

SUMMARY
Lipopolysaccharides (LPS) are essential envelope components in many Gram-negative bacteria and provide intrinsic resistance to antibiotics. LPS molecules are synthesized in the inner membrane and then transported to the cell surface by the LPS transport (Lpt) machinery. In this system, the ATP-binding cassette (ABC) transporter LptB2FGC extracts LPS from the inner membrane and places it onto a periplasmic protein bridge through a poorly understood mechanism. Here, we show that residue E86 of LptB is essential for coupling the function of this ATPase to that of its partners LptFG, specifically at the step where ATP binding drives the closure of the LptB dimer and the collapse of the LPS-binding cavity in LptFG that moves LPS to the Lpt periplasmic bridge. We also show that defects caused by changing residue E86 are suppressed by mutations altering either LPS structure or transmembrane helices in LptG. Furthermore, these suppressors also fix defects in the coupling helix of LptF, but not of LptG. Together, these results support a transport mechanism in which the ATP-driven movements of LptB and those of the substrate-binding cavity in LptFG are bi-directionally coordinated through rigid-body coupling, with LptF’s coupling helix being important in coordinating cavity collapse with LptB dimerization.
Keywords: ABC transporters (D018528), membrane transport proteins (D026901), cell membrane permeability (D002463), Escherichia coli K-12 (D048168), acyltransferases (D000217), lipopolysaccharides (D008070)
ABBREVIATED SUMMARY
The ATP binding cassette (ABC) transporter LptB2FGC extracts LPS from the inner membrane and powers the transport of the glycolipid to the cell surface in Gram-negative bacteria. Our study shows that changes to the LPS structure and the transporter can affect how the function of the cytoplasmic LptB ATPase is coupled to the extraction of LPS by LptFG by either promoting or interfering with the collapse of the substrate-binding cavity.
Graphical Abstract
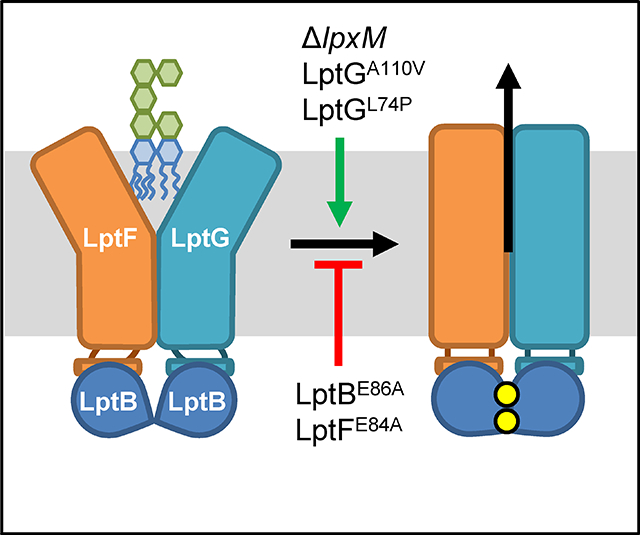
INTRODUCTION
Despite much effort, it is notoriously difficult to develop novel antibiotics effective against Gram-negative bacteria since the structure of their cell envelope provides intrinsic resistance to many antibiotics (Nikaido, 2003). The Gram-negative cell envelope is composed of two membranes, an inner membrane (IM) and outer membrane (OM), that are separated by an aqueous compartment called the periplasm (Silhavy et al., 2010). Unlike the IM, which is a phospholipid bilayer, the OM is an asymmetrical bilayer with an inner leaflet made of phospholipids and an outer leaflet composed of the glycolipid lipopolysaccharide (LPS). The amphipathic nature of LPS creates a potent permeability barrier that is particularly effective in preventing the diffusion of small hydrophobic antibiotics (Nikaido, 2003).
LPS is composed of up to three major components: lipid A, an acylated glucosamine disaccharide; the core oligosaccharide, a nonrepeating set of sugars; and the O antigen, a polysaccharide composed of a highly variable repeating oligosaccharide (Raetz and Whitfield, 2002, Bertani and Ruiz, 2018). LPS synthesis begins in the cytoplasm with the precursor molecule uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). The enzymes in the Raetz pathway convert this UDP-GlcNAc into Kdo2-lipid A (Fig. S1). The core oligosaccharide biosynthesis pathway follows, which ligates and phosphorylates the core sugars. The resulting molecule is then flipped from the inner leaflet to the outer leaflet of the IM by the ATP-binding cassette (ABC) transporter MsbA (Zhou et al., 1998, Mi et al., 2017). In organisms that produce the O antigen, this component is synthesized independently of core-lipid A and is ligated to the core by the WaaL ligase in the outer leaflet of the IM (Klena et al., 1992, Kalynych et al., 2014).
After synthesis is complete, LPS is transported from the IM to the outer leaflet of the OM by the lipopolysaccharide transport (Lpt) machinery (Fig. 1A) (Okuda et al., 2016). The Lpt machinery is typically made of the seven essential proteins, LptB2FGCDE, that span every compartment of the cell (Sherman et al., 2018). First, LPS is extracted from the IM by the ABC transporter, LptB2FGC (Ruiz et al., 2008, Sperandeo et al., 2007, Sperandeo et al., 2008, Okuda et al., 2012). Next, LPS is passed from LptF to the first member of the periplasmic bridge, LptC (Owens et al., 2019). The periplasmic bridge consists of the periplasmic domain of LptC, LptA, and the N-terminal domain of LptD (Freinkman et al., 2012). These bridge components share a conserved β-jellyroll structure, which contains a hydrophobic groove that shields the hydrophobic acyl chains of lipid A from the hydrophilic periplasm (Suits et al., 2008, Tran et al., 2010, Qiao et al., 2014). LptF and LptG also contain one of these conserved β-jellyroll domains in the periplasm, although only that of LptF has been shown to interact with LPS (Owens et al., 2019). Through repeated rounds of LPS extractions powered by ATP by the LptB2FGC ABC transporter, LPS molecules are thought to move as a stream along the periplasmic bridge by pushing previously extracted LPS molecules towards the OM translocon, LptDE (Okuda et al., 2012, Okuda et al., 2016, Sherman et al., 2018). This plug-and-barrel complex allows LPS to transverse the OM and be placed into the outer leaflet of the OM (Chng et al., 2010, Freinkman et al., 2011, Dong et al., 2014). Although the members of the Lpt complex have been known for years, many aspects of the transport process still remain poorly defined. Particularly unclear is the mechanism of LPS extraction from the outer leaflet of the IM by LptB2FGC.
Figure 1: Q-loop residue E86 of LptB is essential for function.

A) Model for LPS transport by the LptB2FGCADE machinery. B) Top-down (membrane) view of the surface-rendered crystal structure of the E. coli LptB dimer (PDB 6MBN) bound to ATP (green). Residues that could be substituted (as shown in panel C) by Simpson et al. (2016) without affecting function are colored blue; those that when substituted (as in panel C) resulted in decreased LptB function are colored orange. Residue E86 is labeled and colored in red. C) Table summarizing phenotypes of lptB mutants in which Q-loop and groove residues were changed as indicated. Plasmid-borne lptB alleles were assessed for their ability to complement chromosomal ΔlptB. Viable mutants were also assessed for OM permeability defects. Functional refers to substitutions that confer no defects, partial LOF (loss of function) refers to those that confer OM-permeability defects, conditional LOF refers to those that prevent complementation of chromosomal ΔlptB in LB but not in minimal media, and total LOF refers to those that do not complement chromosomal ΔlptB under any condition tested. Substitutions from this study are in bold. D) OM-permeability defects conferred by plasmid-encoded lptB(E86) alleles in haploid cells carrying a chromosomal lptB deletion as determined by sensitivity to antibiotics via disc-diffusion assay. The diameter (in mm) of zones of total inhibition of visible growth are shown outside of parenthesis, while those of zones of partial clearing of growth (i.e. hazy zone) are shown in parenthesis. This partial clearing of cell growth reflects cells with Lpt-dependent OM-permeability defects that encounter a concentration of antibiotic that is sub-lethal but high enough to slow cell growth. Bac refers to bacitracin, Nov refers to novobiocin, Ery refers to erythromycin and Rif refers to rifampicin. Data shown are representative of at least three independent experiments.
