

Abstract
Histone H3 arginine 2 (H3R2) is post-translationally modified in three different states by “writers” of the protein arginine methyltransferase (PRMT) family. H3R2 methylarginine isoforms include PRMT5-catalyzed monomethylation (me1) and symmetric dimethylation (me2s), and PRMT6-catalyzed me1 and asymmetric dimethylation (me2a). WD-40 repeat-containing protein 5 (WDR5) is an epigenetic “reader” protein that interacts with H3R2. Previous studies suggested that H3R2me2s specified a high-affinity interaction with WDR5. However, our prior biological data prompted the hypothesis that WDR5 may also interact with H3R2me1. Here, using highly accurate quantitative binding analysis combined with high-resolution crystal structures of WDR5 in complex with unmodified (me0) and me1/me2s L-Arginine amino acids and in complex with H3R2me1 peptide, we provide a rigorous biochemical study and address long-standing discrepancies of this important biological interaction. Despite modest structural differences at the binding interface, our study supports an interaction model regulated by a binary arginine methylation switch: H3R2me2a prevents interaction with WDR5, whereas H3R2me0/me1/me2s are equally permissive.
Keywords: WDR5, histone H3, arginine methylation, chromatin, isothermal titration calorimetry, crystal structure
Introduction
Arginine methylation (Rme) is a prevalent histone post-translational modification (PTM) that influences the ability of epigenetic “reader” proteins to engage with chromatin 1. By members of the protein arginine methyltransferase (PRMT) family, arginine can be monomethylated (Rme1, also MMA), asymmetrically dimethylated (Rme2a, also aDMA), or symmetrically dimethylated (Rme2s, also sDMA) 2. All PRMTs can generate Rme1; Type I PRMTs (isozymes 1–4,6,8) further catalyze Rme2a; Type II PRMTs (isozymes 5 and 9) further catalyze Rme2s; the sole Type III PRMT7 catalyzes only Rme1 3. While each methylation event does not influence the arginine sidechain positive charge, it does reduce hydrogen bond (H-bond) capacity and alters the electronic distribution of the guanidino group by withdrawing electrons through hyperconjugation, thereby increasing hydrophobicity (Figure 1a) 4.
Figure 1.

Overview of arginine methylation and WDR5. a. Guanidino group methylarginine isoforms which occur in vertebrates are depicted and annotated with their respective PRMT writer enzymes. Modeled surface areas and predicted guanidino group pKa values are shown (Maestro Schrödinger software) b. Surface representation of WDR5, depicting the WIN site (green), WBM site (orange), and the amino (blue) and carboxy (red) termini; characterized WIN motifs (underlined) are aligned c. Purified recombinant WDR5 proteins used in this study: full-length (FL) for binding assays; 1–20aa amino terminal truncation (ΔN20) for structural studies.
Although Rme has long been considered an irreversible PTM, emerging evidence suggests that a subset of lysine demethylase Jumonji-domain containing proteins catalyze arginine demethylation in vitro 5, suggesting that arginine methylation is reversible in vivo, where it is utilized to facilitate or disrupt protein-protein and protein-nucleic acid interactions that regulate diverse cellular processes 6. Histone arginine methylation is perhaps the most extensively studied methylarginine PTM for its role–in combination with other histone PTMs (e.g. lysine acetylation/methylation, serine/threonine phosphorylation)–in tuning chromatin state 6.
WD-40 repeat-containing protein 5 (WDR5) is a ubiquitously expressed and promiscuous adaptor protein that interacts with histone H3; it is also present in numerous chromatin-modifying complexes, including histone acetylases, deacetylases, and methylases 7. WDR5 is a globular protein with a narrow channel extending through the center of its seven WD-repeat β-propeller fold. Binding partners interact with WDR5 via two distinct and non-overlapping sites on opposite faces of the protein: the WDR5-interaction (WIN) site and the WDR5 binding motif (WBM) site. Proteins that have been structurally characterized to interact with the WIN site contain a WIN motif: a four amino acid long polypeptide with consensus sequence A-R-[A/C/S/T]-[E/K/R] (Figure 1b, adapted from 7). The first four residues of histone H3 (A-R-T-K) comprise a WIN motif, and it is the only one known to be post-translationally modified with arginine methylation (PRMT6: me1, me2a 8; PRMT5: me1, me2s 9). Intriguingly, the sequence surrounding H3R8 (A-R-K-S) closely resembles and is hypothesized to constitute a second WIN motif on the H3 tail 10–12 that is also modified with arginine methylation (PRMT2: me1, me2a 13; PRMT5: me1, me2s 14).
Previously published structural studies revealed that WDR5 binds both unmodified H3R2 (H3R2me0) 11, 15–17 and H3R2me2s 18 peptides. It was previously reported that WDR5 is a high-affinity reader of H3R2me2s (Kd~100nM) 18 and, conversely, that H3R2me2a does not interact with WDR5 18–20. As both H3R2me0 and H3R2me2s interact with WDR5, it follows that H3R2me1 can also be accommodated. However, in our search of the literature, we found only one study characterizing interaction with H3R2me1 that reported very weak affinity for WDR5 (Kd >500μM), which essentially excludes any biologically relevant interaction 16. Although Rme1 has long been regarded as a non-functional transient species in the ‘obligatory’ pathways to either Rme2a or Rme2s 21, 22, numerous proteomic analyses demonstrate that Rme1 is an abundant PTM, suggesting it is functionally significant in the cell 23–26. Furthermore, our previous biological data led to a model that WDR5 is in fact a reader of H3R2me1 9.
To fully elucidate the biochemical details of this important biological relationship, the conflicting results in both biological and quantitative binding studies suggest that a more rigorous assessment of the WDR5/H3 interaction is warranted. Here, we present a complete interrogation of the H3/WDR5 interaction. Using extremely pure H3 peptides and highly accurate and precise peptide concentrations, as measured by 1D 1H nuclear magnetic resonance (NMR), we performed isothermal titration calorimetry (ITC) to accurately measure the thermodynamic binding properties of the H3/WDR5 interaction. We show that full-length WDR5 (Figure 1c) specifically interacts with H3R2 and does not interact with H3R8. Furthermore, we provide unambiguous evidence that WDR5 indeed interacts with H3R2me1 and that it has comparable affinity for H3R2me0, H3R2me1, and H3R2me2s; additionally, we confirm no WDR5 interaction with H3R2me2a peptides. To assess structural differences at the WIN site, we solved high-resolution crystal structures of ΔN20-WDR5 (Figure 1c) in complex with H3R2me1 peptide and in complex with methylated isoforms of L-Arginine amino acid. We propose that the WDR5/H3R2 interaction is regulated by a binary methylation switch mechanism, such that H3R2me0/me1/me2s are equally permissive for, whereas H3R2me2a is excluded from, interaction with WDR5.
