

Abstract
Background:
Combined angiotensin receptor/neprilysin inhibition with sacubitril/valsartan has emerged as a therapy for heart failure (HF). The presumed mechanism of benefit is through prevention of natriuretic peptide (NP) degradation, leading to increased cGMP-dependent protein kinase (PKG) signaling. However, the specific requirement of PKG for sacubitril/valsartan effects remains untested.
Methods and Results:
We examined sacubitril/valsartan treatment in mice with mutation of the PKGIα leucine zipper domain, which is required for cGMP-PKGIα anti-remodeling actions in vivo. WT or PKG Leucine Zipper Mutant (LZM) mice were exposed to 56-day LV pressure overload by moderate (26 Gauge) transaortic constriction (TAC). At day 14 post-TAC, mice were randomized to vehicle or sacubitril/valsartan by oral gavage. TAC induced the same degree of LV pressure overload in WT and LZM mice, which was not affected by sacubitril/valsartan. Though LZM mice, but not WT, developed LV dilation post-TAC, sacubitril valsartan improved cardiac hypertrophy and LV fractional shortening to the same degree in both the WT and LZM TAC mice.
Conclusion:
These ladings indicate beneficial effects of sacubitril/valsartan on LV structure and function in moderate pressure overload. The unexpected finding that PKGIα mutation does not abolish the sacubitril/valsartan effects on cardiac hypertrophy and on LV function suggests that signaling other than NP-cGMP-PKG mediates the therapeutic benefits of neprilysin inhibition in HF.
Introduction
Inhibition of the neutral endopeptidase enzyme neprilysin (NEP) has emerged as a novel treatment strategy for heart failure (HF). Specifically, the combination of the NEP inhibitor sacubitril with the angiotensin receptor antagonist valsartan improved survival in HF patients with reduced LV ejection fraction (HFrEF)1, and improved surrogate markers in HF patients with preserved ejection fraction (HFpEF)2, compared with enalapril or valsartan, respectively. The success of adding NEP inhibition to RAAS antagonism in these and other studies3 has therefore raised interest in better understanding the biological effects of NEP inhibition in the cardiovascular system.
NEP degrades multiple circulating peptides, including the natriuretic peptides (NPs) ANP, BNP, and CNP4. NPs ameliorate a numbe, of cardiovascular disease processes5. Therefore, the improved outcomes observed in HF patients receiving sacubitril/valsartan have been postulated to occur through prevention of NP degradation, resulting in augmented NP actions2, 4, 6. Circulating NPs affect target tissues chiefly by activating membrane guanylate cyclases to generate intracellular cGMP7. In the heart and cardiovascular system, cGMP activates the cGMP-dependent protein kinase I (PKGI). Multiple preclinical studies support that cGMP-PKG signaling normally opposes cardiac remodeling and dysfunction7. Further, clinical8 and preclinical6, 9,10 studies of sacubitril/valsartan demonstrate increases in mature NPs and circulating and urinary cGMP, providing correlative evidence that the benefits of NEP inhibition on outcomes in HF might occur through the NP-cGMP-PKG axis. Despite these associations, however, the specific mechanisms through which NEP inhibition adds benefit in HF remain unknown. Though sacubitril increases PKG substrate phosphorylation in vivo in heart failure models and in cultured fibroblasts11, the degree to which PKGI is required for the cardiovascular effects of sacubitril/valsartan has not been investigated experimentally. Understanding this mechanism is of clinical interest as multiple PKGIα activating drugs remain under active investigation in HF.
In the current study we examined the requirement of PKGI in mediating the therapeutic effects of sacubitril/valsartan (Sac/Val) in LV pressure overload. We used mice with discrete mutations in the PKGI alpha isoform, the predominant PKG isoform in the heart. These PKGIα leucine zipper mutant (LZM) mice harbor mutations in the leucine zipper domain of PKGIα, so that interactions with leucine zipper-dependent substrates become disrupted12. Thus, despite retained kinase function the mutant protein fails to phosphorylate critical substrates. The LZ domain mediates functional compensation to LV pressure overload,13 and LZ mutation abolishes the functional efficacy of the cGMP-generating molecule sildenafil in pressure overload13. Therefore, in the present study, we examined the effects of Sac/Val in wild type and PKGIα LZM mice subjected to chronic transaortic constriction.