ABC transporters share a conserved structure composed of two sets of domains, the nucleotide-binding domains (NBDs) and transmembrane domains (TMDs) (Davidson et al., 2008, Locher, 2016, Hollenstein et al., 2007). The NBDs are responsible for binding and hydrolyzing ATP, whereas the TMDs directly interact with the substrate to transport it. The ATP-driven movement of the NBDs is coupled to the TMDs via direct protein-protein interactions. Each NBD contains a groove region that interacts with a short helix, known as the coupling helix, in one of the TMDs. In LptB2FGC, ATP binding and hydrolysis by LptB2 (NBDs) control movements of the LptFG (TMDs) through these direct interactions in order to extract LPS and reset the transporter to the resting state (Simpson et al., 2016, Sherman et al., 2014, Tang et al., 2019, Li et al., 2019). In addition, recent structural studies have revealed that a lateral gate located between transmembrane (TM) 1 of LptG and TM5 of LptF allows LPS molecules to enter from the outer leaflet of the IM into a V-shaped cavity formed by the TM helices of LptFGC (Owens et al., 2019). The bottom of this cavity is hydrophobic and interacts with the acyl chains of LPS, while charged residues at the periplasmic rim of the cavity interact with the phosphates and glucosamines of lipid A (Tang et al., 2019, Li et al., 2019, Bertani et al., 2018). The elucidation of the LptB2FGC complex structure also revealed that the single TM helix of the bitopic protein LptC is part of this proposed gate, as it is sandwiched between TM1 and TM5 of LptG and LptF, respectively (Li et al., 2019, Owens et al., 2019, Tang et al., 2019). The presence of this additional helix is unprecedented in ABC transporters, and its functional role in vivo is unclear, as well as how it enters and exits the transporter during the cycle of ATP hydrolysis.
Recent genetic and biochemical studies have largely clarified how the ATP cycle (i. e. ATP binding and hydrolysis) of LptB is connected to the cycle that the LptFG complex undergoes to transport LPS from the IM to the Lpt periplasmic bridge (Simpson et al., 2019, Owens et al., 2019). Briefly, the transporter NBDs (LptB dimer) and TDMs (LptFG) are in an open conformation when there is no nucleotide bound (apo state). LPS can enter the open V-shaped cavity of the transporter in a nucleotide-independent manner (Owens et al., 2019). Then, ATP binding causes the closure of the LptB dimer around two ATP molecules and the concomitant closure or collapse of the LptFG cavity, propelling LPS onto the periplasmic bridge. Subsequent ATP hydrolysis causes the opening of the LptB dimer, resetting the transporter back to its open conformation (Simpson et al., 2019, Owens et al., 2019). Structural studies are in agreement with this proposed model (Li et al., 2019, Tang et al., 2019). Despite having identified the site of interaction between LptB2 (i. e. groove region) and LptFG (i. e. coupling helices) and the general steps of the transport cycle, few mechanistic details of LPS transport are understood. In fact, the precise mechanism of coupling ATP binding and hydrolysis to transport of substrate is still unclear for ABC transporters (Seeger, 2018).
To better understand how LptB’s function is coupled to transport of LPS by LptFG, we further characterized the groove region of LptB that accommodates the coupling helices of LptFG. Here, we show that residue E86 of LptB is essential for coupling LptB’s function to LPS transport. Through suppressor selections, we found that defective lptB(E86) alleles can be suppressed by reducing the acylation of lipid A, or altering residues in TM helices of the TMD LptG. These suppressors also alleviated defects conferred by a substitution in the coupling helix of LptF, but did not suppress the equivalent substitution in the coupling helix of LptG. From these results, we propose that NBD-TMD coupling in LptB2FGC is bi-directional and affected by substrate binding. Our data also suggest that LPS extraction results from the concerted movement of LptFG’s TM helices, and the coupling helix in LptF is specifically involved in this step.
RESULTS
Q-loop residue E86 of LptB is essential for LPS transport
In order to gain insight into how the function of the LptB ATPase is coupled to that of its TMD partners, LptF and LptG, we focused this study on the groove region of LptB (Fig. 1B). This groove region is located at the LptF- and LptG-LptB interfaces, accommodates the LptF/G coupling helices, and contains LptB’s Q loop, a motif that is conserved in NBDs of ABC transporters (Simpson et al., 2016, Luo et al., 2017). The coupling helices and Q loop are thought to be essential for coupling NBD-TMD function in ABC transporters (Davidson et al., 2008). Supporting the essential role of the groove region, we previously identified a cluster of three groove residues (F90, L93, and R150) that is critical for LPS transport (Fig. 1B–C) (Simpson et al., 2016, Sherman et al., 2014). Here, we focused on the uncharacterized Q-loop residue glutamate 86 (E86) because: 1) it is conserved among distantly related LptB orthologues but not in NBDs of other ABC transporters (Fig. S2A–B), suggesting it might be specifically important for the function of LptB2FGC; 2) the side of chain of E86 points up towards LptFG and shows direct contacts with their coupling helices in several structures (Fig. S2C–D); and, 3) E86 is adjacent to the eponymous glutamine (Q85 in LptB) in the Q loop, which interacts with Mg2+ and the γ–phosphate of ATP at the ATP-binding sites in ABC transporters (Sherman et al., 2014, Oswald et al., 2006, Schmitt et al., 2003, Karpowich et al., 2001, Hollenstein et al., 2007). To investigate if E86 is important for function, we generated several plasmid-born lptB alleles encoding changes at residue E86 and tested their ability to complement an lptB chromosomal deletion. As previously described, cells with fully functional Lpt machinery exhibit wild-type growth and resistance to hydrophobic antibiotics; as Lpt function decreases, cells become increasingly sensitive to hydrophobic antibiotics and, eventually, are unable to grow; in addition, severely defective mutants can be conditional lethal, as they are viable in slow-growth conditions (i.e. minimal medium), but not in fast-growth conditions (i.e. LB medium) (Yao et al., 2012).
We found that residue E86 is essential for LptB function and not folding, since substituting it with alanine (LptBE86A) abolished its ability to complement the loss of wild-type LptB (LptBWT) without affecting protein levels (Fig. 1B–C and S3A). In addition, we determined that the negative charge and polarity of E86 are crucial for Lpt function: a conservative substitution retaining the negative charge (LptBE86D) did not affect function; reversing its charge to positive abolished function, as lptB(E86R) haploid cells could not survive; and, a structurally conservative substitution eliminating the negative charge while conserving polarity (LptBE86Q) partially reduced function, as lptB(E86Q) haploid cells were viable but sensitive to antibiotics (Fig. 1C–D). These results suggest that the negative charge of E86 in LptB mediates contacts that are essential for the function of the LptB2FGC transporter. Some of the structures solved to date show residue E86 interacting with combinations of the backbone of Q-loop residues Q85 and S88 in LptB (PDB 6MIT, 5X5Y), and the sidechains of coupling-helix residues S83 in LptF and S87 in LptG (PDB 6MI8, 6S8G) (Owens et al., 2019, Li et al., 2019, Luo et al., 2017). The sidechain of E86 is generally oriented towards the coupling helices, but we did not observe a consistent pattern of contacts even when comparing structures in the same step of the transport cycle. We changed S83 in LptF and/or S87 in LptG to alanine and saw no defects in LPS transport (Simpson et al., 2016). Therefore, although we cannot rule out other interactions not revealed by structures, it is likely that the essential role of E86 stems from its interactions with the backbones of Q85 and S88. Such interactions could be important for maintaining the proper structure of the Q loop and consequently proper NBD-TMD coupling.