Materials and Methods
Expression and Purification of WDR5
Plasmids encoding human full-length WDR5 (2GNQ, Addgene) or a 20aa N-terminal truncation (ΔN20, prerequisite for crystallization; cloned into pRUTH5 expression vector)—both fused to an N-terminal 6x-Histidine affinity tag and Tobacco Etch Virus (tev) protease site—were transformed into E. coli BL21(DE3)pLysS. Cultures were grown in LB medium containing appropriate antibiotics at 37°C to OD600 ~0.7 then induced with 1mM IPTG and incubated for 4 hours at 37°C. Per 1L worth of pelleted cells, lysis was performed by thorough resuspension in 35mL lysis/wash (LW) buffer (50mM Tris-HCl pH 8.0, 1M NaCl, 5mM β-mercaptoethanol, 5mM imidazole, 1mM PMSF). BugBuster (Millipore) was added to 1X and incubated at 25°C for 15min. This was followed by a sonication gradient: 2×30sec pulses at 35% and 40% amplitude and 2×10sec pulses at 45% and 50% amplitude with at least 30sec rest periods between pulses. Insoluble material was pelleted by centrifugation at 14,000rpm for 45min at 4°C. The soluble fraction was incubated with 1.25mL Ni-NTA affinity resin (Thermo) for 1hr with at 4°C. Ni-NTA gravity-flow columns were packed in 20mL disposable chromatography columns (BioRad) and washed with 50mL of buffer LW, 25mL LW+15mM imidazole, and then eluted with 5mL LW+360mM imidazole. Elution fractions containing 6xHis(tev)WDR5 were pooled, and 6x-Histidine-Tobacco Etch Virus protease catalytic domain was added at a 1:50 mass ratio (TEV:WDR5). TEV-cleavage was performed simultaneously with dialysis against 1L TD-buffer (50mM Tris-HCl pH 8.0, 200mM NaCl, 5mM ß-mercaptoethanol) for 16 hours at 4°C. TEV protease and the 6xHis affinity tag were then removed by subtractive Ni-NTA chromatography. To eliminate remaining contaminants, anion exchange chromatography was utilized (MonoQ, GE). In TD-buffer at pH 8.0, contaminants interact with MonoQ resin, whereas WDR5 (pI=8.1) does not; WDR5 was thus collected in the “unbound” fraction and contaminants eluted with a one-step increase to TD-buffer containing 1M NaCl. WDR5 was then polished by size exclusion chromatography (Superdex 75 Increase, GE) in TD-buffer containing 1M NaCl. Fractions containing pure WDR5 were pooled and buffer exchanged into storage buffer (20mM Tris-Cl pH 8.0, 150mM NaCl, 1mM TCEP). Full length WDR5 was concentrated to 500μM; ΔN20-WDR5 was concentrated to 660μM for crystallization experiments. Aliquots were stored at −80°C.
Molecular Modeling and pKa Prediction of Methylarginine Isoforms
Maestro (Shrödinger) was used to model electrostatic surface potential maps and predict pKa’s of methylarginine isoforms. First, a G-R-G peptide was modeled using the “Build” feature in the software. At methylated positions of the arginine side chain, guanidino protons were changed to methyl groups using the “Set element” feature. Guanidino group atoms were then selected and electronic maps were generated using the “Poisson-Boltzmann ESP” task. Electrostatic potentials were displayed from −10 to 10 kBT/e (kB, Boltzmann constant; T, temperature (Kelvin); e, electron charge). Surface transparencies were set to 30% front and 10% back from the “Surface display options” menu. pKa values were predicted using Epik by selecting the “Emprical pKa” task.
Histone H3 Peptide Pulldown Assay
10μg of C-terminal biotinylated histone H3 1–21aa unmodified, H3R2me1, H3R2me2s, or H3R2me2a peptide (Anaspec) were immobilized on 20μL Streptavidin sepharose resin (Thermo) in 200μL pulldown buffer (50mM Tris-Cl pH 8.0, 150mM NaCl, 5mM β-mercaptoethanol). The peptide-bound resin was washed three times with 500μL pulldown buffer then incubated with 10μM WDR5 full-length protein in 200μL buffer for 1hr with end-over-end rotation at 4°C. The resin was washed 8 times with 500μL buffer and eluted with 20μL 2X Laemelli Buffer and heat denatured at 90°C for 5 minutes. 15μL of eluate was loaded on 15% SDS-PAGE and visualized by Coomassie stain.
Purification and Quantitative Analysis of Peptide Reagents
Histone H3 1–12aa unmodified, H3R2K, H3R8K, H3R2KR8K and H3 1–7aa unmodified, H3R2me1, H3R2me2s, and H3R2me2a peptides were obtained from GenScript USA. Extra care was exercised to ensure the highest purity and most accurate H3 peptide concentrations were obtained for quantitative binding measurements with WDR5. To this end, the peptides were again purified by high performance liquid chromatography (HPLC), using a 250 × 4.6mm 5μm Luna C18-(2) column (Phenomenex; #00G-4252-E0) and a gradient separation with water (0.1% TFA; Phase A) and 90% acetonitrile (0.1% TFA; Phase B). All H3 1–12aa peptides were separated using the same protocol: 0–10 min (1 mL/min, 100% A), 10–40 min (1 mL/min, linear ramp to 75% A), 40–44 min (linear ramp to 100% B and 2 mL/min) with hold until 49 min, 49–53 min (2 mL/min, linear ramp to 100% A) with hold until 60 min. The H3 1–7aa peptides were separated following different protocols. Protocol #1 for H3R2me1 and H3R2me2s: 0–10 min (1 mL/min, 100% A), 10–90 min (1 mL/min, linear ramp to 85% A), 90–94 min (linear ramp to 100% B and 2 mL/min) with hold until 99 min, 99–103 min (2 mL/min, linear ramp to 100% A) with hold until 110 min. Protocol #2 for H3R2me0 and H3R2me2a: 0–10 min (1 mL/min, 100% A), 10–40 min (1 mL/min, linear ramp to 95% A), 40–44 min (linear ramp to 100% B and 2 mL/min) with hold until 49 min, 49–53 min (2 mL/min, linear ramp to 100% A) with hold until 60 min. Combined fractions of purified peptides were lyophilized and concentrations were determined using 1H NMR using an adenosine internal standard (D2O, 600 MHz, δ ppm: anomeric proton, ~6.2, 1H, d, J=6.2 Hz)27.
Isothermal Titration Calorimetry
ITC experiments were performed using a PEAQ-ITC calorimeter (Malvern Panalytical). WDR5 aliquots were pooled and dialyzed against storage buffer. Lyophilized histone peptides and WDR5 were diluted into the filtered dialysate. WDR5 (100μM) was titrated with histone H3 peptide (1.5mM), deploying 19 injections (1×0.4μL, 18×2μL) at 25°C with a reference power of 2.5μcal/sec. Heat integration, baseline correction, and curve fitting were performed using the software accompanying the PEAQ-ITC. Stoichiometry was fixed at N=1 (one-site binding model), allowing for ΔH, Kd, and displacement to vary for initial fitting iterations; the final iterations were performed with all variables free for determination of best fit. The best fit data were then imported into Prism to plot Langmuir isotherms.
Crystallization of apo WDR5 and Liganded complexes
The crystallization of apo WDR5 was performed using sitting drop vapor diffusion method at 22˚C. WDR5 was screened around previously published crystallization conditions 18. The crystallization drops were set up in 96-well INTELLI plates (Art Robbins) using the Gryphon crystallization robot (Art Robbins). Each crystallization drop contained 0.5μL of [28mg/mL] WDR5 and 0.5μL of well solution (0.1M Bis-Tris, 56.4mM ammonium sulfate, 32.5% PEG-3350). The volume of the well solution was 70μL. Diffraction quality crystals were obtained within one week. All liganded WDR5 complexes (unmodified, me1, me2s, and histone H3R2me1 1–21aa peptide) were obtained using crystal soaking experiments. The apo crystals of WDR5 were incubated for 30min at 25°C in soaking/cryoprotection buffer (well solution plus 20% glycerol), containing 2mM of L-Arg species or H3R2me1 1–7aa peptide. The crystallization and crystal handling processes are summarized in Table S2.