Methods
Surgical Models
All animal studies were in accordance with and approved by the Institutional Animal Care and Use Committee of Tufts University School of Medicine and Tufts Medical Center. LV pressure overload was induced as described13 by transaortic constriction (TAC). We used a 26G moderate TAC14 in order to ensure high survival, as LZM mice display early and profound mortality after severe (27G) TAC13. We performed TAC, or control sham surgery on 10–12 week-old male C57/Bl6 mice and littermate LZM mice.
Experimental Design
The experimental protocol is outlined in Supplemental Figure 1. Mice were subjected to LV pressure overload or sham by transaortic constriction (TAC), followed by initiation of drug (randomized to vehicle or sacubitril/valsartan) on day 14. At day 56 after pressure overload, mice underwent echocardiography, invasive hemodynamic assessment, and organ harvest. Detailed methods are described in the Supplemental Methods.
Statistical Analysis
We compared multiple groups by 1-way ANOVA with Sidak’s post-test for multiple comparisons. For nonparametric data, we used Kruskal-Wallis testing with Dunn’s multiple comparisons test. For time-dependent comparisons within genotypes, we used Two Way Repeated Measures ANOVA with Sidak’s multiple comparisons test. For comparison of response to therapy with 0% change as comparator variable (Supplemental material) we used one-sample T testing. P ≤ 0.05 was considered statistically significant. We analyzed data and performed statistical analysis in GraphPad Prism (Version 8.3.0).
Results