Changes to Q-loop residue E86 affect the ATP-dependent closure of the LptB dimer
The NBDs of ABC transporters cycle between a closed- and open-dimer state driven by ATP binding and hydrolysis, respectively (Hollenstein et al., 2007). Recently, we identified two types of substitutions in LptB that have opposite effects on the ATP-driven dimerization cycle that this ATPase must undergo to transport LPS. Specifically, dimers of LptBR144H favor the open-dimer state and are defective in ATP binding and dimer closing, while LptBF239A dimers favor the closed-dimer state and are defective in ATP hydrolysis and dimer opening (Simpson et al., 2019). Residue R144 is located in the signature helix of LptB, which constitutes part of the bottom of the groove that accommodates the LptF/G coupling helices (Simpson et al., 2019, Simpson et al., 2016). In addition, the signature helix immediately follows the signature motif, which constitutes one half of each ATP-binding site in ABC transporters (Schmees et al., 1999, Hoof T, 1994, Bakos et al., 1997, Shyamala et al., 1991). Residue F239 is localized in an essential C-terminal domain (CTD) that is unique to LptB (Simpson et al., 2019). Remarkably, although neither LptBR144H nor LptBF239A can sustain viability on their own, cells only producing LptBR144H/F239A behave like wild type. Thus, each of these substitutions suppresses the other’s lethal effect because they exert opposite effects that compensate each other in the LptB dimerization cycle. We also discovered other ways to suppress the effects of the R144H substitution such as the addition of an E-His8 tag to the CTD of LptB, and sub-lethal concentrations of the antibiotic novobiocin (Fig. 2A) (Simpson et al., 2019, May et al., 2017). We note that although the primary target of novobiocin is DNA gyrase, this antibiotic also binds to one edge of the LptB groove region, increasing LPS transport even in wild-type complexes (May et al., 2017). How novobiocin suppresses R144H is still under investigation.
Figure 2: Both novobiocin and changes to the CTD of LptB suppress permeability defects conferred by lptB(E86Q).

A) Table summarizing previously reported phenotypes of haploid strains carrying the lptB(R144H), lptB-EHis8 (encoding His8-tag at C terminus) or lptB/(R144H)-EHis8 alleles (Simpson et al., 2019). Permeability defects refers to increased sensitivity to antibiotics resulting from defective LPS transport. B) Sublethal concentrations of novobiocin decrease the OM permeability of haploid lptB(E86Q) mutants in a dose-dependent manner. OM permeability of lptB+ and lptB(E86Q) was measured by sensitivity to bacitracin via disc diffusion assay on LB containing increasing, but sublethal, concentrations of novobiocin. The clear zone (dark bars) refers to the diameter around the bacitracin disc without any visible growth, while hazy zone (light bars) refers to partial inhibition of growth surrounding the disc. The dashed line indicates the diameter of the antibiotic disc. C) Combining lptB(E86Q) and lptB-EHis8 results in suppression of both single mutant alleles. Disc diffusion assay to bacitracin was performed on LB. The haploid lptB(R144H) mutant is not viable on LB and is indicated by the asterisk. WT refers to strain NR4446. Data in (B) and (C) represent the average of three independent experiments with error bars showing the standard deviation. No error bar indicates a standard deviation value of 0.
Here, we found that lptB(E86Q) haploid mutants are sensitive to several hydrophobic antibiotics, yet are as resistant to novobiocin as wild type (Fig. 1D). This finding made us wonder whether the effects of the E86Q substitution resembled those conferred by the aforementioned R144H change, albeit to lesser extent (May et al., 2017). To explore this further, we first tested whether novobiocin affected the OM permeability of lptB(E86Q) cells. As we previously reported for haploid cells carrying lptB(R144H-EHis8), growing lptB(E86Q) cells in LB medium containing sub-lethal concentrations of novobiocin decreased sensitivity to other hydrophobic antibiotics such as bacitracin in a dose-dependent manner (Fig. 2B) (May et al., 2017). These results demonstrate that novobiocin suppresses defects conferred by lptB(E86Q). Furthermore, we also found that novobiocin suppressed the lethality of the stronger lptB(E86A) allele when added to minimal (slow-growth) but not LB (fast-growth) media. Thus, together, novobiocin and slow growth allow the survival of lptB(E86A) haploid mutants.
Next, we tested for co-suppression between mutations that alter the CTD of LptB (i. e. introducing a C-terminal E-His8 tag or the F239A substitution) and the lptB(E86A) or lptB(E86Q) alleles, as we have reported to exist between CTD changes and lptB(R144H). On their own, lptB-EHis8 and lptB(F239A) are partial and total loss-of-function alleles, respectively, and we found that neither CTD alteration could suppress the lethality conferred by lptB(E86A) (Simpson et al., 2019). However, the E86Q change suppressed the lethality of lptB(F239A), and the lptB(E86Q)-EHis8 double mutant was less sensitive to hydrophobic antibiotics than both of its single mutant parents, demonstrating co-suppression between the two mutations (Fig. 2C).
Finding that changes to E86 and R144 result in similar, specific phenotypes suggests that they affect LptB function similarly, albeit to different extents, with the E86Q change causing milder defects than the R144H change, and the E86A substitution causing the most severe defects. This further suggests that changes to E86, like the R144H change, decrease the ability of LptB to attain the closed dimer conformation, which is the step proposed to catalyze LPS transfer from the LptFG cavity to the periplasmic Lpt bridge. Since residue R144 makes polar contacts with the backbone of Q-loop residue P84 in the ATP-bound LptB dimer (PDB 6MBN), it is possible that the positioning of the Q loop is affected similarly by disrupting either the P84-R144 interaction or the aforementioned interactions between E86 and Q-loop residues Q85 and S88 (Simpson et al., 2019).
Removal of an acyl chain in LPS bypasses the essential function of E86 in LptB
To better understand the role of residue E86 in LptB, we selected for suppressors of the lethality caused by lptB(E86A). For this selection, we used a strain harboring a chromosomal lptB deletion, a stable plasmid encoding lptB(E86A), and a partition-defective mini-F plasmid encoding wild-type lptB and lacZY under the control of an IPTG-inducible promoter (Fig. 3A). Growth of this strain is IPTG-dependent (to produce LptBWT), and many cells in the population die when they randomly lose the partition-defective mini-F plasmid encoding wild-type lptB. We first applied a selection by growing this strain in rich medium (LB) without IPTG overnight in liquid culture. Then, to screen out mutants that might have acquired mutations that rendered transcription from the promoter for lptB and lacZY IPTG-independent, we plated a portion of the overnight culture on LB plates containing the LacZ indicator X-gal. Desired suppressors formed white colonies, since they would have lost the partition-defective plasmid encoding wild-type lptB and lacZY. Following this approach, we isolated a suppressor mutant that exhibited near wild-type growth in LB in the absence of wild-type lptB. In this mutant, the suppressor mutation is a missense mutation that changes the histidine at position 195 in LpxM to an aspartate. In the Raetz pathway, LpxM is a myristoyltransferase that adds the sixth and final acyl chain onto lipid A (Fig. 3B and S1) (Raetz and Whitfield, 2002, Clementz et al., 1997). We predicted that the H195D change would abolish LpxM function, since H195 is the invariant histidine in the HX4D/E catalytic site (Dovala et al., 2016). In agreement, a chromosomal lpxM deletion allele (ΔlpxM) suppressed lptB(E86A) to the same extent as lpxM(H195D) (Fig. S4). Therefore, to eliminate the possibility of reversion of lpxM(H195D), we proceeded our studies with the ΔlpxM allele.
Figure 3: ΔlpxM suppresses lptB(E86A) and lptB(R144H).

A) Workflow for selecting and identifying suppressors of lptB(E86A) as indicated in the text. B) The structure of LPS with its three major components labeled. The R3″ acyl chain added onto lipid A by LpxM is colored red. C) ΔlpxM suppresses both lptB(E86A) and lptB(R144H). OM permeability was measured by sensitivity to bacitracin (right) and rifampicin (left) via disc diffusion assay on LB plates. The dashed line represents the diameter of the antibiotic discs. The diameter of complete clearing (dark bars) and partial clearing (light bars) around the antibiotic disc is shown. Asterisks indicate that strains are unable to grow on LB. Data represent the average of three independent experiments with the error bars showing the standard deviation. No error bar indicates a standard deviation value of 0. WT refers to strain NR4446.