Data collection and processing
The diffraction data were collected at LRL-CAT beam line (Argonne National Laboratory, Argonne, IL) at 0.97931Å wavelength. All diffraction data were processed using iMOSFLM 28 and scaled with the AIMLESS program of the CCP4 suite 29. Data quality was analyzed using SFCHECK and XTRIAGE 29, 30. Matthews coefficients (Vm) calculation were used to estimate the number of monomer molecules present the unit cells. The data collection and processing statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics of human WDR5.
| Dataseta | Human WDR5 | ||||
|---|---|---|---|---|---|
| WDR5-apo | WDR5-Rme0 | WDR5-Rme1 | WDR5-Rme2s | WDR5-H3R2me1 | |
| Unit cell data | |||||
| Space group | P21212 | P21212 | P21212 | P21212 | C2221 |
| Cell parameters (Å, °) | a = 81.57, b = 86.10, c = 39.96 α, β, γ = 90 |
a = 81.19, b = 86.70, c = 40.96 α, β, γ = 90 |
a = 81.07, b = 86.22, c = 40.76 α, β, γ = 90 |
a = 81.05, b = 86.29, c = 40.87 α, β, γ = 90 |
a = 78.29, b = 98.63, c = 80.26 α, β, γ = 90 |
| Vm (Å3/Dalton) | 2.1 | 2.1 | 2.1 | 2.1 | 2.3 |
| Number of subunits in the asymmetric unit | 1 | 1 | 1 | 1 | 1 |
| Data collection | |||||
| Beamline | LRL-CAT | LRL-CAT | LRL-CAT | LRL-CAT | LRL-CAT |
| Wavelength (Å) | 0.97931 | 0.97931 | 0.97931 | 0.97931 | 0.97931 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| Resolution range (Å) | 59.22 – 1.85 (1.89 – 1.85) |
59.26 – 1.92 (1.97 – 1.92) |
59.06 – 1.68 (1.71 – 1.68) |
59.08 – 1.68 (1.71 – 1.68) |
61.32 – 1.66 (1.69 – 1.66) |
| Total number of observed reflections | 313354 (19367) | 282012 (13838) | 240304 (12262) | 240032 (12417) | 263601 (13719) |
| Number of unique reflections | 24681 (1516) | 22592 (1272) | 33408 (1685) | 33411 (1701) | 37045 (1856) |
| Rmerge (%)b | 7.5 (92.2) | 15.9 (93.3) | 7.7 (93.4) | 6.9 (105.5) | 17.5 (85.2) |
| Rpim (%)c | 2.2 (26.4) | 4.8 (28.7) | 3.1 (37.2) | 2.8 (41.7) | 7.1 (33.5) |
| CC1/2 (%) | 0.99 (0.94) | 0.99 (0.75) | 0.99 (0.88) | 0.99 (0.89) | 0.99 (0.80) |
| < I/σ(I)>d | 13.2 (2.0) | 10.7 (3.1) | 14.5 (2.0) | 15.7 (1.9) | 8.2 (2.0) |
| Completeness (%) | 99.6 (99.8) | 99.0 (84.7) | 100 (99.9) | 99.8 (99.9) | 100 (100) |
| Multiplicity | 12.7 (12.8) | 12.5 (10.9) | 7.2 (7.3) | 7.2 (7.3) | 7.1 (7.4) |
| Wilson B-factor (Å2) | 37.9 | 21.1 | 23.5 | 22.7 | 13.6 |
| Refinement | |||||
| Rwork (%)e | 21.0 | 16.0 | 19.7 | 18.1 | 17.2 |
| Rfree (%)f | 25.0 | 20.1 | 23.6 | 22.3 | 20.2 |
| No. of atoms | 2458 | 2629 | 2495 | 2591 | 2766 |
| Protein atoms | 2346 | 2339 | 2339 | 2339 | 2371 |
| Ligand atoms | - | 12 | 13 | 14 | 35 |
| Solvent atoms | 112 | 278 | 143 | 238 | 360 |
| Model quality | |||||
| RMS deviation from ideal value | |||||
| Bond length (Å) | 0.006 | 0.010 | 0.010 | 0.009 | 0.009 |
| Bond angle (°) | 1.17 | 1.45 | 1.45 | 1.43 | 1.45 |
| Average B-factor | |||||
| Protein atoms (Å2) | 45.0 | 25.6 | 24.2 | 26.1 | 13.9 |
| Ligand atoms (Å2) | - | 37.1 | 26.4 | 35.4 | 28.5 |
| Waters (Å2) | 46.4 | 52.0 | 39.2 | 52.1 | 42.9 |
| Ramachandran plotg | |||||
| Most favored regions (%) | 95.4 | 95.7 | 95.4 | 95.7 | 95.4 |
| Allowed regions (%) | 4.6 | 4.3 | 4.6 | 4.3 | 4.6 |
| Outlier regions (%) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| PDB ID entry | 6OFZ | 6OI0 | 6OI1 | 6OI2 | 6OI3 |
Values in parentheses refer to the highest resolution shell.
Rmerge= (ΣhklΣi|Ii(hkl) - < I(hkl)>|)/ΣhklΣi<Ii(hkl)>, where Ii(hkl) is the intensity of the ith measurement of reflection (hkl) and < I(hkl) > is its mean intensity.
Rpim = (Σhkl[1/(Nhkl-1)]1/2Σi|Ii(hkl) - < I(hkl) >|) / ΣhklΣi<Ii(hkl)>, where Ii(hkl) is the intensity of the ith measurement of reflection (hkl), < I(hkl) > is its mean intensity and N is the number of measurements.
I is the integrated intensity and σ(I) is its estimated standard deviation.
Rwork = (Σhkl|Fo-Fc|)/ΣhklFo where Fo and Fc are the observed and calculated structure factors.
Rfree is calculated as for Rwork but from a randomly selected subset of the data (5%), which were excluded from the refinement calculation.
Calculated by MOLPROBITY
Structure determination
The crystal structures of WDR5 complexes were determined by molecular replacement using PHASER 31. Chain-A of wild-type WDR5 (PDB entry: 2H14) was used as the initial phasing template. The model obtained from PHASER was manually adjusted and completed using the graphics program COOT 32. Structure refinement was performed with the REFMAC5 program, using standard protocols for NCS refinement 33. Ligand molecules were omitted from the models in initial stages of building and refinement. After building all the water into the structures, L-Arg species molecules were fitted in their respective electron density features. The final refinement statistics of the structures are summarized in Table 1.
Structure analysis
WDR5 subunit A from each structure was used as representative for all analyses and comparisons (A chain and B chain protein and ligand features were closely superimposable). Except for the images depicting modeled (methyl)arginine surfaces and ligand electron densities, which were generated using PyMOL (Schrödinger), all structural measurements and molecular graphic images were generated using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081) 34. The geometry analyses of the final models were performed using MolProbity 35.
Results
Thermodynamic properties of WDR5 interaction with histone H3 peptides
Using an array of arginine-mutant and methylarginine-modified H3 peptides, we performed ITC experiments to completely interrogate the H3/WDR5 interaction. To provide the most accurate and precise affinity measurements, all commercially sourced peptides used in this study were: 1) repurified by high performance liquid chromatography (HPLC) and 2) concentrations were measured by NMR (Figure S1–4, and Methods). All previously reported WDR5/H3R2 quantitative binding data were obtained using peptides containing H3R8 11, 15, 16, 18. The amino acid sequence surrounding H3R8 (A-R-K-S) potentially constitutes a second WIN motif on the H3 tail that may have contributed to previously reported binding constants. Therefore, to interrogate WDR5/H3 interaction specificity, we performed replicate ITC experiments with H3 1–12aa (Figure 2a) wild-type and R-to-K mutant peptides: H3R2K, H3R8K, and H3R2KR8K.
