the highest (60.1%).
Figure 1. Genome characteristics of HEF.
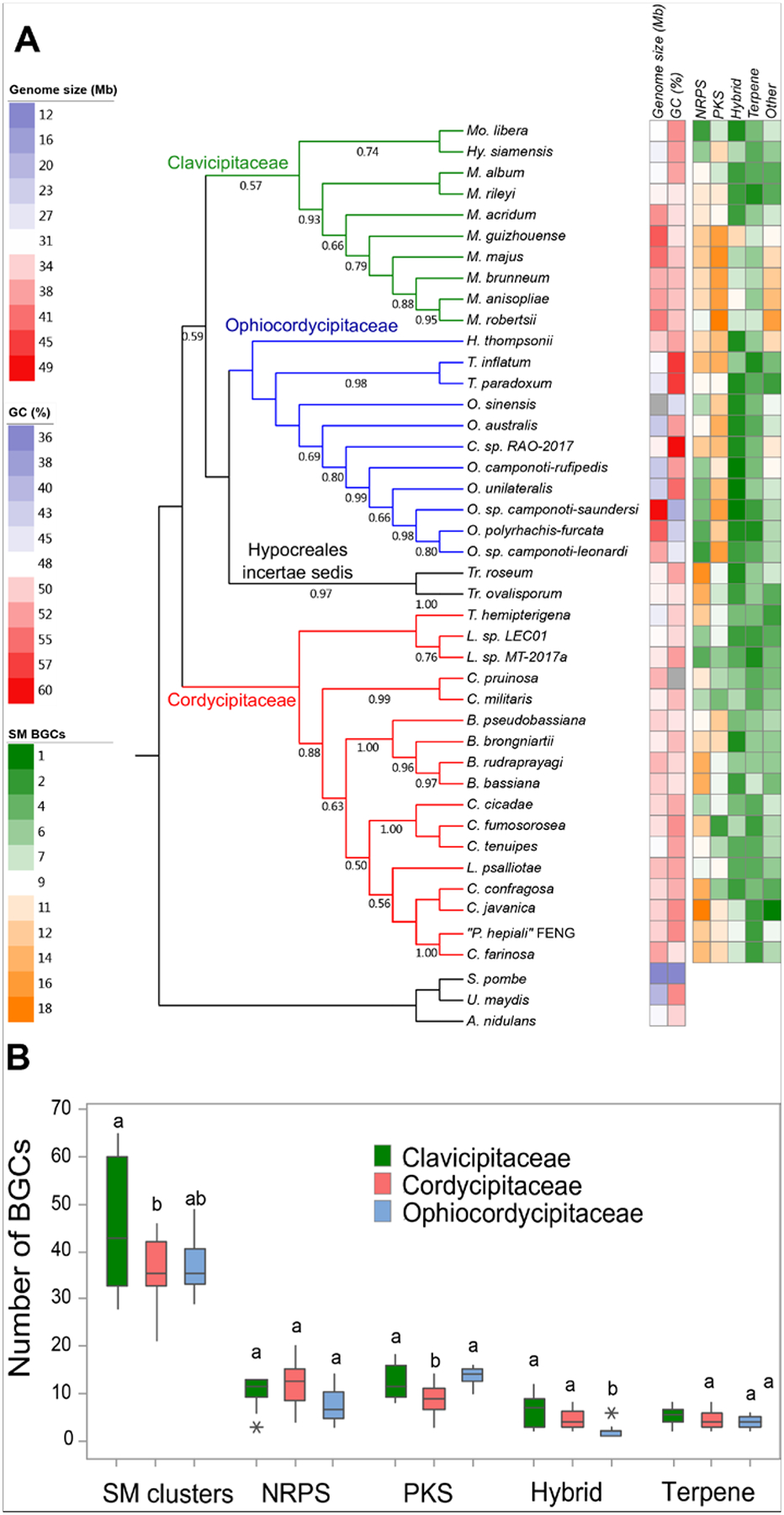
A) Maximum likelihood phylogram and genome statistics of HEF species. The phylogram was constructed from the DNA sequence of the 18S rRNA, the transcription elongation factor EF1α and the RNA polymerase Rpb1 in all genomes. The outgroup includes Aspergillus nidulans, Ustilago maydis and Schizosaccharomyces pombe. Bootstrap support is shown from 1000 replicates. Genome size (Mb) and GC content (%) are from the NCBI database and represented by the first two heatmap columns. The size of the O. sinensis genome is an outlier (112.1 Mb) and is represented in the heatmap in black. The numbers of predicted BGCs in these genomes are shown in the remaining columns of the heatmap by BGC categories. Genomes considered for this analysis are listed in the Supplementary Information. B) Distribution of BGC classes in HEF families. The class ‘Hybrid’ contains BGCs with both PKS and NRPS enzymes (including PKS-like and/or NRPS-like enzymes). In the boxplots, the box indicates the IQR (inter-quartile range) Q1 to Q3, while the whiskers extend to the lowest/highest values that are within 1.5 IQR (inter-quartile range) of the Q1 or Q3 range, respectively. Different letters indicate significant differences among families, and stars represent the outliers.