The loss of LpxM does not alter LPS levels (Fig. S3D), but lpxM mutants produce penta-acylated LPS lacking the R3″ secondary myristoyl group (C14:0) at position 3′ of the distal glucosamine in lipid A (Vorachek-Warren et al., 2002a). On its own, ΔlpxM conferred some sensitivity to hydrophobic antibiotics (Fig. 3C) likely resulting from a decrease in LPS translocation across the IM by the MsbA flippase, which prefers fully acylated LPS as substrate (Zhou et al., 1998, Clementz et al., 1997). Strikingly, analysis of the OM permeability of the suppressor revealed that not only did ΔlpxM allow lptB(E86A) to complement, but it negated nearly all defects conferred by the E86A change, since the OM-permeability defects of the ΔlpxM lptB(E86A) double mutant were only slightly higher than those of the ΔlpxM single mutant (Fig. 3C). Thus, removing the R3″ acyl chain in LPS bypasses the essential role of E86 in LptB. We must note, however, that the lethality conferred by lptB(E86R) was not suppressed by ΔlpxM, suggesting that the charge reversal causes additional defects to those conferred by the loss of the negative charge of E86
The essential role of E86 in LptB is dependent on the presence of fully acylated LPS
To investigate whether suppression of lptB(E86A) was specific to the loss of the R3” acyl chain or if other changes to the lipid A portion of LPS could also suppress lptB(E86A), we built strains lacking one or more of the non-essential acyltransferases in the Raetz pathway. LpxL is the acyltransferase that normally acts immediately upstream of LpxM by adding a lauroyl group (C12:0) to the 2′-hydroxymyristoyl group in KDO2-lipid IVA (Fig. S1 and S5) (Clementz et al., 1996). The LpxP acyltransferase also acts on the 2′-hydroxymyristoyl group in KDO2-lipid IVA, but it adds a palmitoleoyl group (C16:1). LpxP is normally only active at low temperatures (12oC) to increase membrane fluidity but, when LpxL is absent, LpxP can also function at higher temperatures, albeit at a low rate (Fig. S5) (Vorachek-Warren et al., 2002b, Vorachek-Warren et al., 2002a). In addition, in the absence of LpxL, LpxM can acylate both KDO-lipid IVA (at a low rate) and LpxP-modified penta-acylated lipid A (Fig. S5); as such, lpxL mutants produce a mixture of tetra-, penta- and hexa-acylated LPS (Figs. 4A and S5). Lastly, we should also note that mutants lacking these acyltransferases display sensitivity to hydrophobic antibiotics to various extents because translocation of LPS across the IM by MsbA is only optimal for fully hexa-acylated LPS (Zhou et al., 1998).
Figure 4: Decreasing acylation of lipid A suppresses lptB(E86) alleles.

A) Table listing combinations of lpx del. No error bar indicates a standard deviation value of 0.etion alleles, their resulting structure of lipid A as shown by Vorachek-warren, Ramirez et al. (2002), and their phenotype when combined with lptB+ and lptB(E86A) alleles. Except for as noted for the ΔlpxL lptB(E86A) mutant, all phenotypes were observed when growing cells in LB. The length and saturation of the acyl chains at positions R3″ and R2″ are listed. A dash indicates no acylation at that position. Plasmid borne lptB+ and lptB(E86A) alleles were assessed for their ability to complement chromosomal ΔlptB in the context of all combinations of lpx deletions. WT refers to wild-type lpx alleles and phenotypes. Permeability defects refers to an increase in antibiotic sensitivity that results from reduced LPS transport and is qualitatively shown with plus signs. The double mutant ΔlpxL lptB(E86A) is a conditional loss-of-function allele that cannot support viability in LB. B) OM permeability of haploid strains carrying lptB+ (orange bars) or lptB(E86Q) (purple bars) alleles combined with lpx deletions was measured via disc diffusion assay. Sensitivity to bacitracin is used as a representative of all antibiotics tested. Clear zones around the discs are denoted by the dark bars, hazy zones are denoted by the light bars of the corresponding color. Dashed line shows the diameter of the antibiotic disc. Data shown are the average of three independent experiments. Error bars show the standard deviation. Strains with a standard deviation of 0 do not have error bars.
When testing for suppression of lptB(E86A) in various acyltransferase mutants, we found a correlation between suppression and a decrease in levels of hexa-acylated LPS (Fig. 4A and S6A). The ΔlpxL lptB(E86A) mutant, which produces a mixture of tetra-, penta- and hexa-acylated LPS, could only survive under slow-growth conditions (minimal medium) at 37oC, but removing an additional acyltransferase fully suppressed lptB(E86A). We therefore could obtain lptB(E86A) haploid mutants in LB at 37oC, as long as they did not produce hexa-acylated LPS. Moreover, these suppressor mutants lacking hexa-acylated LPS behaved like their lptB+ counterparts with respect to growth and OM permeability (Fig. 4A and S6). The OM-permeability defects conferred by the milder lptB(E86Q) allele could also be suppressed by any allele that decreased (ΔlpxL) or abolished (ΔlpxM, ΔlpxMP, ΔlpxLM, ΔlpxLP, ΔlpxLMP) the production of hexa-acylated LPS (Fig. 4B). As expected, the ΔlpxP allele, which should not alter LPS structure at 37oC, could not suppress either lptB(E86A) or even the milder lptB(E86Q) allele (Fig. 4) (Vorachek-Warren et al., 2002b, Vorachek-Warren et al., 2002a). Thus, the total loss of the R2” and/or R3” chains of LPS suppresses the essential function of E86 in LptB.
The fact that altering the structure of the substrate suppresses defects in LptB2FGC complexes containing LptBE86A of LptBE86Q suggests that LptB residue E86 is essential in a step(s) in the transport cycle in which LPS interacts with the ABC transporter. This would be in agreement with our earlier proposal that changes to E86, like the R144H change, decrease the ability of the LptB dimer to close upon ATP binding, the step in which LPS moves from the LptFG cavity to the periplasmic Lpt bridge. To further probe if changes to residue E86 and the R144H substitution indeed result in similar defects, we tested whether the aforementioned lpx alleles also suppressed defects in LptFGC complexes containing LptBR144H. We found that they did (Fig. 3C and S6B). Since LptBR144H is defective in the dimerization step that triggers the collapse of the LptFG cavity to squeeze out LPS and transfer it to the Lpt periplasmic bridge, our genetic data suggest that reducing the overall size of the hydrophobic region of LPS facilitates LptB dimerization and the collapse of the LptFG cavity that leads to LPS extraction (Simpson et al., 2019). Thus, interactions between the LptFG cavity with its lipid A substrate modulate the function of the LptB ATPase. This implies that NBD-TMD coupling between LptB and LptFG is bidirectional, as suggested for other ABC transporters (Khare et al., 2009, Oldham et al., 2008). Furthermore, these data support a role of Q-loop residue E86 in NBD-TMD coupling, which is in agreement with the proposed function for the Q loop in ABC transporters (Sherman et al., 2014, Oswald et al., 2006, Schmitt et al., 2003, Hollenstein et al., 2007).
ΔlpxM is not a general suppressor of Lpt defects but specifically suppresses defects in LptB-LptF coupling
We reasoned that if, as suggested above, reducing the overall size of the hydrophobic region of lipid A suppresses lptB(E86A) and lptB(R144H) by facilitating the closure of the LptFG cavity and thereby LptB dimerization, then lpx mutations would not be general suppressors of all types of Lpt defects. Instead, they would specifically suppress defects in ATP binding (i. e. LptB dimerization) and coupling between LptB-LptFG. To test their specificity of suppression, we first introduced ΔlpxM into strains with defects in the OM LptDE translocon (lptD4213) and binding of LPS to the LptFG cavity (lptGK34A). Haploid strains producing LptD4213 are highly permeable to hydrophobic antibiotics because they have a reduced number of active Lpt OM translocons owing to an assembly defect of the β–barrel domain of LptD4213 (Sampson et al., 1989, Braun and Silhavy, 2002, Lee et al., 2016). We observed that introducing the ΔlpxM allele did not have an effect on lptD4213 cells (Fig. 5A). LptG residue K34 is localized within the LptFG cavity and contacts the 1-phosphate of lipid A once LPS enters the LptFGC cavity. Therefore, removing the positive charge at this position leads to LPS transport defects (Bertani et al., 2018). We found that introducing ΔlpxM into lptG(K34A) cells led to an increase in OM permeability that appeared additive (Fig. 5B). These results demonstrate that ΔlpxM is not a general suppressor of Lpt defects.
Figure 5: ΔlpxM is not a general suppressor of lpt defects.

A) ΔlpxM cannot suppress defects in the OM translocon caused by the lptD4213 allele. NR754 was used as the parent strain with wild-type lptD and lpxM alleles. B) ΔlpxM cannot suppress defects in LPS-LptG binding conferred by the lptG(K34A) allele. Haploid strains containing either pBAD18LptFG3 or pBAD18LptFG3/LptG/K34A were combined with lpx deletion alleles. NR2761 was used as the parent strain with wild-type lptG and lpxM alleles. Suppression in panels A and B was assessed by changes in OM permeability as measured by sensitivity to bacitracin. Zones of complete clearing (dark bars) and partial clearing (light bars) around the disc were measured in mm. The dashed line represents the size of the bacitracin disc. Data shown are the average of three replicates with the error bars showing the standard deviation. No error bar indicates a standard deviation value of 0. C) ΔlpxM cannot suppress lptB-EHis8, and the two alleles are a synthetically lethal pair. Plasmid-borne LptB-EHis8 does not complement chromosomal ΔlptB in the presence of ΔlpxM. OM permeability defects are indicated qualitatively by the plus signs and are indicative of decreased LPS transport.