Figure 2.

Replicate isothermal calorimetry assays showing WDR5 interacts with specifically with H3R2. a. Histone H3 1–12aa peptide sequence with R to K mutant positions underlined b. Raw power differentials (DP) of peptide titrations c. Integrated heats of binding; error bars represent the standard error between two replicate titrations d. Dissociation constants for replicate titrations determined by fitting heat integrations to a one-site binding model e. Histogram of thermodynamic parameters for interacting peptides (ΔG, Gibbs free energy change; ΔH, enthalpy change; T, temperature in Kelvin; ΔS, entropy change) f. Tabulated thermodynamic parameters for interacting peptides (n.d., not detected). Peptides bound WDR5 with stoichiometries (N-values) between 0.9 and 1.1.
H3 wild-type and H3R8K peptides resulted in similar power differentials when injected into WDR5, whereas there was no indication of interaction with either the H3R2K or H3R2KR8K peptides (Figure 2b). Plotting enthalpy change (ΔH) versus H3 peptide:WDR5 (molar ratio) and fitting of a Langmuir isotherm (one-site binding model; Figure 2c) revealed that WDR5 has comparable equilibrium dissociation constants (Kd; Figure 2d) for H3 (n=2; Kd=10.8±0.9μM, Kd=10.6±0.8μM) and H3R8K (n=2; Kd=10.9±0.9μM, Kd=10.4±1.2μM). Likewise, the thermodynamic parameters ΔG, ΔH, and –TΔS were similar between H3 and HR8K peptides Figure 2e,f). These results clearly demonstrate that WDR5 interacts specifically with H3R2 and does not interact with H3R8.
Next, to characterize the effect arginine methylation has on the H3R2/WDR5 interaction, we performed replicate ITC experiments with H3 1–7aa (Figure 3a) wild type or methylarginine-modified peptides: H3R2me1, H3R2me2s, H3R2me2a. Titration of H3R2me0, H3R2me1, and H3R2me2s 1–7aa peptides into WDR5 produced similar power differentials, whereas no heat was observed in titrations with H3R2me2a peptide (Figure 3b). These results are consistent with observations from our initial pulldown assays using H3 1–21aa peptides (Figure S5) and clearly demonstrate that WDR5 interacts with H3R2me0/me1/me2s but not with H3R2me2a. The data analysis using Langmuir isotherm (one-site binding model; Figure 3c) revealed that WDR5 has comparable equilibrium dissociation constants for H3R2me0 (n=2; Kd =11.0±0.5μM, Kd =11.8±0.5μM); H3R2me1 (n=2; Kd =9.0±0.8μM, Kd =8.1±1.4μM); and H3R2me2s (n=2; Kd =10.6±0.4μM, Kd =10.2±1.2μM), showing a modest increase in affinity for H3R2me1 (Figure 3d). Furthermore, the thermodynamic terms ΔG, ΔH, and -TΔS for each interaction are nearly identical (Figure 3e,f). With respect to H3R2me0/me1/me2s isoforms, our results establish that arginine methylation of H3R2 has a negligible effect on WDR5 interaction affinity and confirm no interaction with H3R2me2a. We were surprised that arginine methylation had such a small effect on WDR5 affinity and were thus compelled to investigate the structural differences between arginine- and methylarginine-liganded WDR5 complexes.
Figure 3.

Replicate isothermal calorimetry assays showing WDR5 interacts with H3, H3R2me1, and H3R2me2s with similar heats and affinities but does not interact with H3R2me2a. a. Histone H3 1–7aa peptide sequence with methylarginine isoforms occurring at Arg2, underlined b. Raw power differentials (DP) of peptide titrations c. Integrated heats of binding; error bars represent the standard error between two replicate titrations d. Dissociation constants for replicate titrations determined by fitting integrated heats to a one-site binding model e. Histogram of thermodynamic parameters for interacting peptides (ΔG, Gibbs free energy change; ΔH, enthalpy change; T, temperature in Kelvin; ΔS, entropy change) f. Tabulated thermodynamic parameters for interacting peptides (n.d., not detected). Peptides bound WDR5 with stoichiometries (N-values) between 0.9 and 1.1.
Structure of WDR5 complexes
WDR5 is a ~37kDa globular protein that adopts a 7-bladed ß-propeller fold (Figure 4a). At one end of the central channel is the WIN site, defined here as residues Ala47, Val48, Ser49, Ala65, Ser91, Glu107, Phe133, Cys134, Phe149, Phe173, Ser175, Tyr191, Tyr260, Cys261, Ile262, Phe263, Ile305, and Leu321 (Figure 1b). The solvent exposed outer surface of the WIN site is slightly acidic and becomes increasingly hydrophobic within the interior of the channel to accommodate the arginine sidechain (Figure S6). All structurally characterized WIN site ligands (Table S1) share a common binding mode: the WIN motif peptidyl arginine extends into the hydrophobic channel, with its guanidinium moiety stacking between aromatic residues Phe133 and Phe263 (Figure S7). As mentioned above, histone H3 is the only WIN site ligand known to be modified with methylarginine marks. Our binding studies establish that WDR5 has comparable affinity for H3R2me0/me1/me2s isoforms but has no interaction with H3R2me2a. To study any structural differences in binding the various methylarginine isoforms, high resolution crystal structures of unliganded (apo)WDR5 and WDR5 in complex with L-arginine (L-Arg) amino acid and its monomethyl (me1-L-Arg) and symmetric dimethyl (me2s-L-Arg) isoforms—completely devoid of additional peptide context—were determined. The structural analyses focused on the region of the WIN site interacting with the arginine side chain. The crystal structure of WDR5 bound to H3R2me1 peptide was also determined in the course of optimizing the system.
Figure 4.

Crystal structures of the WDR5 WIN site detailing ligand guanidino group interactions with arginine and methylarginine isoforms. a. Overview of apo WDR5 with WIN site boxed and magnified. WDR5 complexes with b. L-Arg me0, c. L-Arg me1, d. L-Arg me2s, and e. H3R2me1 peptide ligands. Hydrogen bond (solid line) and p-stack (dashed line) distances are depicted. Water molecule (red sphere). Distances denoted in ångström (Å).
Crystallization of apo-WDR5 and conditions for obtaining WDR5 in complex with L-Arg, me1-L-Arg, and me2s-L-Arg amino acid or with H3R2me1 peptide are summarized in Table S2. To determine the crystal structures of the complexes, molecular replacement using PHASER was performed. WDR5 apo and L-Arg isoform complexed structures yielded data consistent with the P21212 space group, whereas the WDR5/H3R2me1 peptide structure was determined in space group C2221—all structures contained one monomer in the asymmetric unit. The resolution of the structures varied between 1.66Å to 1.92Å. Except for a few amino acid residues at the amino and carboxy termini, electron density was well ordered over the entire WDR5 backbone structure. The sidechain electron density of several surface residues was also not resolved. All ordered amino acid residues were in the most favored or in additionally allowed regions of the Ramachandran plot (Table 1). The electron density corresponding to the L-Arg, me1-L-Arg, and me2s-L-Arg were well resolved, whereas the C-terminal residues of the H3 peptide were not observed in the structure (Figure S8).
