

Abstract
Since the original description of pathogenic germline DICER1 variation underlying PPB, the spectrum of extrapulmonary neoplasms known to be associated with DICER1 has continued to expand and now includes tumors of the ovary, thyroid, kidney, eye and brain among other sites. This report documents our experience with another manifestation: a primitive sarcoma that resembles PPB and DICER1-associated sarcoma of the kidney. These tumors are distinguished by their unusual location in the peritoneal cavity, associated with visceral and/or parietal mesothelium. A total of seven cases were identified through pathology review in children presenting at a median age of 13 years (range 3 to 14 years). Primary sites of origin included the Fallopian tube (4 cases), serosal surface of the colon (one case), and pelvic sidewall (2 cases). One case had pathologic features of type I PPB, another type Ir (regressed) PPB and the remaining 5 had features of type II or III PPB with a mixed primitive sarcomatous pattern with or without cystic elements. All had a pathogenic DICER1 variation identified in germline and/or tumor DNA. PPB-like peritoneal tumors represent a newly described manifestation of DICER1 pathogenic variation whose pathologic features are also recapitulated in DICER1-related renal sarcoma, cervical embryonal rhabdomyosarcoma, and some Sertoli-Leydig cell tumors with heterologous elements. Tumors arising from the Fallopian tube or elsewhere in the abdomen/pelvis, especially those with heterogeneous rhabdomyosarcomatous and/or cartilaginous differentiation, should prompt consideration of germline and tumor DICER1 testing.
Keywords: Pleuropulmonary blastoma, sarcoma, DICER1, tumor predisposition, cancer genetics
DICER1 germline or somatic mutations predispose to a variety of neoplasms with characteristic pathologic features. Since the initial recognition of the connection between pleuropulmonary blastoma (PPB) and DICER1 in 2009, this neoplasm has been a pathognomonic manifestation of DICER1 mutations. (1–3)
Type I PPB may present as an isolated unilocular or multi-septated cystic lung lesion with a characteristic sub-epithelial layer of primitive malignant mesenchymal cells with or without rhabdomyoblastic and cartilaginous differentiation. Progression in type I PPB is recognized pathologically as the malignant mesenchymal cells expand the septa, eventually forming grossly visible solid nodules with residual cysts (termed type II PPB); complete overgrowth of the cysts is recognized as solid type III PPB. It is now appreciated that not all lung cysts in an individual with germline DICER1 variants progress. Some may regress or fail or progress, resulting in type Ir PPB which lacks a primitive small cell population. [18]
Additional extrapulmonary DICER1-related tumors have emerged, including cystic nephroma and anaplastic sarcoma of kidney, Sertoli-Leydig cell tumor and cervical embryonal rhabdomyosarcoma, ciliary body medulloepithelioma, nasal chondromesenchymal hamartoma, DICER1-related sacrococcygeal tumors (4), and several childhood brain tumors including pituitary blastoma, pineoblastoma and intracranial sarcoma. (5–15) Additional examples of these histogenetically unrelated neoplasms continue to be identified, including a multi-cystic hepatic lesion and poorly differentiated thyroid carcinoma in children. (16–18) In addition, a case of intra-abdominal sarcoma with DICER1 mutation was recently reported. (19)
Many DICER1-related cancers, including PPB, ovarian tumors and other sarcomas are most curable when found at early stage. (20, 21) Previous work has shown that identification of germline pathogenic variants or mosaicism may result in earlier identification of DICER1-related cancers. (12, 21) Testing and surveillance guidelines may result in improved outcomes for probands and relatives. (22)
The present study documents seven tumors with the histologic features of a primitive multi-patterned sarcoma but with primary site of origin in the peritoneal cavity. All had germline and/or tumor DICER1 mutations, a finding with relevance for the individual and family members. We propose a new histologic classification for these tumors with the goal of highlighting the unique molecular pathogenesis and implications for individual and family testing and surveillance.
Materials and Methods
Study subject and clinical data ascertainment:
Individuals in this report were identified when tumor samples were sent for pathology consultation (DAH or LPD). Individuals were then enrolled in the International PPB/DICER1 Registry. This study was approved by the Institutional Review Board at Children’s Minnesota, Children’s National Medical Center, Children’s Healthcare of Atlanta/Emory University and Washington University in St. Louis. In one case, informed consent for publication was obtained through a separate mechanism. Tumors meeting the case definition of intra-abdominal sarcoma with rhabdomyosarcomatous or cartilaginous features were submitted for DICER1 testing. Germline DNA was tested when available. Pedigrees were reviewed. Medical records including operative, pathology and imaging reports and treatment data were reviewed. Follow-up data was requested annually or until death or loss to follow-up.