Mining the 40 HEF genomes for BGCs using antiSMASH returned a total of 1,569 putative BGCs, corresponding to an average of 39.2 BGCs per species (median 36.0), with the highest number of 65 BGCs observed in M. guizhouense ARSEF 977 (Fig. 2). The most abundant class of BGCs contains a PKS as the core enzyme, with 448 in total and an average of 11.2 per species (median 11.0). This is followed by NRPS-based BGCs, with 415 in total and an average of 10.4 per species (median 11.0). Among the hybrid BGCs, the majority (181 of 228) contain PKS-NRPS or NRPS-PKS hybrid enzymes as their core.
Entomopathogenic fungi fall into three major families in the order Hypocreales: Cordycipitaceae, Clavicipitaceae and Ophiocordycipitaceae. Two of the fungi with published whole genome sequences, Trichothecium ovalisporum DAOM 186447 and Trichothecium roseum DAOM 195227 do not belong to these three families and remain unclassified as Hypocreales incertae sedis, thus we elected to exclude the genomes of these strains from our family-level analysis. Based on the currently available data, the sequenced Clavicipitaceae genomes feature more BGCs than those of the other two families of HEF (p = 0.012, Fig. 1B). This suggest that fungi belonging to Clavicipitaceae may be especially promising resources for SM discovery. However, the distribution of the different classes of SMs among the main HEF families seems to be not uniform either. Thus, both Ophiocordycipitaceae and Clavicipitaceae outnumber Cordycipitaceae in the number of BGCs with PKSs as their core enzymes (p = 0.000). However, this seems to be compensated in Cordycipitaceae by a tendency to have a larger number of BGCs with NRPSs as their core enzymes, although the difference is not statistically significant (p = 0.126, Fig. 1B). Although the total number of hybrid BGCs is relatively modest, fungi in Ophiocordycipitaceae seem to be a poor source of this type of clusters and by extension, hybrid SMs (p = 0.000, Fig. 1B). Genus-level analysis is premature at this point and should await the availability of more genome sequences. Nevertheless, the current dataset seems to suggest that Metarhizium spp. genomes encode a higher number of BGCs than other HEF (average 51.6; Fig. 1A), and contain the most BGCs for polyketide SMs.
If such trends for the differential distribution of SMs in different HEF lineages are confirmed upon sequencing an even larger number of whole genomes, then chemical ecologists and “genome miners” will face intriguing questions. For example, do such differences result from the expansion of specific gene families in certain evolutionary lineages, or are they the consequences of the contraction and loss of other gene families in certain groups? Even more speculatively, is the PK chemical space inherently better suited to the ecological needs of the Ophiocordycipitaceae and Clavicipitaceae but less suitable for those of the Cordycipitaceae? If so, then which aspects of the lifestyle and the biotic interactions of these families lead to such a tendency? While such questions are considered, can we use these emerging trends to focus SM drug discovery towards more “talented” subgroups of HEF? Even more intriguingly, can we use such putative correlations between the phylogenomics of SMs and their chemical ecological roles to identify subgroups of HEF especially promising for drug discovery for certain human disease areas?