Binding of WDR5 with L-Arginine and methylarginine isoforms
Analysis of the WIN site of apo- and liganded-WDR5 structures provide details of the guanidino-binding interface. Hydrogen bonding, Van der Waals (VDW) contacts, and π-π stacking interactions were assessed in the structures (Figure 4, Table 2).
Table 2.
VDW contacts and H-bonds of liganded WDR5 complexes
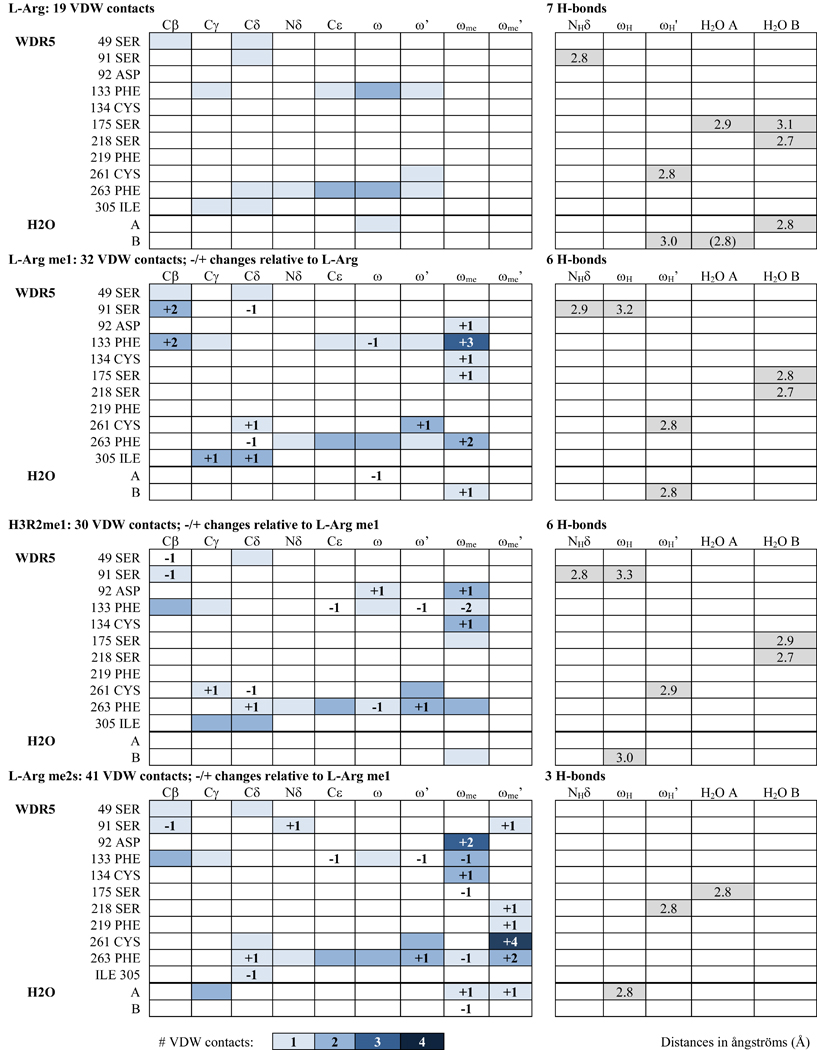 |
In the apo WDR5 structure, Phe133 and Phe263 aromatic sidechains are not in a stacked orientation, and the WIN site is occupied by four water molecules (Figure 4a). When bound to L-Arg ligand, Phe133 and Phe263 engage the guanidino group in π-π stacking interactions, and two waters remain in the WIN site (Figure 4b).These water molecules will be referred to as water A and water B, and the guanidino amino group proximal to water A will be referred to as ω and that proximal to water B as ω’ (Figure 4a, inset). A total of seven hydrogen bonds are formed between the arginine sidechain, structural water molecules, and WIN site residues in the L-Arg liganded structure (Figure 4b, Table 2). Water A forms one H-bond with the Ser175 backbone oxygen (OB) and one with water B. Water B is also hydrogen bonded to Ser175 OB and to Ser218 OB. These four H-bonds are arranged in the same configuration in the apo structure, suggesting their structural significance (Figure 4a). The L-Arg guanidino group engages in three more H-bonds in the WIN site: one between L-Arg ωH’ and water B, another between L-Arg ωH’ and Cys261 OB, and a third between L-Arg NHδ and Ser91 OB.
We next inspected other contacts within VDW-interaction distances (Figure S9); WDR5 residues are listed with corresponding L-Arg atom interactions in Table 2. With respect to L-Arg, 19 VDW contacts are observed in the WIN site: 18 with WDR5 residues and one with water A (Figure S9a).
The aromatic rings of Phe133 and Phe263 engage in π-π stacking interactions with the delocalized electrons of guanidino group atoms. To assess whether arginine methylation affects this interaction, we measured the distance from the center of each phenylalanine aromatic ring to the guanidinium group central carbon (Cε) for each complex (Figure 4). With respect to L-Arg, the distance from Phe133−L-Arg was 4.4Å and Phe263−L-Arg was 4.2Å, giving a total π-π stacking distance of 8.6Å (Figure 4b).
When me1-L-Arg occupies the WIN site, the bulk of the guanidino methyl group (ωme) results in water A being displaced from the binding site (Figure 4c), resulting in a net loss of one hydrogen bond compared to the L-Arg structure. Water B remains engaged in the same 3 H-bonds as described for L-Arg; however, the hydrogen bond between me1-L-Arg ωH’ and water B and that between water B and Ser175 OB are slightly shorter, and thus stronger. The hydrogen bond interaction between Ser91 OB and NHδ is slightly longer, and one additional H-bond is gained between Ser91 OB and me1-L-Arg ωH. 31 VDW contacts are formed between me1-L-Arg and WIN site residues and one with water B—13 more than L-Arg (Table 2, Figure S9b). The overall π-π stacking distance is unchanged (Figure 4c); however, the distance between Phe133−me1-L-Arg (4.6Å) is slightly increased by 0.2Å and the distance between Phe263−me1-L-Arg (4.0Å) is decreased by 0.2Å, compared to L-Arg (4.4Å and 4.2Å, respectively).
When WDR5 binds me2s-L-Arg, the guanidino moiety is rotated 180° about the Nδ-Cε bond, such that the ωme group is arranged perpendicular relative to its position in the me1-L-Arg structure. This arrangement now allows water A to be retained in the WIN site, whereas the bulk of me2s-L-Arg ωme’ results in the displacement of water B (Figure 4d). Overall, this results in a net loss of 3 H-bonds compared to the me1-L-Arg structure. Water A forms a slightly shorter H-bond with Ser175 OB and another with ωH. The two hydrogen bonding interactions between me1-L-Arg ωH and Ser91 OB and between me1-L-Arg NHδ Ser91 OB are lost in the me2s-L-Arg structure. 39 VDW contacts are made between me2s-L-Arg and WDR5 and one with both water A and water B—nine more than compared to the me1-L-Arg structure (Table 2, Figure S9c). The total π-π stacking distance is unchanged at 8.6Å (Figure 4d).