Molecular analyses:
DICER1 gene sequencing was performed on blood and/or saliva and tumor tissue using either Sanger sequencing or a next generation sequencing assay designed to detect base substitutions and small insertions/deletions in both coding and intron/exon flanking regions. (2, 5)
Results
Seven cases of intraperitoneal sarcoma were identified. These individuals presented at a median age of 13 years (range 3 to 14 years). The primary site of origin was determined to be Fallopian tube in 4 cases, including one with multiple other cystic lesions throughout the peritoneum. One appeared to arise from the serosal surface of the colon and two from the pelvic sidewall. One case had pathologic features resembling type I PPB and one was similar to type Ir PPB; the other 5 were virtually identical to type II or III PPB with a primitive mixed sarcomatous pattern with or without cystic elements. All six tumors with available DNA showed biallelic loss of function and RNase IIIb DICER1 mutations. Four of five individuals tested had germline DICER1 mutations. Six individuals received intensive, sarcoma-based therapy. The single individual who did not receive chemotherapy had a benign-appearing multi-cystic lesion of the Fallopian tube whose features were those of Ir PPB. Six of seven children are doing well without evidence of disease at median follow up of 67 months (range10 – 155 months); one child died from progressive disease despite multiple treatment regimens.
Case 1:
A 13-year-old girl with no significant medical history presented with severe, generalized abdominal pain. Computed tomography (CT) scan demonstrated a multi-loculated cystic mass in the pelvic cavity (Fig. 1). Intraoperatively, a hemorrhagic mass involving the left Fallopian tube was excised en bloc. Histologically, the tumor resembled a Type I PPB (Fig. 2A). She was treated with 4 cycles of vincristine, actinomycin D and cyclophosphamide (VAC) followed by 4 cycles of vincristine and actinomycin D (VA) and second look surgery; she remains disease free 30 months following original resection. Additionally, she was found to have Bethesda category II thyroid nodules; fine needle aspiration/biopsy of the thyroid nodule was negative for malignant cells.
Fig. 1:
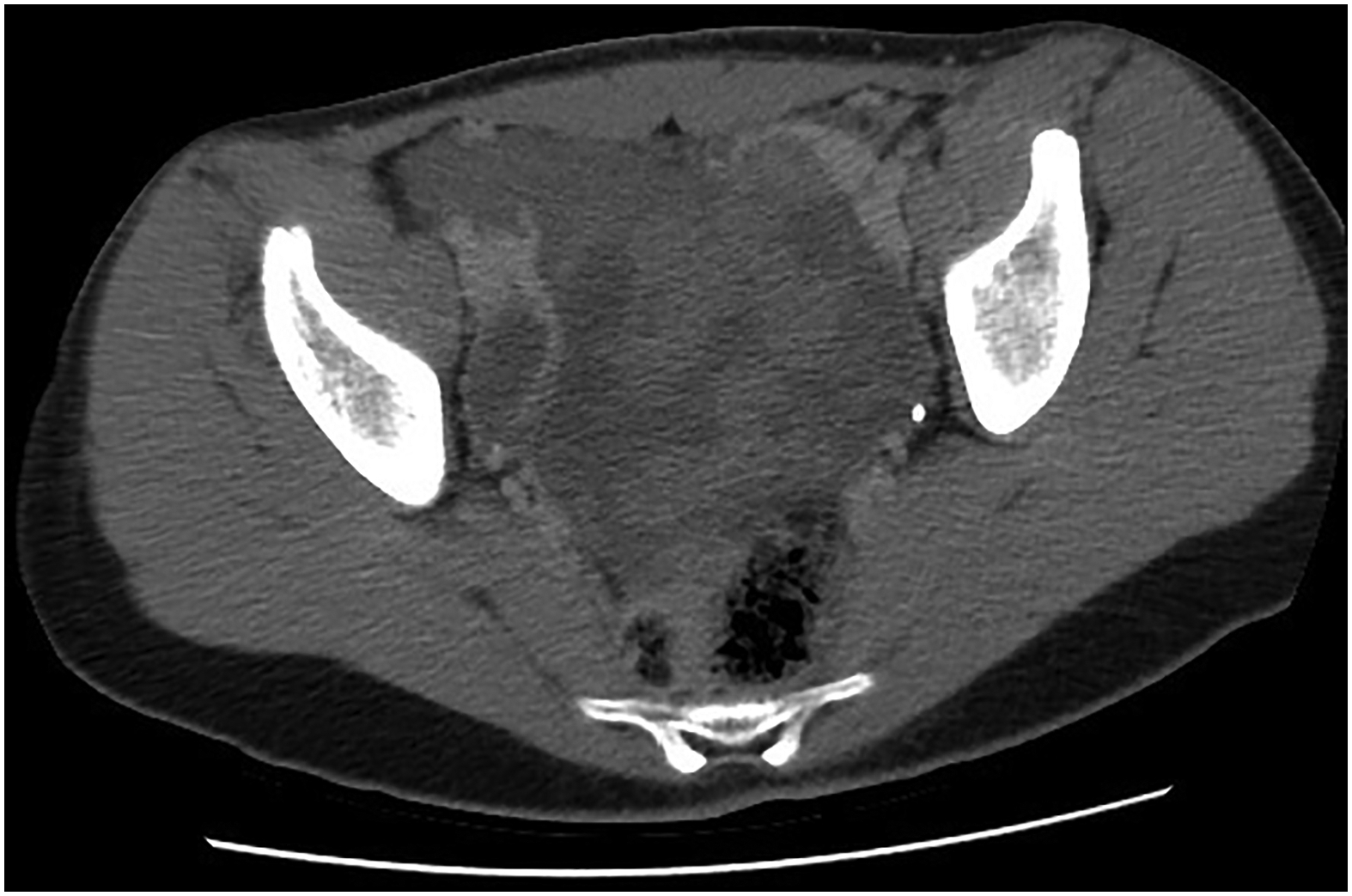
Case 1 - Axial (A) computed tomography images demonstrate a heterogeneous low attenuation intraperitoneal lesion within the pelvis with peripheral areas of decreased attenuation and areas with higher attenuation consistent with solid components, embedded bowel or a combination.