5. Conclusions
The decreasing cost of whole genome sequencing and the consequent explosion in the number of high-quality fungal genome sequences in public databases have revolutionized the cloning and functional characterization of BGCs. PCR with guessmer oligonucleotides, heterologous hybridization, cosmid genome libraries and similar “old school” methods quickly gave way to a new workflow that entails: 1. Draft sequencing of the whole genome of the fungal strain that produces an interesting SM; 2. Bioinformatic prediction of all BGCs in the resulting genome sequence; 3. Comparison of the predicted BGCs with the retrobiosynthetic assessment of the structure of the target SM; 4. Comparative genome analysis with fungi that produce similar SMs and with fungi that are phylogenetically close but are not known to yield the target SM; and 5. Comparative analysis of transcriptomes under SM-producing vs. nonproducing conditions. Such genomics-based methods are now the primary means of finding and delimiting target BGCs. At the same time, the increasing access to genome sequences also allowed the discovery of a very large number of orphan BGCs without a priori knowledge of their products. These orphan BGCs from genome mining may be prioritized by detailed comparisons with functionally characterized BGCs aided by expert systems such as antiSMASH,95 SMURF96 and others. Next, one may select those BGCs that encode the expected biosynthetic genes for a certain SM group, or may focus on those that seem to encode novel enzyme variants or novel enzyme combinations. Improving methods to activate orphan and/or silent candidate BGCs identified by bioinformatics; the use of gene silencing and CRISPR for knocking down / knocking out genes in BGCs to study the biosynthetic steps; and heterologous expression of BGC functional units or entire clusters to produce SM congeners led to major advances in our understanding of the enzymology of chemical complexity generation in fungi, and also allowed the combinatorial biosynthesis of novel SM analogues. All these methods are increasingly applied to HEF, a fascinating group of fungi that has made the evolutionary leap from plant pathogenic, mycoparasitic and saprobiotic trophic modes to insect pathogenesis on several different occasions. HEF developed a variety of ways to gain entry into the insect; subdue their immune system; commandeer their bodies as a nutrient source and in some cases to help fungal dispersal; and defend the insect carcass from competing pathogens or opportunists. At the same time, many HEF retained the ability for symbiosis with plants, and an aptitude to flourish in the soil as a free-living saprobiont. Studying the encoded parvome of HEF will increase our understanding of the multifaceted roles that SMs undoubtedly play in all these biotic and abiotic interactions and will also reveal biologically active natural products that can be exploited for the discovery of human and veterinary drugs or crop protection agents.
Supplementary Material
6. Acknowledgements
Work in the authors’ laboratories are supported by National Transgenic Major Program (2019ZX08010-004 to Q.Y.), the National Key Research and Development Program of China (2018YFA0901800 to Y.X.); the National Natural Science Foundation of China (31870076 and 31570093 to Y.X., 31500079 to L.Z.); the USDA National Institute of Food and Agriculture Hatch project (ARZT-1361640-H12-224 to I.M.); the Higher Education Institutional Excellence Program of the Ministry of Human Capacities in Hungary (NKFIH-1150-6/2019 to I.M.); the U.S. National Institutes of Health (NIGMS 5R01GM114418 to I.M.); and the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP to Y.X. and L.Z.). The authors thank Dr. Richard A. Humber (USDA ARS, Emeritus) for helpful discussions on the taxonomy of “P. hepiali” FENG.
Footnotes
7. Conflicts of interest
I.M. has disclosed financial interests in Teva Pharmaceuticals Works Ltd., Hungary and the University of Debrecen (Hungary) which are unrelated to the subject of the research presented here. All other authors declare no conflicts of interest.