We first tested the requirement of the PKG LZ domain for the biological actions of natriuretic peptides. BNP injection significantly reduced mean arterial pressure in WT mice, but to a lesser extent in LZM mice, supporting that the LZ domain mediates cardiovascular NP effects in vivo (Figure 1). We next performed TAC on WT and on LZM mice treated with vehicle (Veh) or sacubitril/valsartan (Sac/Val) to test the requirement of the PKGIα LZ domain for the effects of NEP inhibition in pressure overload (Supplemental Figure 1). At day 56 post-TAC, LV systolic pressure was increased significantly in the TAC groups of both genotypes, compared with respective sham, indicating significant pressure overload which was not affected by Sac/Val (Figure 2A). In WT mice TAC mice left ventricles, Sac/Val increased phosphorylation at serine 273 of the PKG substrate cardiac myosin binding protein C (cMyBP-C)15, compared with TAC vehicle (Figure 2B). By contrast, Sac/Val did not induce significant phosphorylation of cMyBP-C in LZM TAC LVs, supporting that Sac/Val induces cMyBP-C signaling in the LV through activation of PKGIα. We observed similar results for the known PKGI substrate vasodilator-stimulated phosphoprotein (VASP), in which Sac/Val increased phosphorylation on the PKG-specific serine 239 of VASP in WT TAC, compared to LZM TAC LVs (Figure 3).
Fig. 1.
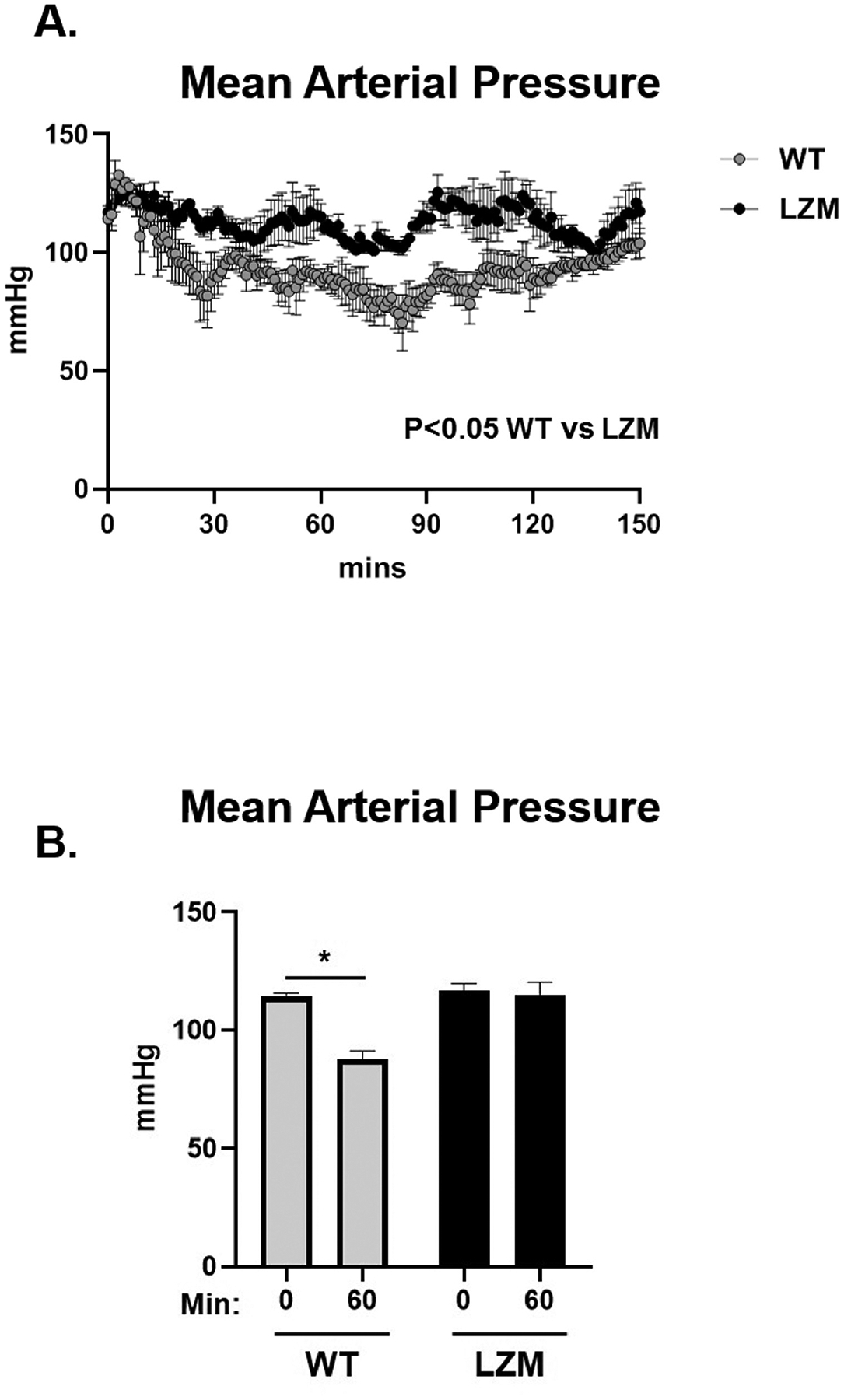
Cyclic guanosine monophosphate-dependent protein kinase I (PKGI)α leucine zipper domain required for hemodynamic effects of brain natriuretic peptide (BNP). (A) Mean arterial pressure (MAP) response to intraperitoneal injection of 0.1 mg/kg BNP in male wild-type (WT) and PKGIα leucine zipper mutant (LZM) littermates, implanted with aortic telemetry. Mice injected at 0 minutes. (B) Summary data of MAP at 60 minutes after injection. 0 minutes, before injection; 60 minutes, 60 minutes after injection. n = 4 WT, 3 leucine zipper mutant. *P < .05.
Figure 2. PKGIα leucine zipper domain mutation does not affect degree of TAC-induced pressure overload but induces LV dilation after chronic pressure overload.