Fig. 2:
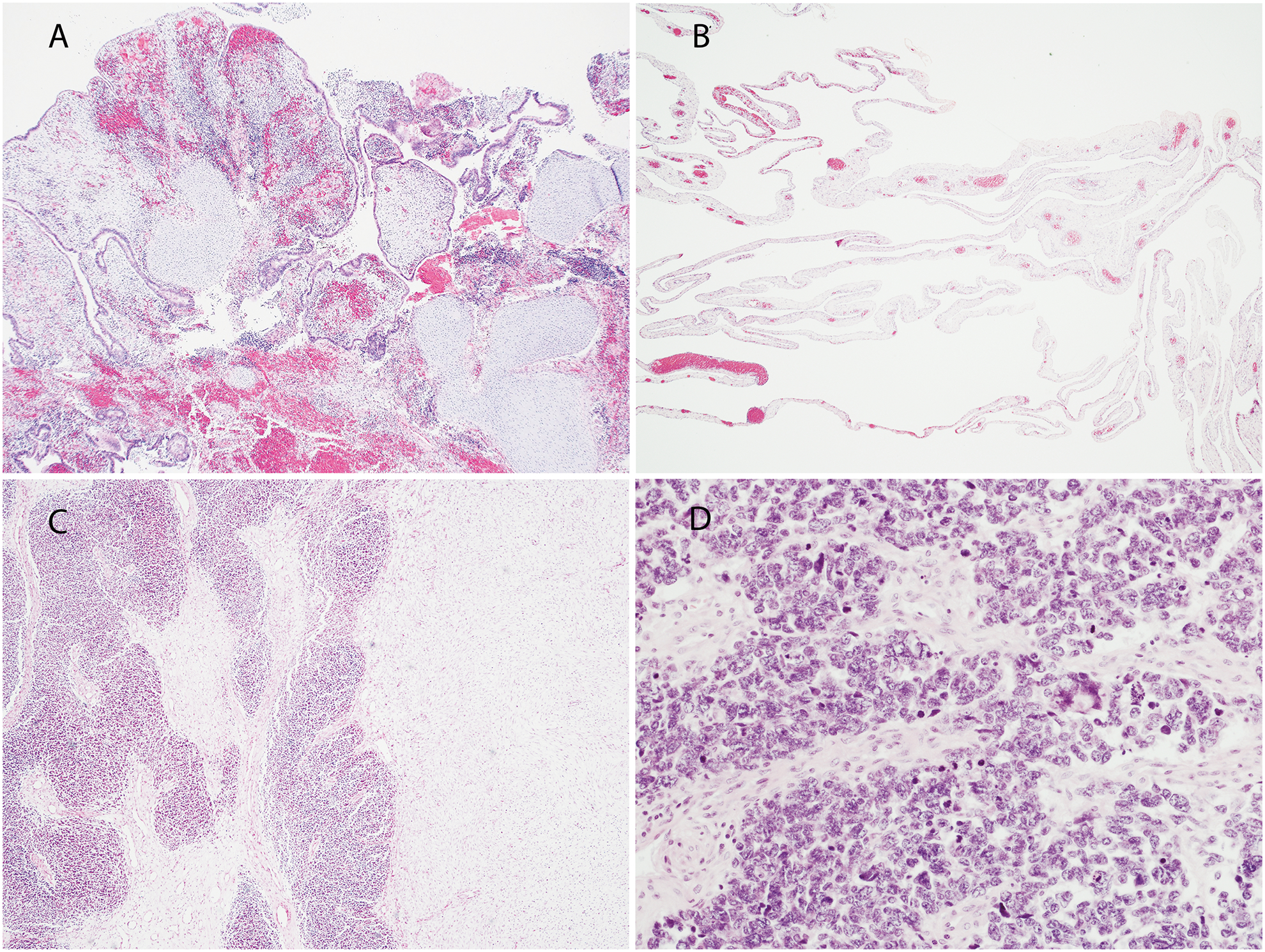
A Shows a cystic and solid mass arising in the Fallopian tube with histologic patterns of embryonal RMS with a botryoid growth pattern and nodules of cartilage (Case 1). B Shows multi-locular peritoneal cysts, without a primitive cellular component, resembling type Ir “regressed” PPB (Case 2). C Shows a low power view of blastemal nodules on left and embryonal RMS on the right (Case 6). D Shows nodules of primitive blastemal cells with anaplasia in a solid mass (Case 7).