Since our results are consistent with a model in which lptB(E86A) and lptB(R144H) cause defects in the coupling of LptB to its partners LptFG, we next tested whether the suppression by ΔlpxM also extends to defects in the coupling helices of LptFG. Each coupling helix of LptF and LptG interacts with the groove region of LptB (Fig. 6A) and contains a conserved glutamate (residues E84 in LptF and E88 in LptG) that is functionally important since substituting each to alanine results in LPS transport defects (Simpson et al., 2016, Luo et al., 2017). Here, we found that ΔlpxM suppressed the OM-permeability defects of the lptF(E84A) mutant, but not those caused by the lptG(E88A) mutant Fig. 6B). Next, we tested the effects of ΔlpxM in the lptF(E84A) lptG(E88A) double mutant, whose viability is conditional so that it can grow in minimal media (i.e. slow growth) but not in LB (i.e. fast growth) (Simpson et al., 2016). Introducing the ΔlpxM allele restored growth of lptF(E84A) lptG(E88A) cells in LB. Furthermore, the OM permeability defects of the ΔlpxM lptF(E84A) lptG(E88A) mutant were equivalent to those of the ΔlpxM lptG(E88A) mutant, indicating that ΔlpxM only abolished the defects conferred by lptF(E84A) (Fig. 6B). Together, these analyses show that ΔlpxM suppresses defects caused by changes to the conserved glutamate in the coupling helix of LptF, but not the equivalent change in the coupling helix of LptG. Notably, a cryo-EM structure of the E. coli LptB2FG complex bound to LPS shows that the R3″ chain of LPS, which is added by LpxM, interacts with LptF (PDB 6MHU) (Li et al., 2019). Therefore, we propose that LptFGC complexes containing either LptBE86A or LptBR144H are defective in LptB-LptF coupling and likely cannot properly induce the necessary movement of the LptF cavity TM helices that are required to close it in order to squeeze out LPS; somehow, reducing the number of acyl chains in lipid A restores cavity collapse.
Figure 6: ΔlpxM suppress defects in the coupling helix of LptF.

A) Top: Top-down view from the membrane of the cartoon rendition of the Vibrio cholerae LptB2FGC structure (PDB 6MJP) cut away showing only the coupling helices of LptF (green) and LptG (light blue) and the LptB dimer. Residues E84 of LptF, E88 of LptG, and E86 of LptB are shown as spheres. Bottom: Drawing of the top-down view of the LptB dimer with the coupling helices (CH) pictured. ATP is bound between the signature (Sig) and Walker A (WA) motifs. B) ΔlpxM suppresses defects conferred by lptF(E84A) but not lptG(E88A). Plasmid-borne lptFG alleles were assessed for their ability to complement chromosomal ΔlptFG alleles in strains with chromosomal wild-type and deletion alleles of lpxM. OM permeability of viable mutants was assessed by sensitivity to bacitracin as measured by disc diffusion assay on LB plates. The haploid strain harboring the double glutamate mutant lptF(E84A) lptG(E88A) is unable to complement on LB, therefore it is not shown. The dashed line represents the diameter of the bacitracin disc. Data shown are the average of three independent experiments with error bars representing the standard deviation. NR2761 was used as the parent strain with wild-type lptFG and lpxM alleles. An unpaired two-tailed Student’s t-test was used to assess the significance of difference between lptF(E84A) and lptF(E84A) ΔlpxM. * indicates P < 0.05.
Changes in transmembrane helices 1 and 2 of LptG suppress lptB(E86A)
If LptB2FGC complexes containing LptBE86A/Q are defective because their LptFG cavity cannot properly collapse owing to a defect in LptB-LptF coupling and this defect can be suppressed by changing the structure of LPS, we reasoned that we might identify residues in LptFG that are involved in promoting cavity collapse by isolating mutations in lptFG that suppress lptB(E86A). Therefore, we selected for spontaneous suppressor mutations of the lethality caused by lptB(E86A) and focused on those mapping to the lptFG chromosomal locus (Fig. 7A). Using this approach, we identified two independent changes, L74P and A110V, in two residues located in TM helices 2 and 3 of LptG, respectively (Fig. 7B). Specifically, a recent cryo-EM structure of the E. coli LptB2FG complex revealed that residues L74 and A110 are localized to a plane that is normal to the membrane just below where the acyl chains of LPS reside in the LptFG cavity prior to transport (Fig. 7B) (Li et al., 2019).
Figure 7: Changes to TM helices 2 and 3 in LptG suppress lptB(E86A).

A) Strategy to identify lptFG-linked suppressors of lptB(E86A). Tet refers to tetracycline, sup refers to suppressor. B) Cartoon representation of the cryo-EM structure of E. coli LptB2FG in a nucleotide-free, LPS-bound state LPS (PDB 6MHU). Lipid A is shown as magenta spheres with the R3″ and R2″ acyl chains colored orange and blue, respectively. Residues L74 and A110 in LptG are shown as red spheres. C) Substitutions L74P and A110V in LptG suppress lptB(E86A). Sensitivity to bacitracin was measured by disc diffusion assay. The dashed line indicates the diameter of the antibiotic disc. Clear and hazy zones of inhibitions are measured in mm and represented by dark and light bars respectively. The single lptB(E86A) mutant is not viable and is indicated by the asterisk. Data shown are the average of three independent experiments. Error bars represent the standard deviation. No error bar indicates a standard deviation value of 0. Strain NR4831 was used as the parental wild-type lptB lptG strain.
The lptB(E86A) lptG(L74P) and lptB(E86A) lptG(A110V) suppressor strains were viable, but still exhibited some OM-permeability defects (Fig. 7C). We also generated the single lptG(L74P) and lptG(A110V) mutants and observed that while the lptG(L74P) mutant was as resistant as the wild-type strain to bacitracin, the lptG(A110V) mutant showed increased OM permeability reflective of LPS transport defects (Fig. 7C). In addition, we tested the ability of the L74P and A110V changes in LptG to suppress the conditional lethality of lptB(R144H) and the OM-permeability defects caused by lptB(E86Q). While lptG(L74P) suppressed the inability of lptB(R144H) cells to grow in rich LB medium, lptG(A110V) did not (Fig. S7A–B). However, both lptG alleles decreased the OM-permeability defects of lptB(E86Q) cells (Fig. S7C). Thus, lptG(L74P) suppresses LPS transport defects caused by lptB(E86A/Q) and lptB(R144H), but lptG(A110V) can only suppress defects caused by lptB(E86A/Q). Given that structures suggest that TM helices 2 and 3 in LptG undergo significant movement when the LptFG cavity collapses in order to translocate LPS to the Lpt periplasmic bridge (Fig. 8 A–B), and that both the L74P and A110V changes are predicted to lower the stability of α-helices (see Discussion), these changes likely suppress lptB(E86A) by destabilizing TM2 and TM3 helices in LptG, and thus promoting closure of the LptFG cavity (Owens et al., 2019, Li et al., 2019).
Figure 8: Model for LPS extraction from the IM by LptB2FGC.