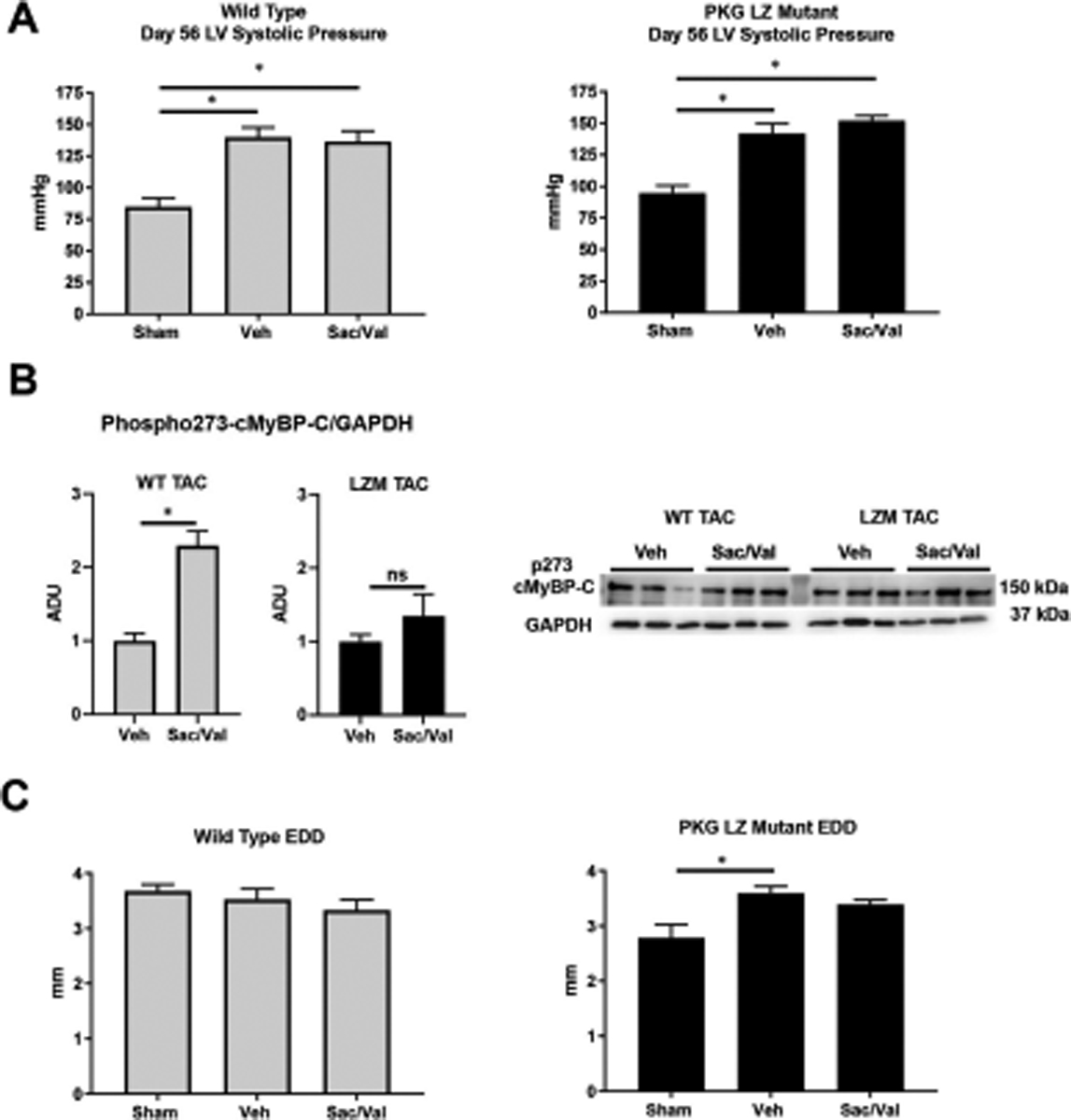
A. LV systolic pressure at day 56 of pressure overload by 26-gauge transaortic constriction (TAC) in WT (left) and LZM (right). n=5 WT sham, 6 WT TAC Vehicle, 8 WT TAC sacubitril/valsartan; 3 LZM sham, 6 LZM TAC vehicle, 8 LZM TAC sacubitril/valsartan. B. Western blot for phosphorylated cardiac myosin binding protein C (cMyBP-C) on serine 273. Summary densitometry of p273 normalized to GAPDH is shown. n=7 WT TAC vehicle, 6 WT TAC sacubitril/valsartan; 6 LZM TAC vehicle, 7 LZM TAC sacubitril/valsartan. C. LV end diastolic diameter obtained by echocardiography at day 56 after pressure overload in WT (left) and LZM (right) mice. n=5 WT and 5 LZM sham; 8 WT TAC vehicle; 8 WT TAC sacubitril/valsartan; 11 LZM vehicle; 10 LZM TAC sacubitril/valsartan. *, p<0.05. Veh, vehicle; Sac/Val, sacubitril/valsartan. ADU, arbitrary densitometric units. EDD, end diastolic diameter.