Case 2:
A 13-year-old girl with history of Type II PPB at age 5, thyroid carcinoma at age 8, nasal chondromesenchymal hamartoma at age 13 and known germline DICER1 pathogenic variation presented with lower abdominal pain. Ultrasound demonstrated bilateral pelvic masses. Intraoperative examination showed multi-loculated cystic masses arising from the bilateral round ligaments with unilateral ovarian torsion. The cysts were lined by mesothelium. The septa were collagenous with scattered inflammatory cells. No primitive mesenchymal component was identified (Fig. 2B). No adjuvant therapy was administered. Subsequent to surgical excision of these peritoneal cysts, she presented with ovarian Sertoli-Leydig cell tumor. She is alive and well 5 years after the diagnosis of the peritoneal multi-locular cyst. (23)
Case 3:
A 13-year-old girl with history of Wilms tumor at age 5 (treated with chemotherapy and radiation therapy) presented with abdominal pain, urinary frequency and fevers. Ultrasound demonstrated an abdominal mass which intraoperatively appeared to be arising from the Fallopian tube. Histology of the solid portions of the abdominal mass showed a high grade sarcoma with pleomorphic and hyperchromatic nuclei. Histology of the cystic portions of the tumor showed areas with features of adenosarcoma. She initially was treated with 4 cycles of VAC followed by gross total resection, abdominal radiation and further VAC. She remains alive and well 7 years following resection.
Case 4:
A 14-year old previously healthy female presented with abdominal pain and urinary incontinence. Computed tomography scan showed a large mass arising from the right Fallopian tube with abdominal and pelvic implants (Fig. 3). The pathologic features included embryonal rhabdomyosarcoma with heterologous cartilage and anaplasia. Further testing of the tumor tissue confirmed biallelic pathogenic mutations in DICER1. She received chemotherapy with 2 cycles of VAC followed by therapy per Children’s Oncology Group protocol ARST08P1 with irinotecan/temozolomide and vincristine, doxorubicin and cyclophosphamide alternating with ifosfamide and etoposide followed by abdominal radiation. She has no evidence of disease 10 months following diagnosis.
Fig. 3:
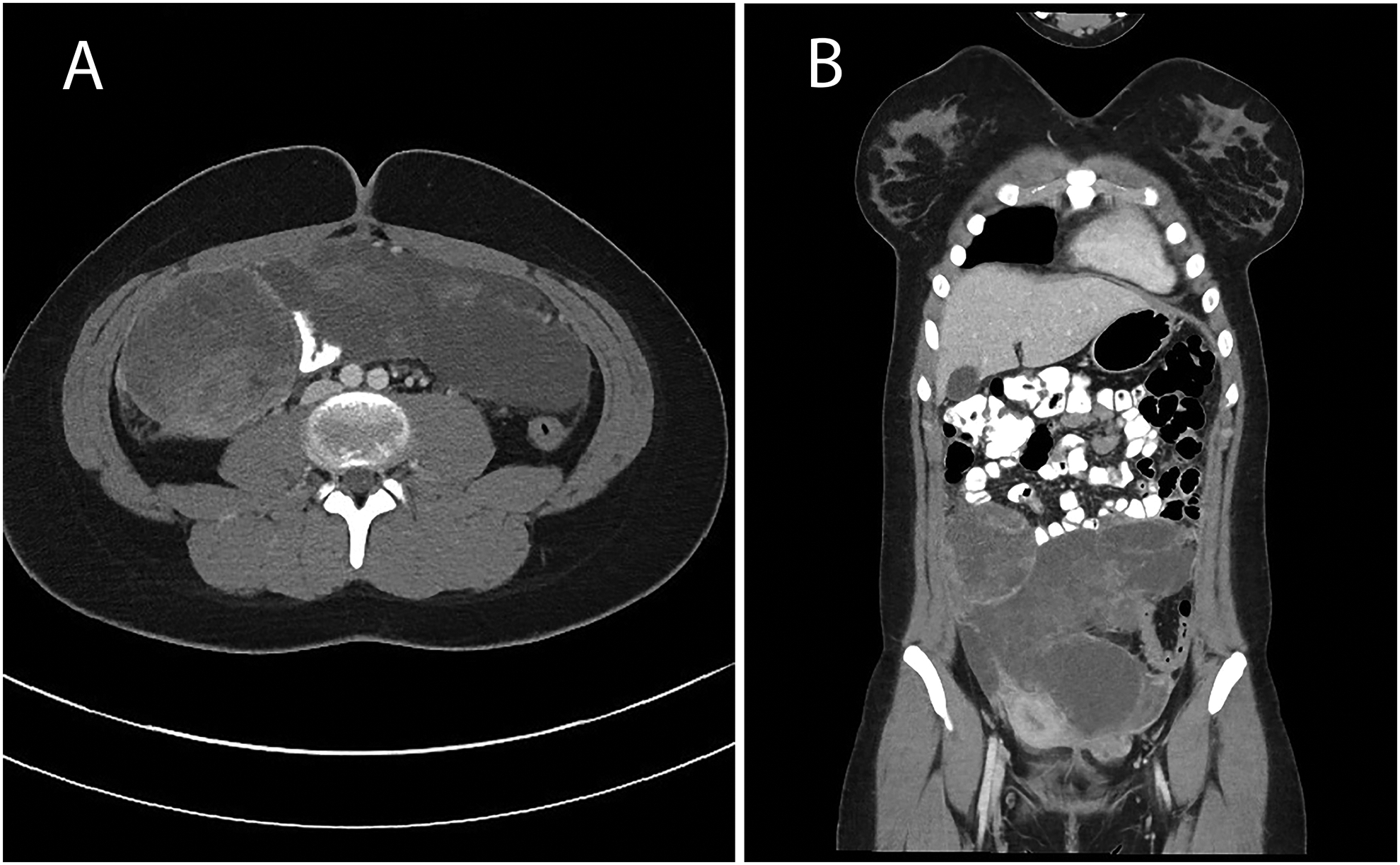
Case 4 - Axial (A) and coronal (B) computed tomography images demonstrate a heterogeneous low attenuation lobulated intraperitoneal mass with nonenhancing regions suggesting intratumoral necrosis extending from the level of the umbilicus to the lower pelvis in appearance to a type III PPB.