8. References
- 1.Molnár I, Gibson DM and Krasnoff SB, Nat. Prod. Rep, 2010, 27, 1241–1275. [DOI] [PubMed] [Google Scholar]
- 2.Gibson DM, Donzelli BGG, Krasnoff SB and Keyhani NO, Nat. Prod. Rep, 2014, 31, 1287–1305. [DOI] [PubMed] [Google Scholar]
- 3.Beemelmanns C, Guo HJ, Rischer M and Poulsen M, Beilstein J. Org. Chem, 2016, 12, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pedrini N, Fungal Biol, 2018, 122, 538–545. [DOI] [PubMed] [Google Scholar]
- 5.Olatunji OJ, Tang J, Tola A, Auberon F, Oluwaniyi O and Ouyang Z, Fitoterapia, 2018, 129, 293–316. [DOI] [PubMed] [Google Scholar]
- 6.Cimermancic P, Medema MH, Claesen J, Kurita K, Brown LCW, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, Birren BW, Takano E, Sali A, Linington RG and Fischbach MA, Cell, 2014, 158, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lyu H-N, Liu H-W, Keller NP and Yin W-B, Nat. Prod. Rep, 2020, 37, 6–16. [DOI] [PubMed] [Google Scholar]
- 8.Wang B, Kang Q, Lu Y, Bai L and Wang C, Proc. Natl. Acad. Sci. U.S.A, 2012, 109, 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiemann P, Guo CJ, Palmer JM, Sekonyela R, Wang CCC and Keller NP, Proc. Natl. Acad. Sci. U.S.A, 2013, 110, 17065–17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sbaraini N, Guedes RLM, Andreis FC, Junges A, de Morais GL, Vainstein MH, de Vasconcelos ATR and Schrank A, BMC Genomics, 2016, 17, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rokas A, Wisecaver JH and Lind AL, Nat. Rev. Microbiol, 2018, 16, 731–744. [DOI] [PubMed] [Google Scholar]
- 12.Donzelli BGG, Krasnoff SB, Sun-Moon Y, Churchill ACL and Gibson DM, Curr. Genet, 2012, 58, 105–116. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y, Orozco R, Wijeratne EMK, Gunatilaka AAL, Stock SP and Molnár I, Chem. Biol, 2008, 15, 898–907. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Orozco R, Wijeratne EMK, Espinosa-Artiles P, Gunatilaka AAL, Stock SP and Molnár I, Fungal Genet. Biol, 2009, 46, 353–364. [DOI] [PubMed] [Google Scholar]
- 15.Moon YS, Donzelli BGG, Krasnoff SB, McLane H, Griggs MH, Cooke P, Vandenberg JD, Gibson DM and Churchill ACL, Appl. Environ. Microbiol, 2008, 74, 4366–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng P, Shang Y, Cen K and Wang C, Proc. Natl. Acad. Sci. U.S.A, 2015, 112, 11365–11370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yakasai AA, Davison J, Wasil Z, Halo LM, Butts CP, Lazarus CM, Bailey AM, Simpson TJ and Cox RJ, J. Am. Chem. Soc, 2011, 133, 10990–10998. [DOI] [PubMed] [Google Scholar]
- 18.Heneghan MN, Yakasai AA, Halo LM, Song Z, Bailey AM, Simpson TJ, Cox RJ and Lazarus CM, Chembiochem, 2010, 11, 1508–1512. [DOI] [PubMed] [Google Scholar]
- 19.de Bekker C, Ohm RA, Loreto RG, Sebastian A, Albert I, Merrow M, Brachmann A and Hughes DP, BMC Genomics, 2015, 16, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin H, Lyu H, Zhou S, Yu J, Keller NP, Chen L and Yin W-B, Org. Biomol. Chem, 2018, 16, 4973–4976. [DOI] [PubMed] [Google Scholar]
- 21.Janevska S, Arndt B, Baumann L, Apken LH, Marques LMM, Humpf HU and Tudzynski B, Toxins (Basel), 2017, 9, E126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clevenger KD, Bok JW, Ye R, Miley GP, Verdan MH, Velk T, Chen C, Yang K, Robey MT, Gao P, Lamprecht M, Thomas PM, Islam MN, Palmer JM, Wu CC, Keller NP and Kelleher NL, Nat. Chem. Biol, 2017, 13, 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Netzker T, Fischer J, Weber J, Mattern DJ, König CC, Valiante V, Schroeckh V and Brakhage AA, Front. Microbiol, 2015, 6, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anyaogu DC and Mortensen UH, Front. Microbiol, 2015, 6, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu G, Zhou H, Zhang P, Wang X, Li W, Zhang W, Liu X, Liu H, Keller NP, An Z and Yin WB, Org. Lett, 2016, 18, 1832–1835. [DOI] [PubMed] [Google Scholar]