Figure 3: PKGI substrate phosphorylation in LVs of vehicle and sacubitril/valsartan treated mice subjected to pressure overload.
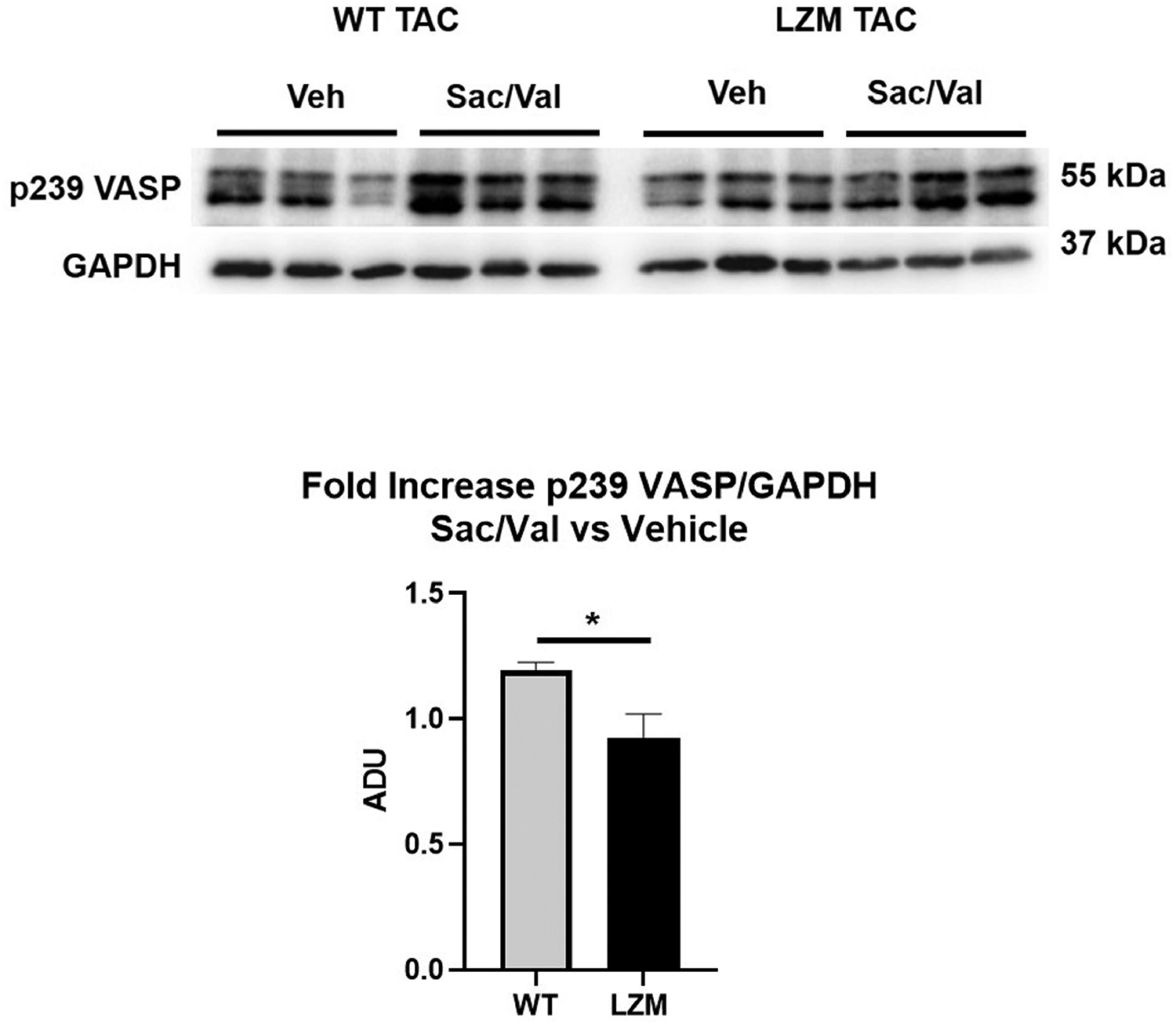
Western blot for vasodilator-stimulated phosphoprotein (VASP) phosphorylation on the PKGI-specific serine 239, and for loading control with GAPDH. Also shown is fold-increase of VASP phosphorylation on serine 239 normalized to GAPDH, in sacubitril/valsartan-treated TAC left ventricles, normalized to vehicle treated TAC of the same genotype. n=7 WT TAC vehicle, 6 WT TAC sacubitril/valsartan; 6 LZM TAC vehicle, 7 LZM TAC sacubitril/valsartan. *, p<0.05. ADU, arbitrary densitometric units.