Case 5:
A 6-year-old girl was found to have a solid mass arising from the left pelvic sidewall. Pathology was consistent with embryonal rhabdomyosarcoma. She was treated with vincristine, doxorubicin and cyclophosphamide, alternating with ifosfamide and etoposide and pelvic radiation. Five years later, she presented with recurrence. Embryonal rhabdomyosarcoma with extensive cartilaginous differentiation and foci of pleomorphism were noted on microscopic examination. Therapy was initiated with temsirolimus, vinorelbine, and cyclophosphamide but stopped after one cycle due to cardiac toxicity. She then received 13 cycles of vincristine, oral irinotecan and cyclophosphamide with resection after cycle 6 and pelvic proton beam radiation during cycle 8 and 9. Surveillance imaging demonstrated evidence of a local recurrence with a 2 cm pelvic tumor within the radiation therapy field. Therapy with irinotecan/bevacizumab was initiated, resulting in complete remission after 6 cycles. During month 9 of chemotherapy, she was found to again have local recurrence and was started on dasatinib and ganitumab. After one month of treatment, therapy was withdrawn and she was placed on hospice. She died of disease 80 months after initial diagnosis.
Case 6:
A 3-year-old boy presented with abdominal pain. Computed tomography scan demonstrated a large intra-abdominal mass. Intraoperatively, a large mass arising from the serosal surface of the colon was identified. Pathology showed rhabdomyosarcoma with mixed pattern with predominance of embryonal histology but with notable solid blastemal components and brisk mitotic activity, extensive necrosis and anaplasia (Fig. 2C). Studies for typical translocations seen in alveolar rhabdomyosarcoma, synovial sarcoma, desmoplastic small round cell tumor and Ewing sarcoma were all negative. He underwent complete resection followed by intensive chemotherapy and high dose chemotherapy with autologous stem cell rescue. During treatment, a lung cyst was resected and identified as Type Ir PPB. At 12 years following initial diagnosis, routine surveillance ultrasound showed thyroid nodules. A total thyroidectomy was performed and the thyroid showed papillary carcinoma (follicular variant) limited to the thyroid gland. He remains well 12 years following initial diagnosis.
Case 7:
A 14-year-old girl presented with a solid abdominal mass arising from the left Fallopian tube. The resected specimen was interpreted as malignant mesenchymoma (Fig. 2D). Histologically, the tumor was described as a primitive malignant mesenchymal neoplasm with a multi-patterned appearance composed of areas of embryonal rhabdomyosarcoma, blastema, malignant cartilage, and spindle cell carcinoma. Areas of anaplasia characterized by large cells with large hyperchromatic nuclei and atypical mitotic figures were identified. She was treated with vincristine, doxorubicin and cyclophosphamide alternating with ifosfamide and etoposide followed by abdominal radiation. Biallelic loss of function and RNase IIIb (hotspot) mutations were identified in the tumor tissue. She has no evidence of disease more than 13 years following diagnosis.
Discussion
Shortly after the initial report of PPB as a distinct entity in 1989, familial aspects of this neoplasm began to emerge. (24) It has also been clear that pathologic manifestations are not restricted to the lung. Originally thought to be in the histogenetic lineage of Wilms tumor; cystic nephroma was identified in nearly 10% of kindreds with PPB. (25) In 2009, Hill and associates reported linkage of familial PPB with heterozygous germline mutations in DICER1. As additional families and individual patients underwent DICER1 germline and somatic testing of tumors, it was revealed that a phenotypically diverse spectrum of neoplasms arising in the central nervous system, thyroid, eye, ovary and uterine cervix were in fact genetically related. (26) The present report documents yet another example of this seemingly ubiquitous capacity for the development of this broad range of tumor types. There is, as observed in PPB, a progression from a simple multi-loculated cyst to a high grade multi-patterned primitive sarcoma.
It is tempting to offer a more generic diagnosis such as “DICER1-associated sarcoma.” However, we believe the terminology PPB-like peritoneal tumor/sarcoma is justified as it reflects the relevant histologic similarities between these two entities. Recognition of this pattern should prompt consideration of DICER1 pathogenic variation. This is important as although molecular testing is becoming standard in some settings, availability is still limited by resource constraints in other settings. Identification of DICER1 pathogenic variation impacts clinical care and individual and family surveillance.