Binding of WDR5 with histone H3R2me1 peptide
The crystal structure of WDR5 in complex with H3R2me1 1–21aa peptide was determined at 1.66Å resolution. The four N-terminal residues of the peptide are clearly visible at the binding pocket, whereas the remaining carboxy terminal residues were not resolved and presumably disordered. The arginine sidechain of the H3R2me1 peptide interacts with WDR5 much like Rme1 with only modest differences (Figure 4e). Peptidyl H3R2me1 makes 29 VDW contacts with WDR5 and one with water B—two fewer compared to me1-L-Arg (Table 2, Figure S9d). The π-π stacking interaction distance totals 8.7Å; the distance between Phe133−H3R2me1 is unchanged, whereas the distance between Phe263−H3R2me1 is slightly increased by 0.1Å to 4.1Å (Figure 4e).
Structural comparisons
The overall Cα-Cα RMSD between apo and liganded structures is 0.381Å (L-Arg), 0.359Å (L-Arg me1), 0.324Å (L-Arg me2s), and 0.326Å (H3R2me1 peptide). Additionally, the RMSD between common interacting residues (Ser49, Ser91, Asp92, Phe133, Cys134, Ser175, Ser218, Phe219, Cys261, and Phe263) of liganded structures are all less than 0.22Å, indicating strong agreement with very little deviation between structures. Comparing B-factors of the WDR5 WIN site bound to the L-Arg isoforms shows that each complex is of similar rigidity with B-factors ranging between 15–20Å2 (Figure S10). The miniscule differences in RMSD values and B-factors are consistent with our binary methylarginine switch interaction model: WDR5 does not discriminate between unmodified, mono-, and symmetric dimethyl-arginine ligands (Figure 5).
Figure 5.

Superimposition of L-Arg(me) isoforms- and H3R2me1 peptide-liganded WDR5 complexes showing very modest differences between structures. WDR5 backbone and Arg-interacting residues are shown, with water molecules (spheres) colored accordingly. L-Arg (tan); L-Arg me1 (light blue); L-Arg me2s (green); H3R2me1 peptide (purple).
Consistent with our observations that Rme2a does not interact with WDR5, we did not observe electron density in the WIN site of WDR5 crystals after soaking with asymmetric dimethyl-L-arginine (Rme2a). Modelling Rme2a into the WDR5 WIN site revealed clashes that potentially prevent interaction (Figure S11). Our attempts to crystallize WDR5 bound to H3R8 using the H3R2me2a 1–21aa peptide were also unsuccessful.
Discussion
For many years, the WDR5/H3 interaction has been of considerable interest for its role in regulating gene expression and development 12. However, previously published biological 9 and quantitative binding 16 data disagree with respect to which H3R2 methylarginine isoforms interact with WDR5. To answer this open question, we provide a thorough and rigorous biochemical assessment of the WDR5/H3 N-terminal tail interaction. We unequivocally demonstrate that WDR5 binding is: 1) specific for H3R2, showing no interaction with H3R8; and 2) regulated by a binary methylarginine switch mechanism. In this binary switch, H3R2me0/me1/me2s all have near equal affinity for WDR5, whereas H3R2me2a is not compatible for interaction.
We utilized ITC with a variety of H3 peptides to interrogate the thermodynamics of interaction with WDR5. A major technical challenge to quantitatively probing histone tail interactions is the lack of aromatic residues, making measurement of peptide concentration by A280 impossible. Furthermore, although peptide concentrations can be measured by A205, this method relies either on a standard 36 or predicted 37, 38 molar extinction coefficient—both of which are estimated and introduce inaccuracies in final reported quantitative binding constant values. Additionally, we wanted to ensure that our peptides were of the utmost purity in our experiments. To address these concerns, we first repurified the peptides by HPLC over a much longer elution gradient than that normally used by peptide suppliers. We then measured peptide concentration using NMR, which is considerably more sensitive than A205 and allows for both highly accurate and precise concentration measurements.
WDR5 interacting proteins contain a WIN-motif—a four amino acid peptide with a consensus sequence of A-R-(A/C/S/T)-(E/K/R). The first two WIN motif residues are completely conserved with some plasticity at positions three and four. Histone H3 possesses a WIN motif at 1ARTK4 and is hypothesized to carry another at 7ARKS1010–12—both of which are modified by PRMT 5 and 6 and PRMT 2 and 5, respectively. Therefore, to delineate which H3 arginine residues interact with WDR5, H3 1–12aa peptides were synthesized and each arginine was systematically mutated to lysine followed by ITC measurements to quantify binding. Our results demonstrate that WDR5 only interacts with peptides containing H3R2; when H3R2 is mutated to lysine, no interaction is detected although H3R8 remains available for binding. These results define WIN motif requirements, i.e. position three requires a small R-group amino acid, and position four requires a charged sidechain. Furthermore, this offers a remarkable example of how a simple substitution of residues at WIN motif positions three and four imparts WDR5 binding specificity without post-translational modification. To define the most permissible WIN-motif, a point for future study would be to test how residues flanking WIN-motif arginine’s affect interaction with WDR5.
WDR5 has been reported as interacting with H3R2me0 11, 15–17 and H3R2me2s 18, with the latter described as a high affinity interaction in vitro 18. Both H3R2me1 16 and H3R2me2a 18–20 had previously been reported not to interact with WDR5. However, more recent biological data from our group suggested that WDR5 does interact with H3R2me1 9. Therefore, to address the discrepancies between biological and biochemical reports, we synthesized H3 1–7aa peptides containing the full array of H3R2 methylarginine isoforms and performed ITC to determine which isoforms interact with WDR5. Our results clearly show that WDR5 indeed interacts with H3R2me1 and, furthermore, that H3R2me0/me1/me2s all bind WDR5 with near equal affinity, whereas H3R2me2a is excluded. We highlight the rigorous analytical chemistry methodology we undertook to obtain ultra-pure peptides and highly accurate and precise peptide concentrations for use in our qualitative binding assays. Inadequate peptide purification and/or inefficacious concentration measurement are two main sources of inaccuracies in quantitative binding measurements; these may have contributed to the prior findings that WDR5 had very weak affinity (~500μM) for H3R2me1. Nonetheless, in line with findings that WDR5 indeed interacts with monomethylarginine, small molecule WIN site inhibitors have been designed that utilize a monomethyl-like appendage to engage WDR5 39, 40. For all interacting H3 peptide ligands, the energy required is ~90% entropic, indicating that binding is predominantly driven by a relatively large solvent gain upon rearrangement of the WDR5 WIN site and ligand arginine hydration shells (i.e. hydrophobic effect) with a small change in bond enthalpy. These results reveal that WDR5/H3R2 interactions are governed by a binary methylarginine switch, such that H3R2me0/me1/me2s are equally favorable for binding, whereas H3R2me2a is not.
As unmodified arginine and its methylated isoforms are structurally distinct, we attempted to assess the differences among the ligands when complexed with WDR5. To that end, we determined high-resolution crystal structures of WDR5 in complex with unmodified and methylated (me1, me2s) L-Arg amino acids and in complex with H3R2me1 peptide. Unmodified L-Arg interacts with WDR5 by utilizing two structural water molecules, seven H-bonds, and 19 VDW contacts. When me1-L-Arg binds WDR5, one water molecule is displaced which results in the net loss of one H-bond, but the bulk of the methyl group increases VDW contacts to 32. When me2s-L-Arg binds WDR5, the opposite water molecule is displaced which results in the loss of three more H-bonds; with two methyl groups present, VDW contacts are increased to 41. Arginine methylation had no effect on the π-π stacking interaction distance (8.6Å) made with Phe133 and Phe263. Additionally, comparison of the RMSD values and B-factors of each liganded WDR5 complex shows that no major differences are brought about by arginine methylation. The structural findings within our minimized system of WDR5 in complex with single-residue L-Arg are consistent with previously reported structures of WDR5 in complex with unmodified H3R2 peptide (seven H-bonds, 25 VDW contacts, π-π interaction distance 8.6Å, RMSD 0.389Å; PDB: 2H13)16 and H3R2me2s peptide (four H-bonds, 47 VDW contacts, π-π interaction distance 8.5Å, RMSD 0.300Å, and displacement of water molecule A; PDB: 4A7J)18.