In contrast to WT mice, in LZM mice TAC induced a significant increase in end diastolic diameter compared with sham, indicating LV dilation and more severe LV response to TAC in LZM mice (Figure 2C). In WT mice cardiac hypertrophy developed in the TAC vehicle group, compared with sham (Figure 4A), but heart mass normalized to tibia length in Sac/Val treated WT TAC mice did not differ significantly from sham, supporting prevention of hypertrophy with drug treatment. As with WT mice, LZM TAC mice treated with vehicle displayed cardiac hypertrophy at day 56, but again heart mass normalized to tibia length did not differ between LZM sham and LZM TAC-Sac/Val groups, supporting an equivalent effect of sacubitril/valsartan on cardiac hypertrophy in the LZM mice. Sac/Val reduced cardiac hypertrophy to the same degree in WT and in LZM mice (Supplemental Figure 2).
Figure 4. Sacubitril/valsartan (Sac/Val) prevents cardiac hypertrophy and improves left ventricular systolic function after pressure overload to the same degree in the presence of wild type or mutant PKGIα.
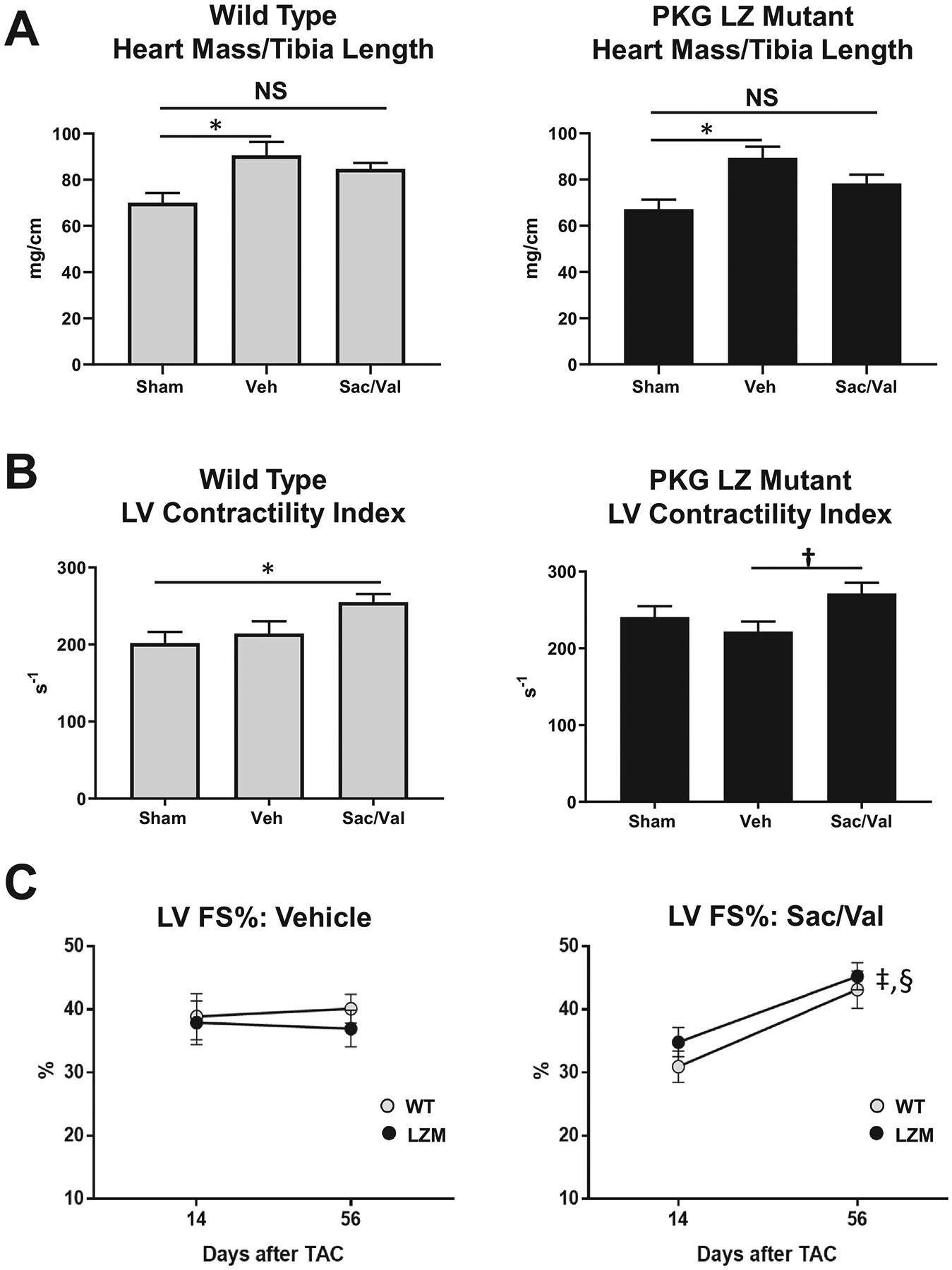
A. Summary data of heart mass normalized to tibia length in wild type and PKG Leucine Zipper (LZ) mutant mice on day 56 after transaortic constriction. B. LV Contractility Index (dP/dt normalized to instantaneous pressure) obtained by invasive hemodynamics at Day 56. C. LV Fractional shortening percentage (FS%) on Day 14 and Day 56 in WT and PKG LZ mutant mice treated with vehicle (Veh), or sacubitril/valsartan (Sac/Val). *, p<0.05; †, p=0.05; ‡, p<0.01 WT Day 14 vs WT Day 56; §, p<0.01 LZM Day 14 vs LZM Day 56. n=5 per sham group, 8 WT TAC Veh, 8 WT TAC Sac/Val; 11 LZM TAC Veh, 10 LZM TAC Sac/Val. A-B, data analyzed by One Way ANOVA with Tukey’s multiple comparisons test. C, data analyzed by Two Way Repeated Measures ANOVA with Sidak’s multiple comparisons test.
In WT mice, Sac/Val, but not vehicle, increased the LV contractility index (dP/dt normalized to instantaneous pressure). In LZM TAC mice, Sac/Val also improved the LV contractility index compared with LZM TAC vehicle (Figure 4B).