PPB-like peritoneal sarcoma is the latest recognized manifestation of DICER1 pathogenic variation. The pathologic features of this new entity are very similar to those of types I through III PPB. Specifically, the range of histologic features includes a multi-loculated cyst without sarcomatous elements with a resemblance to type Ir PPB to a cystic/solid and/or purely solid multi-patterned sarcoma with rhabdomyosarcomatous and chondroid differentiation. This same pattern is seen in DICER1-related renal sarcoma, ovarian DICER1-related sarcomas, cervical embryonal rhabdomyosarcoma and Sertoli-Leydig cell tumor with heterologous elements especially rhabdomyosarcoma. These newly recognized tumors in the abdomen are distinguished, however, by their unusual origin in the peritoneal cavity.
Intriguingly, both PPB and PPB-like peritoneal sarcoma share the distinct cambium-like pattern of a mesenchymal proliferation beneath a cell layer – epithelium in the former and mesothelium in the latter. One mouse model showed that upregulation of fibroblast growth factor 9 (FGF9) in lung epithelium during early development results in pulmonary mesenchymal hyperplasia and multicystic growth, mimicking type I PPB. (27) FGF9 was found to be overexpressed both in Dicer1 knockout mice, as well as in human PPB tissue samples. Of particular interest is the localization of FGF9 to both the epithelium and the mesothelium in early lung development, suggesting that there could be mesothelial-mesenchymal signaling in these tumors analogous to the epithelial-mesenchymal signaling responsible for traditional pulmonary PPB tumorigenesis. (28)
Prior to re-evaluation and genetic testing, these tumors were diagnosed as malignant mesenchymoma, “sarcoma not otherwise specified” or embryonal rhabdomyosarcoma with cartilaginous features. It was thought that those neoplasms may be a type of malignant mixed Müllerian tumor (carcinosarcoma) or adenosarcoma, but without a carcinomatous component. Adenosarcoma and the extrauterine DICER1 peritoneal sarcoma share in common not only pathologic features but pathogenic hotspot DICER1 mutations. (29, 30) A recently reported case of DICER1-related abdominal sarcoma is likely within this same category. (19) In general, the differential diagnosis is malignant mixed Müllerian tumor (carcinosarcoma), which is typically seen in women 40 years of age and older. In younger girls and rarely boys, tumors arising in the abdomen/pelvis with heterogeneous rhabdomyosarcomatous and/or cartilaginous differentiation should prompt consideration of germline and tumor DICER1 testing.
Importantly, this tumor description, reflecting uncommon sites of origin, also broadens the differential diagnosis for an individual with a predisposing DICER1 variant who presents with a pelvic mass. Although Sertoli-Leydig cell tumor or gynandroblastoma are more common manifestations of an underlying DICER1 mutation (10), a mass arising from the Fallopian tube or elsewhere in the peritoneum cannot be assumed to be primary ovarian or metastatic disease. Likewise, some Fallopian tube cysts may represent a regressed tumor analogous to thoracic type Ir PPB, and their presence may be a clue to an underlying DICER1 mutation. In our series, this tumor occurred primarily in children and adolescents and more often in females than males. Nearly all individuals (with the exception of the individual with Fallopian tube cysts analogous to type Ir PPB) received intensive, sarcoma-based therapies and 6/7 are alive at time of this report.
The distinction between primary and metachronous or metastatic disease is critical in determining optimal therapy and prognosis. (12) Fortunately, in situations where the clinical picture is difficult to elucidate, for example when a new pelvic mass is identified in a young woman with an underlying DICER1 mutation and a remote history of ovarian neoplasm, genetic testing of the tumor samples, in particular sequencing of the “second hit” in the RNase IIIb domain, will generally differentiate between recurrent and metachronous disease. It should be noted, however, that in the rare instance of a predisposing mutation in the RNase IIIb domain, the loss of function mutation would instead vary between different tumors.
If a germline DICER1 mutation is identified, individual surveillance strategies and family testing are available to maximize the chance to find additional tumors in their earliest and most curable form. (31, 32) Additionally, as increased recognition and increased molecular testing lead to an increase in this diagnosis, it will be important to continue to collate and analyze clinical information including treatment and outcome data to determine optimal therapies for this unique tumor type.
Table 1.
Clinical characteristics of individuals with peritoneal PPB in this report.