A) Cartoon representation of the cryo-EM structure of the LPS-bound cavity formed by the TM helices of LptF (green) and LptG (light blue). The structure was generated from E. coli LptB2FG complexes containing nucleotide-free LptB and LPS-bound cavity (PDB 6MHU). Shown is the periplasmic view of the cavity with lipid A represented as magenta sticks with the R3″ and R2″ acyl chains colored blue and orange, respectively. TM helices 1–3 of LptFG are labeled. Residues L74 (TM2) and A110 (TM3) in LptG are shown as red spheres. B) Alignment of structures showing the LptFG cavity region of cryo-EM structures of E. coli LptB2FG complexes in nucleotide-free (LptF in orange and LptG in yellow) and vanadate-trapped (LptF in green and LptG in blue) conformations (PDB 6MHU and 6MHZ, respectively). Pictured is the periplasmic view of the cavity with residues L74 and A110 in LptG shown as spheres colored red in the nucleotide-free structure and cyan in the vanadate bound structure. C) Model for LPS transport by LptB2FG. For simplicity, LptC and the β–jelly roll domains of LptFG are not shown. 1) LPS enters the LptFG cavity when LptB2 is in a nucleotide-free (apo), open-dimer state. 2) LptB2 binds ATP (yellow circles) causing simultaneous closure of the LptB dimer and the LptFG cavity through a rigid-body movement. LPS is transferred to the periplasmic bridge (not shown) 3) After ATP hydrolysis, ADP (red) and Pi (black) are released, resetting the transporter back at step 1. Both LptBE86A and LptFE84A are defective at proceeding through step 2 but can be suppressed by ΔlpxM, LptGA110V or LptGL74P, which increase the rate of step 2.
Combining mutations that suppress lptB(E86A/Q) leads to synthetic lethality
We have reported three types of mutations that suppress defects in LPS transport caused by alterations to residue E86 in LptB. These suppressors result in the addition of a C-terminal –EHis8 tag to LptB, the loss of the R2″ and/or R3″ acyl chains in LPS, and changes to TM2 and TM3 helices in LptG. These different mutations must achieve the same final effect on the LptB2FGC complex, which we hypothesize is to facilitate LptFG cavity closure to move LPS onto the Lpt bridge. However, it is unclear if all three types of mutations suppress through an identical mechanism. If two mutations act through different mechanisms, their effects should be additive when combined. However, when two mutations affect the same mechanism, their effects should be non-additive when combined, resulting in either epistasis or synthetic effects. To investigate if the three types of suppressors acted through the same mechanism, we tried to combine the ΔlpxM, lptB-EHis8, and lptG(L74P) or lptG(A110V) alleles in strains in which E86 remained unchanged. We found that, although the respective single mutants were viable, and in some cases behaved similarly to the wild-type strain, the ΔlpxM lptB-EHis8, and lptB-EHis8 lptG(L74P) combinations were synthetic lethal, and the lptB-EHis8 lptG(A110V), ΔlpxM lptG(L74P), and ΔlpxM lptG(A110V) combinations were synthetic defective (Fig. S8). These synthetic interactions, together with the fact that these mutations suppress the defect caused by changes to E86A in LptB, indicate that these changes to LptB, LptG, and LPS alter the activity of LptB2FGC complex by affecting the same function or step but in different ways that, when combined, break the LPS transport cycle.
Mutations that suppress lptB(E86A/Q) specifically suppresses defects in coupling mediated by LptF
Our observation that lptG(L74P) or lptG(A110V) suppress lptB(E86A) and are synthetic defective when combined with ΔlpxM implied a similar mechanism of suppression. As noted before, ΔlpxM suppresses a defect in the coupling helix of LptF but not in that of LptG. We wondered if the alterations to TM2 and TM3 of LptG would also show a similar pattern of suppression. We therefore introduced mutations resulting in E84A and E88A changes in the coupling helices of LptF and LptG, respectively, in plasmid-borne lptG(L74P) and lptG(A110V) alleles.
The plasmid-based lptG(L74P) and lptG(A110V) single mutants displayed some OM-permeability defects (Fig. S9). Likewise, the coupling helix lptF(E84A) and lptG(E88A) single mutants were also defective (Fig. S9). However, lptF(E84A) and either lptG(L74P) or lptG(A110V) suppressed one another (Fig. S9). In contrast, introducing lptG(E88A) into either lptG(L74P) or lptG(A110V) increased OM-permeability in additive fashion (Fig. S9). We also observed that the inability of the double glutamate mutant lptF(E84A) lptG(E88A) to grow in rich LB media was suppressed by either lptG(L74P) or lptG(A110V) (Fig. S9). Thus, these data demonstrate that, like ΔlpxM, the L74P and A110V changes to TM2 and TM3 of LptG cannot suppress the coupling defect in LptG, but suppress defects with LptF-LptB coupling. Since our genetic data show that the defect in the coupling helix of LptF caused by lptF(E84A) is suppressed by mutations that promote LptFG cavity closure [i. e. ΔlpxM, lptG(L74P), and lptG(A110V)], we propose that the conserved glutamate in the coupling helix of LptF (i. e. residue E84), but not that in LptG (i. e. residue E88), is important for coordinating the collapse of the substrate-binding cavity of the LptB2FGC transporter.
DISCUSSION
ABC transporters constitute a large family of proteins present in all domains of life (Davidson et al., 2008, Hollenstein et al., 2007). Although their overall architecture and ATP dependence is conserved, ABC transporters differ in function, as they can import, export, and flip substrates across, and extract substrates from membranes. Given this diversity in mechanism of transport, it is not surprising that ABC transporters also differ in how they couple the function of NBDs and TMDs. In fact, the molecular mechanisms of NBD-TMD coupling through long distances remain poorly understood. Here, we investigated NBD-TMD coupling in LptB2FGC, an atypical ABC transporter that extracts the glycolipid LPS from the IM of Gram-negative bacteria to load it onto a periplasmic protein complex. We identified a novel residue (E86) localized in the Q loop of LptB that is essential for LPS transport. Based on previous structural work (Owens et al., 2019, Li et al., 2019, Tang et al., 2019), and our functional analyses, we believe that E86 is important in facilitating the proper structure of the Q loop, likely through its contacts with the backbone of the essential Q-loop residue Q85. Genetic suppression analyses revealed that changes either to the structure of the LPS substrate or the TM helices of LptG that form the substrate-binding cavity suppress the lethality caused by an E86A change in LptB.
Changes in LPS structure suppress alterations to E86 in LptB, indicating that this residue’s role in coupling is critical in a step in the transport cycle in which LptB2FGC interacts with its substrate. Changes to cavity TM helices in LptG also suppress defects caused by changes to E86 in LptB. Together, these findings lead us to propose that residue E86 is important in the step in the transport cycle in which LPS is squeezed out of the LptFG cavity, the step that is triggered when the LptB dimer closes upon binding ATP (Simpson et al., 2019). This model is supported by our genetic data, which shows that defects caused by substitutions at residue E86 resemble those caused by the R144H change in the signature helix of LptB. Previously, we demonstrated that LptBR144H dimers are defective in attaining the closed-dimer state that is induced by ATP binding(Simpson et al., 2019). We also showed that these defects can be fixed by promoting closure of the LptB dimer through alterations to the CTD of LptB. Thus, the suppressors altering LPS structure and TM helices in LptG we report here specifically overcome problems caused by the inability of the transporter to properly undergo, upon binding ATP, the coordinated closure of both the LptB dimer and the LPS-binding cavity of LptFG (Fig. 8C). But, how?
For the closure of the LPS-bound LptFG cavity to occur, hydrophobic interactions between lipid A and residues in the LptFG cavity must be replaced by cavity-to-cavity hydrophobic interactions. Therefore, for LPS to move out of the transmembrane cavity, binding affinity of the LptFG cavity to its substrate must decrease while binding affinity between cavity residues increases. Reducing the number of acyl chains by removing LpxM, LpxL, or any combination of the LpxLMP acyltransferases could therefore suppress by reducing the hydrophobic contacts between LPS and residues in LptF. This change in LPS structure would be expected to reduce the energy required for moving LPS out of the cavity, possibly also speeding up this extraction. In addition, structures of the LptB2FGC complex also suggest that TM helices 1–3 of each LptF and LptG must move towards the center of the cavity to collapse the substrate-binding site and thereby cause LPS translocation (Li et al., 2019, Tang et al., 2019). Movement of these helices are thought to be coupled with the ATP-dependent closure of the LptB dimer through the aforementioned rigid-body coupling mechanism involving interactions between the groove of LptB, and the coupling helices of LptF and LptG. Notably, the coupling helices are localized between TM helices 2 and 3 in LptF and LptG. We therefore propose that the L74P and A110V substitutions in LptG alter the structure of TM helices 2 and 3, respectively, so that they favor the inward movement required for cavity collapse. By reducing the energy required for the transition of the LPS-binding cavity from the open to the closed state, these suppressors promote the concomitant LPS extraction in the defective coupling mutants.