- 26.Keller NP, Nat. Rev. Microbiol, 2019, 17, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutledge PJ and Challis GL, Nat. Rev. Microbiol, 2015, 13, 509–523. [DOI] [PubMed] [Google Scholar]
- 28.Přichystal J, Schug KA, Lemr K, Novák J and Havlíček V, Anal. Chem, 2016, 88, 10338–10346. [DOI] [PubMed] [Google Scholar]
- 29.Gomes NGM, Pereira DM, Valentão P and Andrade PB, J. Pharmaceut. Biomed. Anal, 2018, 147, 234–249. [DOI] [PubMed] [Google Scholar]
- 30.Hautbergue T, Jamin EL, Debrauwer L, Puel O and Oswald IP, Nat. Prod. Rep, 2018, 35, 147–173. [DOI] [PubMed] [Google Scholar]
- 31.Covington BC, McLean JA and Bachmann BO, Nat. Prod. Rep, 2017, 34, 6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo Z, Ren H, Mousa JJ, Rangel DEN, Zhang Y, Bruner SD and Keyhani NO, Environ. Microbiol, 2017, 19, 788–802. [DOI] [PubMed] [Google Scholar]
- 33.Asai T, Yamamoto T and Oshima Y, Tetrahedron Lett, 2011, 52, 7042–7045. [Google Scholar]
- 34.Fan A, Mi W, Liu Z, Zeng G, Zhang P, Hu Y, Fang W and Yin WB, Org. Lett, 2017, 19, 1686–1689. [DOI] [PubMed] [Google Scholar]
- 35.Chung Y-M, El-Shazly M, Chuang D-W, Hwang T-L, Asai T, Oshima Y, Ashour ML, Wu Y-C and Chang F-R, J. Nat. Prod, 2013, 76, 1260–1266. [DOI] [PubMed] [Google Scholar]
- 36.Asai T, Chung YM, Sakurai H, Ozeki T, Chang FR, Wu YC, Yamashita K and Oshima Y, Tetrahedron, 2012, 68, 5817–5823. [Google Scholar]
- 37.Donzelli BGG and Krasnoff SB, in Genetics and Molecular Biology of Entomopathogenic Fungi, eds. Lovett B and StLeger RJ, Elsevier Academic Press Inc, San Diego, 2016, vol. 94, pp. 365–436. [Google Scholar]
- 38.Bischoff JF, Rehner SA and Humber RA, Mycologia, 2009, 101, 512–530. [DOI] [PubMed] [Google Scholar]
- 39.Rao Z, Cao L, Wu H, Qiu X, Liu G and Han R, Insects, 2019, 11, E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Y, Wang W, Wang L, Pang F, Guo L, Song L, Liu G and Feng C, Genome Announc, 2016, 4, e00606–00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inglis PW and Tigano MS, Genet. Mol. Biol, 2006, 29, 132–136. [Google Scholar]
- 42.Luangsa-ard JJ, Hywel-Jones NL and Samson RA, Mycologia, 2004, 96, 773–780. [DOI] [PubMed] [Google Scholar]
- 43.Luangsa-Ard JJ, Hywel-Jones NL, Manoch L and Samson RA, Mycol. Res, 2005, 109, 581–589. [DOI] [PubMed] [Google Scholar]
- 44.El Basyouni SH and Vining LC, Can. J. Biochem, 1966, 44, 557–565. [DOI] [PubMed] [Google Scholar]
- 45.Schroeckh V, Scherlach K, Nutzmann HW, Shelest E, Schmidt-Heck W, Schuemann J, Martin K, Hertweck C and Brakhage AA, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 14558–14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaehle C, Gressler M, Shelest E, Geib E, Hertweck C and Brock M, Chem. Biol, 2014, 21, 719–731. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen SH, Frandsen RJ, Nielsen KF, Lysoe E, Sondergaard TE, Wimmer R, Giese H and Sorensen JL, Fungal. Genet. Biol, 2014, 70, 24–31. [DOI] [PubMed] [Google Scholar]