Thus, although arginine methylation decreases WIN site hydrogen bonding interactions, the loss of associated binding energy is compensated by an increase in VDW contacts. This compensation is consistent with the low change in enthalpy observed in our quantitative binding analysis and further supports that guanidino-group ligand interactions with WDR5 are primarily entropic: binding is driven by hydrophobic π-π interactions between Phe133, Phe263, and the ligand guanidino moiety and by desolvation of the arginine sidechain and WIN site pocket. Liberation of a water molecule from a protein cavity is predicted to contribute −0.46 to −2.67 kcal/mol of entropy (-TΔS) to binding interactions, with higher values associated with water molecules near charged residues 41. As the interior WIN site cavity is hydrophobic and the unbound arginine sidechain, although charged, is not buried, the entropy gained by liberation of each water molecule is likely toward the lower end of this scale. Our data is consistent with the displacement of 2–3 water molecules from the WIN site and another handful from the arginine sidechain upon WDR5/H3R2 interaction.
WDR5 interacts with transcription factors, such as MYC, and is present in numerous chromatin-modifying complexes, such as KANSL (lysine Acetyltransferase Non-Specific Lethal); NuRD (Nucleosome Remodeling and Deacetylase); and MLL 7. Moreover, WDR5 is expressed at levels considerably higher than other components of these complexes in vivo. It is enticing to speculate that WDR5 is stored on and maintains open chromatin at sites of unmodified, monomethyl, and symmetric dimethylarginine. As epigenetic complexes generally carry multiple reader subunits that stabilize interactions with chromatin, it raises the possibility that chromatin bound WDR5 may serve as a homing beacon for partially assembled complexes, thereby completing assembly and activating complexes as WDR5 disengages chromatin. Consistent with this hypothesis, two recent cryo-EM structures of MLL were determined in complex with the nucleosome core particle: one structure showed WDR5 engaged with the MLL WIN motif 42, whereas the WDR5 WIN site is unoccupied in the other 43. In all published liganded WDR5 structures, sidechains of residues outside of the interacting WIN-motif are predominantly disordered and not well resolved, suggesting that they do not influence WDR5 interaction. As other subunit components of WDR5-containing complexes contain epigenetic reader domains, such as plant homeodomain finger motifs and bromodomains, it follows that that recruitment of WDR5-containing complexes is orchestrated by additional subunit interactions, complimenting that of WDR5. This brings about another excellent point for future study: determine how other subunits interact with the flanking residues of WIN-motif-containing ligands, which would shed some much-needed light on the physiological mechanisms underlying WDR5/H3 interaction.
Conclusion
This study clearly demonstrates that WDR5 interacts with unmodified, monomethyl-, and symmetric dimethyl-arginine with near equal binding affinity but does not interact with asymmetric dimethylarginine. Moreover, this defines a novel binary arginine methylation switch mechanism which suggests that the purpose of installing either dimethyl isoform is to prevent installation of the other isoform. The binary switch mechanism supports a model in which dimethylarginine isoforms function in opposition to either maintain or block docking sites for interacting proteins, which may serve as a global regulatory mechanism for WDR5-containing and/or other methylarginine-reading complexes.
Supplementary Material
ACKNOWLEDGMENT
We thank Maxim Maron for thoroughly reading and commenting on the final manuscript.
Funding Sources
This work was supported by NIH R01GM108646 and the American Lung Association (LCD-564723) (both to D.S.). The Albert Einstein Crystallographic Core X-Ray diffraction facility is supported by NIH Shared Instrumentation Grant S10 OD020068. Data collection also utilized resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. The Bruker Avance IIIHD 600 MHz system was purchased using funds from the National Institutes of Health (1S10OD016305).
ABBREVIATIONS
- H3
histone H3
- HPLC
high performance liquid chromatography
- ITC
isothermal titration calorimetry
- K4
lysine 4
- Kd
equilibrium dissociation constant
- KANSL
lysine acetyltransferase non-specific lethal
- MLL
mixed lineage leukemia
- NuRD
nucleosome remodeling and deacetylase
- PRMT
protein arginine methyltransferase
- R2
arginine 2
- Rme1/MMA
monomethylarginine
- Rme2s/sDMA
symmetric dimethylarginine
- Rme2a/aDMA
asymmetric dimethylarginine
- RMSD
root mean squared deviation
- WBM
WDR5 binding motif
- WDR5
WD-40 repeat-containing protein 5
- WIN
WDR5 interacting
Footnotes
ACCESSION NUMBERS
The structural data supporting this study have been deposited in the PBD. The accession numbers for each structure are: apo-WDR5 (6ofz); L-Arg/WDR5 (6oi0); me1-L-Arg/WDR5 (6oi1); me2s-L-Arg/WDR5 (6oi2); and H3R2me1/WDR5 (6oi3). The UniProtKB accession number for human WDR5 is P61964.
ASSOCIATED CONTENT
The following files are available free of charge.
Supporting Information:
Supplementary Figures 1–11 (PDF)
HPLC chromatograms and NMR spectra of H3 peptides; peptide pulldown assays; WDR5 surface maps: electrostatic potential and hydropathicity; superimposition of published WDR5 structures; L-Arg and H3R2me1 peptide ligand density maps; depiction of VDW contacts; WIN site surface maps for B-factor analysis; modeled WDR5/H3R2me2a clashes
Conflict of interest: None declared.