Together, our suppressor data provide the first in vivo evidence supporting a model in which coupling between NBDs-TMDs in the LptB2FGC transporter is bi-directional, as expected if movement of the LptB dimer and the LptFG cavity were coupled through rigid-body mechanism. Furthermore, the fact that changes in two different TM helices of LptG or in LPS structure facilitate the collapse of the substrate-binding cavity also supports the idea that this collapse results from the concerted movement of TM helices and substrate. Additionally, since all of the suppressors we have identified in our study also fix defects conferred by altering the conserved glutamate in the coupling helix of LptF, but not those resulting from the equivalent change in the coupling helix of LptG, our results strongly suggest that the glutamate in the coupling helix of LptF, and not that in LptG, is necessary for properly promoting the closure of the LptFG cavity that leads to LPS movement to the Lpt bridge.
EXPERIMENTAL PROCEDURES
Strains and growth conditions
For a list of strains used in this study refer to Table S1. Strains were grown at 37°C unless otherwise noted in either LB or M63 supplemented with 0.2% (wt/vol) glucose. Liquid cultures were grown with aeration or on 1.5% agar plates. When appropriate, growth media were supplemented with ampicillin (125 μg/mL), chloramphenicol (20 μg/mL), novobiocin (5 μg/mL), kanamycin (30 μg/mL), tetracycline (25 μg/mL), 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-Gal, 33 μg/mL), spectinomycin (100 μg/mL), or isopropyl-β-D-1-thiogalactopyranoside (IPTG, 0.16 mM).
Strain construction and analysis of functionality
lpt mutant alleles were constructed by site-directed mutagenesis (Quick change method) using pET21/22LptB or pBAD18LptFG3 plasmid as a template (Simpson et al., 2016, Sherman et al., 2014) using primers listed in Table S2 and PfuTurbo polymerase (Agilent Technologies, Inc.) or Phusion High-Fidelity polymerase (New England Biolabs). Functionality of lpt mutants was assayed via complementation in a pRC7-containing strain as previously described (Simpson et al., 2016, Sherman et al., 2014). Complementation was assessed on LB, M63 minimal media, and LB supplemented with novobiocin. The lpxLMP deletion alleles from the Keio collection (Baba et al., 2006) were introduced to strains via P1vir transduction and selecting for transductants on media supplemented with kanamycin. Removal of the kanamycin-resistance cassette was accomplished using the temperature-sensitive PCP20 plasmid that encodes Flp recombinase as previously described (Cherepanov and Wackernagel, 1995). The lptG(A110V) yjgN::tet and lptG(L74P) yjgN::tet alleles were introduced into strains using P1vir transduction and selecting for transductants on tetracycline (25μg/mL) supplemented LB plates. lptG was PCR amplified and sequenced to confirm the presence of these alleles. For strains with a chromosomal lptFG deletion lptG(A110V) and lptG(L74P) were introduced via site-directed mutagenesis of the pBAD18LptFG3 plasmid as described above.
Antibiotic sensitivity testing
100μl of overnight culture grown in M63 supplemented with 0.2% glucose, or 50ul grown in LB were used for disc diffusion assays as previously described (Sherman et al., 2014). OM permeability was assessed using bacitracin, novobiocin, erythromycin, and rifampicin discs. Data is representative of at least 3 overnight samples. To assess effect of novobiocin on OM permeability, the LB plates (1.5% agar) and LB top agar (0.75% agar) were supplemented with 0.2, 2, and 20 μM of novobiocin.
Suppressor selection and mapping
An overnight culture of NR4607 [lptB(E86A)/pRC7KanLptB merodiploid] was grown in 5 mL of LB supplemented with 0.16 mM IPTG. Cells were washed then streaked on LB agar. White colonies arose from cells that lost pRC7KanLptB and therefore survived because they acquired a suppressor of the total loss of function of lptB(E86A). A suppressor was mapped to lpxM using P1vir co-transduction frequency to tetracycline-resistant markers as previously described (Ruiz et al., 2006). To identify the suppressor mutation, chromosomal lpxM was amplified by PCR and sequenced.
An overnight culture of strain NR4745 [lptB(E86A)/pRC7KanLptB merodiploid], which carries yjgN::tet, a mini-Tntet transposon chromosomal insertion located between bp 34 and 35 of yjgN that has a 83% linkage to the lptFG locus, was grown in LB without IPTG. Cells that grew overnight without IPTG should have acquired suppressor mutations. Lysates were generated from these sup+ tet+ overnight cultures and used to transduce the sup− tet− strain NR4607. Transductants were plated on LB media supplemented with tetracycline, X-gal, and IPTG. All surviving transductants received the yjgN::tet marker that is 83% linked to lptFG. Colonies that were white, and therefore complemented, contained suppressors. The chromosomal lptFG locus was amplified via PCR and sequenced to identify suppressors.
Immunoblotting
Whole cell lysates were prepared from overnight or exponential phase cultures and normalized by cell density as previously described (Sherman et al., 2014). Samples were probed with rabbit anti-LptB (1:10,000 dilution) or mouse anti-LPS (1:10,000 dilution; Bio-Rad 4329–5004) primary antibodies. Following this, exposure to horseradish peroxidase-conjugated secondary antibodies anti-rabbit (1:10,000 dilution; GE Amersham) or anti-mouse (1:10,000 GE Healthcare Life Sciences) were used, respectively. Signal developed using Clarity Western ECL substrate (Bio-Rad) and imaged with Chemidoc CRS+ system. ImageLab 5.2.1 software (Bio-Rad) was used to quantify band intensity in LPS blots.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Rebecca M. Davis for the construction of pET2342LptB(E86A) plasmid and its initial functional study, and members of the Ruiz laboratory for the helpful discussions about this study. This work was supported by the National Institute of General Medical Sciences award GM100951 (to N.R.).
Footnotes
The authors do not have a conflict of interest to declare.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology 2: 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakos E, Klein I, Welker E, Szabo K, Muller M, Sarkadi B, and Varadi A (1997) Characterization of the human multidrug resistance protein containing mutations in the ATP-binding cassette signature region. Biochemical Journal 323 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani B, and Ruiz N (2018) Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani BR, Taylor RJ, Nagy E, Kahne D, and Ruiz N (2018) A cluster of residues in the lipopolysaccharide exporter that selects substrate variants for transport to the outer membrane. Molecular Microbiology 109: 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, and Silhavy TJ (2002) Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Molecular Microbiology 45: 1289–1302. [DOI] [PubMed] [Google Scholar]
- Cherepanov PP, and Wackernagel W (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158: 9–14. [DOI] [PubMed] [Google Scholar]
- Chng SS, Ruiz N, Chimalakonda G, Silhavy TJ, and Kahne D (2010) Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proceedings of the National Academy of Sciences of the United States of America 107: 5363–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz T, Bednarski JJ, and Raetz CR (1996) Function of the htrB high temperature requirement gene of Escherichia coli in the acylation of lipid A: HtrB catalyzed incorporation of laurate. Journal of Biological Chemistry 271: 12095–12102. [DOI] [PubMed] [Google Scholar]
- Clementz T, Zhou Z, and Raetz CR (1997) Function of the Escherichia coli msbB gene, a multicopy suppressor of htrB knockouts, in the acylation of lipid A. Acylation by MsbB follows laurate incorporation by HtrB. Journal of Biological Chemistry 272: 10353–10360. [DOI] [PubMed] [Google Scholar]
- Davidson AL, Dassa E, Orelle C, and Chen J (2008) Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiology and Molecular Biology Reviews 72: 317–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Xiang Q, Gu Y, Wang Z, Paterson NG, Stansfeld PJ, He C, Zhang Y, Wang W, and Dong C (2014) Structural basis for outer membrane lipopolysaccharide insertion. Nature 511: 52–56. [DOI] [PubMed] [Google Scholar]
- Dovala D, Rath CM, Hu Q, Sawyer WS, Shia S, Elling RA, Knapp MS, and Metzger LE (2016) Structure-guided enzymology of the lipid A acyltransferase LpxM reveals a dual activity mechanism. Proceedings of the National Academy of Sciences of the United States of America 113: E6064–E6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freinkman E, Chng SS, and Kahne D (2011) The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proceedings of the National Academy of Sciences of the United States of America 108: 2486–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freinkman E, Okuda S, Ruiz N, and Kahne D (2012) Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry 51: 4800–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K, Dawson RJ, and Locher KP (2007) Structure and mechanism of ABC transporter proteins. Current Opinion in Structural Biology 17: 412–418. [DOI] [PubMed] [Google Scholar]