| Case ID | Sex | Age at diagnosis (years) | Initial diagnosis | Site | Laterality | Analogous to PPB Type | DICER1: germline result (c.nomenclature, variant allele frequency) | DICER1: somatic result (c.nomenclature, variant allele frequency) | Treatment | Outcome | Associated Conditions | Overall Survival (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 13 | embryonal RMS with botryoid features | Fallopian tube | Left | I | Positive (c.1315_5316delTT 50.33%) | Positive c.1315_5316delTT 50.14%; c.5428G>T 25.3%) | Surgery > VAC / VA | NED | Thyroid nodule- 13y | 30 |
| 2 | F | 13 | Multifocal peritoneal cysts | Fallopian tube | Bilateral | Ir | Positive (ND) | ND | Surgery x2 | NED | PPB Type II– 5y; Thyroid Carcinoma– 8y SLCT and NCMH–13y; | 63 |
| 3 | F | 13 | adenosarcoma with rhabdomyosarcomatous overgrowth | Pelvic side wall | Left | II | Positive (ND) | Positive (ND) | Surgery > VAC > Surgery > VAC / XRT | NED | Wilms’– 6y Thyroid nodules- 14y | 67 |
| 4 | F | 14 | embryonal RMS | Fallopian tube Abdominal, omental and pelvic implant | Right | II / III | Negative | Positive (16 bp deletion in exon 19 splice site; c.5428G>C) | Surgery > VAC > ARST08P1>XRT | NED | None | 10 |
| 5 | F | 6 | embryonal RMS | Pelvic side wall | Left | III | ND | Positive (ND) | Surgery>VAdC / IE / XRT > recur > VOIT / XRT > recur > bevacizumab / irinotecan > dasatinib / ganitumab | DOD | Unknown | 80 |
| 6 | M | 3 | RMS | Serosal surface of colon | Not Specified | III | Positive (c.3540C>A; c.5425G>A) | Positive (c.3540C>A 44%; c.5425G>A 47.4%) | Surgery > intensive chemo> ASCR | NED | PPB Type Ir-3y; Papillary Thyroid Carcinoma-15y | 120 |
| 7 | F | 14 | Malignant mesenchymoma | Fallopian tube | Left | III | ND | Positive (c.5125G>A) | Surgery > VAdC/IE > XRT | NED | None | 155 |
Key: y=years; RMS = rhabdomyosarcoma; VAdC = vincristine/doxorubicin/cyclophosphamide; XRT = radiation therapy; VAC = vincristine/actinomycin D/cyclophosphamide; I = ifosfamide; E =etoposide; VOIT= vincristine/irinotecan/temozolamide; ARST08P1 = irinotecan/temozolamide and vincristine, doxorubicin and cyclophosphamide alternating with ifosfamide and etoposide; ASCR = autologous stem cell rescue; VA= vincristine/actinomycin D; NED= no evidence of disease; AWD= alive with disease
Acknowledgements:
The authors wish to thank the many treating physicians, genetic counselors, and patients and families who collaboratively support the International PPB/DICER1 Registry. The authors also wish to thank Children’s Minnesota, and the Pine Tree Apple Classic Fund, whose volunteers, players and donors have provided more than 30 years of continuous support of PPB research. This work was also funded by National Institutes of Health grant NCI R01CA143167 (Hill) and by the Division of Cancer Epidemiology and Genetics of the National Cancer Institute. The authors also wish to thank Gretchen M. Williams for her contributions to this manuscript and many years of thoughtful work on behalf of children with all types of PPB.
Footnotes
Conflict of Interest/Disclosure Statement: DRS provides telegenetics services for Genome Medical, Inc, in accordance with relevant National Cancer Institute policies. The other authors have no conflicts of interest to disclose. DAH is founder/investor in ResourcePath LLC.
References
- 1.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009;325(5943):965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pugh TJ, Yu W, Yang J, Ambrogio L, Carter SL, Kiezun A, et al. Progressive biallelic loss of TP53 is associated with progression of pleuropulmonary blastoma initiated by germline loss and somatic mutation of DICER1 [abstract]. Cancer Research 2013;73(8 Suppl):3806. [Google Scholar]
- 3.Brenneman M, Field A, Yang J, Williams G, Doros L, Rossi C, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma / DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000Res 2015;4:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakano Y, Hasegawa D, Stewart DR, Schultz KAP, Harris AK, Hirato J, et al. Presacral malignant teratoid neoplasm in association with pathogenic DICER1 variation. Mod Pathol 2019;32(12):1744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doros LA, Rossi CT, Yang J, Field A, Williams GM, Messinger Y, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol 2014;27(9):1267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz KA, Harris A, Messinger Y, Sencer S, Baldinger S, Dehner LP, et al. Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Fam Cancer 2016;15(1):105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rutter MM, Jha P, Schultz KA, Sheil A, Harris AK, Bauer AJ, et al. DICER1 Mutations and Differentiated Thyroid Carcinoma: Evidence of a Direct Association. J Clin Endocrinol Metab 2016;101(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Kock L, Priest JR, Foulkes WD, Alexandrescu S. An update on the central nervous system manifestations of DICER1 syndrome. Acta Neuropathol 2019. [DOI] [PubMed] [Google Scholar]