REFERENCES
- [1].Gayatri S; Bedford MT (2014) Readers of histone methylarginine marks, Biochim Biophys Acta 1839, 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Morales Y; Caceres T; May K; Hevel JM (2016) Biochemistry and regulation of the protein arginine methyltransferases (PRMTs), Arch Biochem Biophys 590, 138–152. [DOI] [PubMed] [Google Scholar]
- [3].Haghandish N; Baldwin RM; Morettin A; Dawit HT; Adhikary H; Masson JY; Mazroui R; Trinkle-Mulcahy L; Cote J (2019) PRMT7 methylates eukaryotic translation initiation factor 2alpha and regulates its role in stress granule formation, Mol Biol Cell 30, 778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fuhrmann J; Clancy KW; Thompson PR (2015) Chemical biology of protein arginine modifications in epigenetic regulation, Chem Rev 115, 5413–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Walport LJ; Hopkinson RJ; Chowdhury R; Schiller R; Ge W; Kawamura A; Schofield CJ (2016) Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases, Nat Commun 7, 11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lorton BM; Shechter D (2019) Cellular consequences of arginine methylation, Cell Mol Life Sci 76, 2933–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Guarnaccia AD; Tansey WP (2018) Moonlighting with WDR5: A Cellular Multitasker, J Clin Med 7(2), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Iberg AN; Espejo A; Cheng D; Kim D; Michaud-Levesque J; Richard S; Bedford T (2008) Arginine methylation of the histone H3 tail impedes effector binding, J Biol Chem 283, 3006–3010. [DOI] [PubMed] [Google Scholar]
- [9].Chen H; Lorton B; Gupta V; Shechter D (2017) A TGFbeta-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression, Oncogene 36, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kim DH; Tang Z; Shimada M; Fierz B; Houck-Loomis B; Bar-Dagen M; Lee S; Lee SK; Muir TW; Roeder RG; Lee JW (2013) Histone H3K27 trimethylation inhibits H3 binding and function of SET1-like H3K4 methyltransferase complexes, Mol Cell Biol 33, 4936–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schuetz A; Allali-Hassani A; Martin F; Loppnau P; Vedadi M; Bochkarev A; Plotnikov AN; Arrowsmith CH; Min J (2006) Structural basis for molecular recognition and presentation of histone H3 by WDR5, Embo j 25, 4245–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wysocka J; Swigut T; Milne TA; Dou Y; Zhang X; Burlingame AL; Roeder RG; Brivanlou AH; Allis CD (2005) WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development, Cell 121, 859–872. [DOI] [PubMed] [Google Scholar]
- [13].Dong F; Li Q; Yang C; Huo D; Wang X; Ai C; Kong Y; Sun X; Wang W; Zhou Y; Liu X; Li W; Gao W; Liu W; Kang C; Wu X (2018) PRMT2 links histone H3R8 asymmetric dimethylation to oncogenic activation and tumorigenesis of glioblastoma, Nat Commun 9, 4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pal S; Vishwanath SN; Erdjument-Bromage H; Tempst P; Sif S (2004) Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes, Mol Cell Biol 24, 9630–9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ruthenburg AJ; Wang W; Graybosch DM; Li H; Allis CD; Patel DJ; Verdine GL (2006) Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex, Nat Struct Mol Biol 13, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Couture JF; Collazo E; Trievel RC (2006) Molecular recognition of histone H3 by the WD40 protein WDR5, Nat Struct Mol Biol 13, 698–703. [DOI] [PubMed] [Google Scholar]
- [17].Avdic V; Zhang P; Lanouette S; Voronova A; Skerjanc I; Couture JF (2011) Fine-tuning the stimulation of MLL1 methyltransferase activity by a histone H3-based peptide mimetic, Faseb j 25, 960–967. [DOI] [PubMed] [Google Scholar]
- [18].Migliori V; Muller J; Phalke S; Low D; Bezzi M; Mok WC; Sahu SK; Gunaratne J; Capasso P; Bassi C; Cecatiello V; De Marco A; Blackstock W; Kuznetsov V; Amati B; Mapelli M; Guccione E (2012) Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance, Nat Struct Mol Biol 19, 136–144. [DOI] [PubMed] [Google Scholar]
- [19].Guccione E; Bassi C; Casadio F; Martinato F; Cesaroni M; Schuchlautz H; Luscher B; Amati B (2007) Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive, Nature 449, 933–937. [DOI] [PubMed] [Google Scholar]
- [20].Hyllus D; Stein C; Schnabel K; Schiltz E; Imhof A; Dou Y; Hsieh J; Bauer UM (2007) PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation, Genes Dev 21, 3369–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lakowski TM; Frankel A (2008) A kinetic study of human protein arginine N-methyltransferase 6 reveals a distributive mechanism, J Biol Chem 283, 10015–10025. [DOI] [PubMed] [Google Scholar]
- [22].Kolbel K; Ihling C; Bellmann-Sickert K; Neundorf I; Beck-Sickinger AG; Sinz A; Kuhn U; Wahle E (2009) Type I Arginine Methyltransferases PRMT1 and PRMT-3 Act Distributively, J Biol Chem 284, 8274–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guo A; Gu H; Zhou J; Mulhern D; Wang Y; Lee KA; Yang V; Aguiar M; Kornhauser J; Jia X; Ren J; Beausoleil SA; Silva JC; Vemulapalli V; Bedford MT; Comb MJ (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation, Mol Cell Proteomics 13, 372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Geoghegan V; Guo A; Trudgian D; Thomas B; Acuto O (2015) Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling, Nat Commun 6, 6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Larsen SC; Sylvestersen KB; Mund A; Lyon D; Mullari M; Madsen MV; Daniel JA; Jensen LJ; Nielsen ML (2016) Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells, Sci Signal 9, rs9. [DOI] [PubMed] [Google Scholar]
- [26].Yakubu RR; Silmon de Monerri NC; Nieves E; Kim K; Weiss LM (2017) Comparative Monomethylarginine Proteomics Suggests that Protein Arginine Methyltransferase 1 (PRMT1) is a Significant Contributor to Arginine Monomethylation in Toxoplasma gondii, Mol Cell Proteomics 16, 567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feng Y; Wang J; Asher S; Hoang L; Guardiani C; Ivanov I; Zheng YG (2011) Histone H4 acetylation differentially modulates arginine methylation by an in Cis mechanism, J Biol Chem 286, 20323–20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Battye TG; Kontogiannis L; Johnson O; Powell HR; Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM, Acta Crystallogr D Biol Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].(1994) The CCP4 suite: programs for protein crystallography, Acta Crystallogr D Biol Crystallogr 50, 760–763. [DOI] [PubMed] [Google Scholar]
- [30].Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ (2007) Phaser crystallographic software, J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Emsley P; Lohkamp B; Scott WG; Cowtan K (2010) Features and development of Coot, Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Murshudov GN; Vagin AA; Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [34].Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis, J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- [35].Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Goldfarb AR; Saidel LJ; Mosovich E (1951) The ultraviolet absorption spectra of proteins, J Biol Chem 193, 397–404. [PubMed] [Google Scholar]
- [37].Anthis NJ; Clore GM (2013) Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm, Protein Sci 22, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Scopes RK (1974) Measurement of protein by spectrophotometry at 205 nm, Anal Biochem 59, 277–282. [DOI] [PubMed] [Google Scholar]
- [39].Grebien F; Vedadi M; Getlik M; Giambruno R; Grover A; Avellino R; Skucha A; Vittori S; Kuznetsova E; Smil D; Barsyte-Lovejoy D; Li F; Poda G; Schapira M; Wu H; Dong A; Senisterra G; Stukalov A; Huber KVM; Schonegger A; Marcellus R; Bilban M; Bock C; Brown PJ; Zuber J; Bennett KL; Al-Awar R; Delwel R; Nerlov C; Arrowsmith CH; Superti-Furga G (2015) Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia, Nat Chem Biol 11, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li DD; Wang ZH; Chen WL; Xie YY; You QD; Guo XK (2016) Structure-based design of ester compounds to inhibit MLL complex catalytic activity by targeting mixed lineage leukemia 1 (MLL1)-WDR5 interaction, Bioorg Med Chem 24, 6109–6118. [DOI] [PubMed] [Google Scholar]
- [41].Huggins DJ (2015) Quantifying the entropy of binding for water molecules in protein cavities by computing correlations, Biophys J 108, 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xue H; Yao T; Cao M; Zhu G; Li Y; Yuan G; Chen Y; Lei M; Huang J (2019) Structural basis of nucleosome recognition and modification by MLL methyltransferases, Nature 573, 445–449. [DOI] [PubMed] [Google Scholar]
- [43].Park SH; Ayoub A; Lee YT; Xu J; Kim H; Zheng W; Zhang B; Sha L; An S; Zhang Y; Cianfrocco MA; Su M; Dou Y; Cho US (2019) Cryo-EM structure of the human MLL1 core complex bound to the nucleosome, Nat Commun 10, 5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.