- Hoof T DA, Hadam MR, Riordan JR, Tummler B. (1994) Cysticfibrosis-type mutational analysis in the ATP-binding cassette transporter signature of human P-glycoprotein MDR1. Journal of Biological Chemistry 269. [PubMed] [Google Scholar]
- Kalynych S, Morona R, and Cygler M (2014) Progress in understanding the assembly process of bacterial O-antigen. FEMS Microbiology Reviews 38: 1048–1065. [DOI] [PubMed] [Google Scholar]
- Karpowich N, Martsinkevich O, Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, and Hunt JF (2001) Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure 9: 571–586. [DOI] [PubMed] [Google Scholar]
- Khare D, Oldham ML, Orelle C, Davidson AL, and Chen J (2009) Alternating access in maltose transporter mediated by rigid-body rotations. Molecular Cell 33: 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klena JD, Ashford RS 2nd, and Schnaitman CA (1992) Role of Escherichia coli K-12 rfa genes and the rfp gene of Shigella dysenteriae 1 in generation of lipopolysaccharide core heterogeneity and attachment of O antigen. Journal of Bacteriology 174: 7297–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Xue M, Wzorek JS, Wu T, Grabowicz M, Gronenberg LS, Sutterlin HA, Davis RM, Ruiz N, Silhavy TJ, and Kahne DE (2016) Characterization of a stalled complex on the beta-barrel assembly machine. Proceedings of the National Academy of Sciences of the United States of America 113: 8717–8722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Orlando BJ, and Liao M (2019) Structural basis of lipopolysaccharide extraction by the LptB2FGC complex. Nature 567: 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher KP (2016) Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nature Structural & Molecular Biology 23: 487–493. [DOI] [PubMed] [Google Scholar]
- Luo Q, Yang X, Yu S, Shi H, Wang K, Xiao L, Zhu G, Sun C, Li T, Li D, Zhang X, Zhou M, and Huang Y (2017) Structural basis for lipopolysaccharide extraction by ABC transporter LptB2FG. Nature Structural & Molecular Biology 24: 469–474. [DOI] [PubMed] [Google Scholar]
- May JM, Owens TW, Mandler MD, Simpson BW, Lazarus MB, Sherman DJ, Davis RM, Okuda S, Massefski W, Ruiz N, and Kahne D (2017) The Antibiotic Novobiocin Binds and Activates the ATPase That Powers Lipopolysaccharide Transport. Journal of the American Chemical Society 139: 17221–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W, Li Y, Yoon SH, Ernst RK, Walz T, and Liao M (2017) Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 549: 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiology and Molecular Biology Reviews 67: 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Freinkman E, and Kahne D (2012) Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science 338: 1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, and Kahne D (2016) Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nature Reviews: Microbiology 14: 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Davidson AL, and Chen J (2008) Structural insights into ABC transporter mechanism. Current Opinion in Structural Biology 18: 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C, Holland IB, and Schmitt L (2006) The motor domains of ABC-transporters. What can structures tell us? Naunyn-Schmiedebergs Archives of Pharmacology 372: 385–399. [DOI] [PubMed] [Google Scholar]
- Owens TW, Taylor RJ, Pahil KS, Bertani BR, Ruiz N, Kruse AC, and Kahne D (2019) Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature 567: 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S, Luo Q, Zhao Y, Zhang XC, and Huang Y (2014) Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 511: 108–111. [DOI] [PubMed] [Google Scholar]
- Raetz CR, and Whitfield C (2002) Lipopolysaccharide endotoxins. Annual Review of Biochemistry 71: 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N, Gronenberg LS, Kahne D, and Silhavy TJ (2008) Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 105: 5537–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N, Wu T, Kahne D, and Silhavy TJ (2006) Probing the barrier function of the outer membrane with chemical conditionality. ACS Chemical Biology 1: 385–395. [DOI] [PubMed] [Google Scholar]
- Sampson BA, Misra R, and Benson SA (1989) Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmees G, Stein A, Hunke S, Landmesser H, and Schneider E (1999) Functional consequences of mutations in the conserved ‘signature sequence’ of the ATP-binding-cassette protein MalK. European Journal of Biochemistry 266: 420–430. [DOI] [PubMed] [Google Scholar]
- Schmitt L, Benabdelhak H, Blight MA, Holland IB, and Stubbs MT (2003) Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. Journal of Molecular Biology 330: 333–342. [DOI] [PubMed] [Google Scholar]
- Seeger MA (2018) Membrane transporter research in times of countless structures. Biochimica et Biophysica Acta - Biomembranes 1860: 804–808. [DOI] [PubMed] [Google Scholar]
- Sherman DJ, Lazarus MB, Murphy L, Liu C, Walker S, Ruiz N, and Kahne D (2014) Decoupling catalytic activity from biological function of the ATPase that powers lipopolysaccharide transport. Proceedings of the National Academy of Sciences of the United States of America 111: 4982–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DJ, Xie R, Taylor RJ, George AH, Okuda S, Foster PJ, Needleman DJ, and Kahne D (2018) Lipopolysaccharide is transported to the cell surface by a membrane-to-membrane protein bridge. Science 359: 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyamala V, Baichwal V, Beall E, and Ames GF (1991) Structure-function analysis of the histidine permease and comparison with cystic fibrosis mutations. Journal of Biological Chemistry 266: 18714–18719. [PubMed] [Google Scholar]
- Silhavy TJ, Kahne D, and Walker S (2010) The bacterial cell envelope. Cold Spring Harbor Perspectives in Biology 2: a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson BW, Owens TW, Orabella MJ, Davis RM, May JM, Trauger SA, Kahne D, and Ruiz N (2016) Identification of Residues in the Lipopolysaccharide ABC Transporter That Coordinate ATPase Activity with Extractor Function. mBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson BW, Pahil KS, Owens TW, Lundstedt EA, Davis RM, Kahne D, and Ruiz N (2019) Combining Mutations That Inhibit Two Distinct Steps of the ATP Hydrolysis Cycle Restores Wild-Type Function in the Lipopolysaccharide Transporter and Shows that ATP Binding Triggers Transport. mBio 10: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandeo P, Cescutti R, Villa R, Di Benedetto C, Candia D, Deho G, and Polissi A (2007) Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. Journal of Bacteriology 189: 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandeo P, Lau FK, Carpentieri A, De Castro C, Molinaro A, Deho G, Silhavy TJ, and Polissi A (2008) Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. Journal of Bacteriology 190: 4460–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suits MD, Sperandeo P, Deho G, Polissi A, and Jia Z (2008) Novel structure of the conserved gram-negative lipopolysaccharide transport protein A and mutagenesis analysis. Journal of Molecular Biology 380: 476–488. [DOI] [PubMed] [Google Scholar]
- Tang X, Chang S, Luo Q, Zhang Z, Qiao W, Xu C, Zhang C, Niu Y, Yang W, Wang T, Zhang Z, Zhu X, Wei X, Dong C, Zhang X, and Dong H (2019) Cryo-EM structures of lipopolysaccharide transporter LptB2FGC in lipopolysaccharide or AMP-PNP-bound states reveal its transport mechanism. Nature Communications 10: 4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AX, Dong C, and Whitfield C (2010) Structure and functional analysis of LptC, a conserved membrane protein involved in the lipopolysaccharide export pathway in Escherichia coli. Journal of Biological Chemistry 285: 33529–33539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorachek-Warren MK, Carty SM, Lin S, Cotter RJ, and Raetz CR (2002a) An Escherichia coli mutant lacking the cold shock-induced palmitoleoyltransferase of lipid A biosynthesis: absence of unsaturated acyl chains and antibiotic hypersensitivity at 12 degrees C. Journal of Biological Chemistry 277: 14186–14193. [DOI] [PubMed] [Google Scholar]
- Vorachek-Warren MK, Ramirez S, Cotter RJ, and Raetz CR (2002b) A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. Journal of Biological Chemistry 277: 14194–14205. [DOI] [PubMed] [Google Scholar]
- Yao Z, Davis RM, Kishony R, Kahne D, and Ruiz N (2012) Regulation of cell size in response to nutrient availability by fatty acid biosynthesis in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 109: E2561–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, White KA, Polissi A, Georgopoulos C, and Raetz CR (1998) Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. Journal of Biological Chemistry 273: 12466–12475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.