- 48.Lu Y, Luo F, Cen K, Xiao G, Yin Y, Li C, Li Z, Zhan S, Zhang H and Wang C, BMC Genomics, 2017, 18, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang G, Liu Z, Lin R, Li E, Mao Z, Ling J, Yang Y, Yin WB and Xie B, PLoS Pathog, 2016, 12, e1005685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Momose I, Onodera T, Doi H, Adachi H, Iijima M, Yamazaki Y, Sawa R, Kubota Y, Igarashi M and Kawada M, J. Nat. Prod, 2019, 82, 1120–1127. [DOI] [PubMed] [Google Scholar]
- 51.Abe H, Kawada M, Sakashita C, Watanabe T and Shibasaki M, Tetrahedron, 2018, 74, 5129–5137. [Google Scholar]
- 52.Kawada M, Inoue H, Ohba SI, Masuda T, Momose I and Ikeda D, Int. J. Cancer, 2010, 126, 810–818. [DOI] [PubMed] [Google Scholar]
- 53.Ishiyama A, Otoguro K, Iwatsuki M, Namatame M, Nishihara A, Nonaka K, Kinoshita Y, Takahashi Y, Masuma R, Shiomi K, Yamada H and Ōmura S, Journal of Antibiotics, 2009, 62, 303–308. [DOI] [PubMed] [Google Scholar]
- 54.Muroi M, Suehara K, Wakusawa H, Suzuki K, Sato T, Nishimura T, Otake N and Takatsuki A, Journal of Antibiotics, 1996, 49, 1119–1126. [DOI] [PubMed] [Google Scholar]
- 55.Ishidoh K, Kinoshita H and Nihira T, Appl. Microbiol. Biotechnol, 2014, 98, 7501–7510. [DOI] [PubMed] [Google Scholar]
- 56.Bushley KE, Raja R, Jaiswal P, Cumbie JS, Nonogaki M, Boyd AE, Owensby CA, Knaus BJ, Elser J, Miller D, Di Y, McPhail KL and Spatafora JW, PLoS Genet, 2013, 9, e1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang X, Feng P, Yin Y, Bushley K, Spatafora JW and Wang C, MBio, 2018, 9, e01211–01218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lawen A and Zocher R, J. Biol. Chem, 1990, 265, 11355–11360. [PubMed] [Google Scholar]
- 59.Weber G and Leitner E, Curr. Genet, 1994, 26, 461–467. [DOI] [PubMed] [Google Scholar]
- 60.di Salvo ML, Florio R, Paiardini A, Vivoli M, D’Aguanno S and Contestabile R, Arch. Biochem. Biophys, 2013, 529, 55–65. [DOI] [PubMed] [Google Scholar]
- 61.Hoffmann K, Schneider-Scherzer E, Kleinkauf H and Zocher R, J. Biol. Chem, 1994, 269, 12710–12714. [PubMed] [Google Scholar]
- 62.Sanglier JJ, Traber R, Buck RH, Hofmann H and Kobel H, Journal of Antibiotics, 1990, 43, 707–714. [DOI] [PubMed] [Google Scholar]
- 63.Offenzeller M, Santer G, Totschnig K, Su Z, Moser H, Traber R and Schneider-Scherzer E, Biochem, 1996, 35, 8401–8412. [DOI] [PubMed] [Google Scholar]
- 64.Donzelli BGG, Gibson DM and Krasnoff SB, Fungal Genet. Biol, 2015, 82, 56–68. [DOI] [PubMed] [Google Scholar]
- 65.Liu L, Zhang J, Chen C, Teng J, Wang C and Luo D, Fungal Genet. Biol, 2015, 81, 191–200. [DOI] [PubMed] [Google Scholar]
- 66.Heneghan MN, Yakasai AA, Williams K, Kadir KA, Wasil Z, Bakeer W, Fisch KM, Bailey AM, Simpson TJ, Cox RJ and Lazarus CM, Chem. Sci, 2011, 2, 972–979. [Google Scholar]
- 67.Eley KL, Halo LM, Song Z, Powles H, Cox RJ, Bailey AM, Lazarus CM and Simpson TJ, Chembiochem, 2007, 8, 289–297. [DOI] [PubMed] [Google Scholar]
- 68.Kramer GJ and Nodwell JR, BMC Genomics, 2017, 18, 912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halo LM, Marshall JW, Yakasai AA, Song Z, Butts CP, Crump MP, Heneghan M, Bailey AM, Simpson TJ, Lazarus CM and Cox RJ, Chembiochem, 2008, 9, 585–594. [DOI] [PubMed] [Google Scholar]
- 70.Santos FM, Latorre AO, Hueza IM, Sanches DS, Lippi LL, Gardner DR and Spinosa HS, Phytomedicine, 2011, 18, 1096–1101. [DOI] [PubMed] [Google Scholar]
- 71.Cook D, Donzelli BGG, Creamer R, Baucom DL, Gardner DR, Pan J, Moore N, Krasnoff SB, Jaromczyk JW and Schardl CL, G3 (Bethesda), 2017, 7, 1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hino M, Nakayama O, Tsurumi Y, Adachi K, Shibata T, Terano H, Kohsaka M, Aoki H and Imanaka H, Journal of Antibiotics, 1985, 38, 926–935. [DOI] [PubMed] [Google Scholar]