- 9.Stewart DR, Best AF, Williams GM, Harney LA, Carr AG, Harris AK, et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J Clin Oncol 2019;37(8):668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz KA, Pacheco MC, Yang J, Williams GM, Messinger Y, Hill DA, et al. Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol 2011;122(2):246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doros L, Schultz KA, Stewart DR, Bauer AJ, Williams G, Rossi CT, et al. DICER1-Related Disorders In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al. , editors. GeneReviews(R). Seattle (WA): University of Washington, Seattle University of Washington, Seattle; All rights reserved.; 2015. [Google Scholar]
- 12.Schultz KAP, Harris AK, Finch M, Dehner LP, Brown JB, Gershenson DM, et al. DICER1-related Sertoli-Leydig cell tumor and gynandroblastoma: Clinical and genetic findings from the International Ovarian and Testicular Stromal Tumor Registry. Gynecol Oncol 2017;147(3):521–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Priest JR, Williams GM, Manera R, Jenkinson H, Brundler MA, Davis S, et al. Ciliary body medulloepithelioma: four cases associated with pleuropulmonary blastoma--a report from the International Pleuropulmonary Blastoma Registry. Br J Ophthalmol 2011;95(7):1001–5. [DOI] [PubMed] [Google Scholar]
- 14.Stewart DR, Messinger Y, Williams GM, Yang J, Field A, Schultz KA, et al. Nasal chondromesenchymal hamartomas arise secondary to germline and somatic mutations of DICER1 in the pleuropulmonary blastoma tumor predisposition disorder. Hum Genet 2014;133(11):1443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dehner LP, Messinger YH, Schultz KA, Williams GM, Wikenheiser-Brokamp K, Hill DA. Pleuropulmonary Blastoma: Evolution of an Entity as an Entry into a Familial Tumor Predisposition Syndrome. Pediatr Dev Pathol 2015;18(6):504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apellaniz-Ruiz M, Segni M, Kettwig M, Glüer S, Pelletier D, Nguyen VH, et al. Mesenchymal Hamartoma of the Liver and DICER1 Syndrome. N Engl J Med 2019;380(19):1834–42. [DOI] [PubMed] [Google Scholar]
- 17.Vargas SO, Perez-Atayde AR. Mesenchymal Hamartoma of the Liver and DICER1 Syndrome. N Engl J Med 2019;381(6):586–7. [DOI] [PubMed] [Google Scholar]
- 18.Chernock RD, Rivera B, Borrelli N, Hill DA, Fahiminiya S, Shah T, et al. Poorly differentiated thyroid carcinoma of childhood and adolescence: a distinct entity characterized by DICER1 mutations. Mod Pathol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warren M, Hiemenz MC, Schmidt R, Shows J, Cotter J, Toll S, et al. Expanding the spectrum of dicer1-associated sarcomas. Mod Pathol 2020;33(1):164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witt RG, Baldini EH, Raut CP. Screening populations at high risk for soft tissue sarcoma and surveillance following soft tissue sarcoma resection. J Surg Oncol 2019;120(5):882–90. [DOI] [PubMed] [Google Scholar]
- 21.Messinger YH, Stewart DR, Priest JR, Williams GM, Harris AK, Schultz KA, et al. Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer 2015;121(2):276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz KA, Harris A, Williams GM, Baldinger S, Doros L, Valusek P, et al. Judicious DICER1 testing and surveillance imaging facilitates early diagnosis and cure of pleuropulmonary blastoma. Pediatr Blood Cancer 2014;61(9):1695–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz KA, Yang J, Doros L, Williams GM, Harris A, Stewart DR, et al. DICER1-pleuropulmonary blastoma familial tumor predisposition syndrome: a unique constellation of neoplastic conditions. Pathol Case Rev 2014;19(2):90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Priest JR, Watterson J, Strong L, Huff V, Woods WG, Byrd RL, et al. Pleuropulmonary blastoma: a marker for familial disease. J Pediatr 1996;128(2):220–4. [DOI] [PubMed] [Google Scholar]
- 25.Boman F, Hill DA, Williams GM, Chauvenet A, Fournet JC, Soglio DB, et al. Familial association of pleuropulmonary blastoma with cystic nephroma and other renal tumors: a report from the International Pleuropulmonary Blastoma Registry. J Pediatr 2006;149(6):850–4. [DOI] [PubMed] [Google Scholar]
- 26.Dehner LP, Schultz KA, Hill DA. Pleuropulmonary Blastoma: More Than a Lung Neoplasm of Childhood. Mo Med 2019;116(3):206–10. [PMC free article] [PubMed] [Google Scholar]
- 27.Yin Y, Castro AM, Hoekstra M, Yan TJ, Kanakamedala AC, Dehner LP, et al. Fibroblast Growth Factor 9 Regulation by MicroRNAs Controls Lung Development and Links DICER1 Loss to the Pathogenesis of Pleuropulmonary Blastoma. PLoS Genet 2015;11(5):e1005242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development 2001;128(11):2095–106. [DOI] [PubMed] [Google Scholar]
- 29.Bean GR, Anderson J, Sangoi AR, Krings G, Garg K. DICER1 mutations are frequent in müllerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol 2019;32(2):280–9. [DOI] [PubMed] [Google Scholar]
- 30.de Kock L, Yoon JY, Apellaniz-Ruiz M, Pelletier D, McCluggage WG, Stewart CJR, et al. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol 2020. [DOI] [PubMed] [Google Scholar]
- 31.Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res 2018;24(10):2251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Engelen K, Villani A, Wasserman JD, Aronoff L, Greer MC, Tijerin Bueno M, et al. DICER1 syndrome: Approach to testing and management at a large pediatric tertiary care center. Pediatr Blood Cancer 2018;65(1). [DOI] [PubMed] [Google Scholar]