- 73.He X, Zhang M, Guo YY, Mao XM, Chen XA and Li YQ, Org. Lett, 2018, 20, 6323–6326. [DOI] [PubMed] [Google Scholar]
- 74.Chiang YM, Szewczyk E, Davidson AD, Entwistle R, Keller NP, Wang CC and Oakley BR, Appl. Environ. Microbiol, 2010, 76, 2067–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee JC, Lobkovsky E, Pliam NB, Strobel G and Clardy J, J. Org. Chem, 1995, 60, 7076–7077. [Google Scholar]
- 76.Kato H, Tsunematsu Y, Yamamoto T, Namiki T, Kishimoto S, Noguchi H and Watanabe K, Journal of Antibiotics, 2016, 69, 561–566. [DOI] [PubMed] [Google Scholar]
- 77.Araki Y, Awakawa T, Matsuzaki M, Cho R, Matsuda Y, Hoshino S, Shinohara Y, Yamamoto M, Kido Y, Inaoka DK, Nagamune K, Ito K, Abe I and Kita K, Proc. Natl. Acad. Sci. U.S.A, 2019, 116, 8269–8274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berry D, Mace W, Rehner SA, Grage K, Dijkwel PP, Young CA and Scott B, Environ. Microbiol, 2019, 21, 928–939. [DOI] [PubMed] [Google Scholar]
- 79.Rowan DD, Dymock JJ and Brimble MA, J. Chem. Ecol, 1990, 16, 1683–1695. [DOI] [PubMed] [Google Scholar]
- 80.Tanaka A, Tapper BA, Popay A, Parker EJ and Scott B, Mol. Microbiol, 2005, 57, 1036–1050. [DOI] [PubMed] [Google Scholar]
- 81.Xia Y, Luo F, Shang Y, Chen P, Lu Y and Wang C, Cell Chem. Biol, 2017, 24, 1479–1489. [DOI] [PubMed] [Google Scholar]
- 82.Xiang L, Li Y, Zhu Y, Luo H, Li C, Xu X, Sun C, Song J, Shi L, He L, Sun W and Chen S, Genomics, 2014, 103, 154–159. [DOI] [PubMed] [Google Scholar]
- 83.Liu T, Liu Z, Yao X, Huang Y, Qu Q, Shi X, Zhang H and Shi X, R. Soc. Open Sci, 2018, 5, 181247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pang F, Wang L, Jin Y, Guo L, Song L, Liu G and Feng C, Genomics, 2018, 110, 162–170. [DOI] [PubMed] [Google Scholar]
- 85.St Leger RJ and Wang CS, Appl. Microbiol. Biotechnol, 2010, 85, 901–907. [DOI] [PubMed] [Google Scholar]
- 86.Shang Y, Xiao G, Zheng P, Cen K, Zhan S and Wang C, Genome Biol. Evol, 2016, 8, 1374–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Conlon BH, Mitchell J, de Beer ZW, Caroe C, Gilbert MT, Eilenberg J, Poulsen M and de Fine Licht HH, Data Brief, 2017, 11, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kobmoo N, Wichadakul D, Arnamnart N, De La Vega RCR, Luangsa-ard JJ and Giraud T, Mol. Ecol, 2018, 27, 3582–3598. [DOI] [PubMed] [Google Scholar]
- 89.de Bekker C, Ohm RA, Evans HC, Brachmann A and Hughes DP, Sci. Rep, 2017, 7, 12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wichadakul D, Kobmoo N, Ingsriswang S, Tangphatsornruang S, Chantasingh D, Luangsa-ard JJ and Eurwilaichitr L, BMC Genomics, 2015, 16, 881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agrawal Y, Narwani T and Subramanian S, BMC Genomics, 2016, 17, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quandt CA, Patterson W and Spatafora JW, Mycologia, 2018, 110, 104–117. [DOI] [PubMed] [Google Scholar]
- 93.Lin R, Zhang X, Xin B, Zou M, Gao Y, Qin F, Hu Q, Xie B and Cheng X, Appl. Microbiol. Biotechnol, 2019, 103, 7111–7128. [DOI] [PubMed] [Google Scholar]
- 94.Li Y, Hsiang T, Yang RH, Hu XD, Wang K, Wang WJ, Wang XL, Jiao L and Yao YJ, J. Microbiol. Methods, 2016, 128, 1–6. [DOI] [PubMed] [Google Scholar]
- 95.Blin K, Pascal Andreu V de los Santos ELC Del Carratore F, Lee SY, Medema MH and Weber T, Nucleic Acids Res, 2019, 47, D625–D630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH and Fedorova ND, Fungal Genet. Biol, 2010, 47, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.