In vehicle-treated TAC mice, LV fractional shortening did not change significantly from day 14 (start of drug) to day 56 (end of study) in either genotype (Figure 4C). In Sac/Val treated mice, however, LV fractional shortening improved from day 14 to day 56 in WT TAC mice, but also improved significantly, and to the same degree, in the LZM TAC mice, indicating that mutation of the PKGIα LZ domain did not alter the beneficial effects of Sac/Val on LV function (Figure 4C).
Discussion
This study examined the effects of sacubitril/valsartan (Sac/Val) in a chronic, moderate pressure overload model in WT mice and LZM mice harboring mutations in PKGIα, the postulated downstream effector of sacubitril11. We observed that treatment with Sac/Val: 1) improved cardiac hypertrophy and LV systolic function; and 2) these benefits occurred to the same degree between WT and LZM mice. We interpret these findings to indicate that Sac/Val induces improvements in LV hypertrophy and systolic function in the setting of moderate pressure overload and that these benefits do not require a functional PKGIα LZ interaction domain.
Sacubitril prevents NEP degradation of natriuretic peptides, allowing increased circulating ANP and BNP to activate NP receptor synthesis of cGMP and activation of PKG. This mechanism has been proposed to explain the improved outcomes observed in the PARADIGM-HF study of HFrEF patients8 and the surrogate outcome improvements in the PARAMOUNT study in HFpEF2 Further, experimental studies have identified a requirement of PKG for the effect of the cGMP-generating drug sildenafil on LV function13 and fibrotic gene expression16. Based on these observations, we hypothesized that mutation of PKG would prevent or attenuate the cardiovascular effects of chronic Sac/Val treatment. However, in the current study vehicle treated LZM mice developed LV dilation as evidenced by increased EDD, yet Sac/Val treatment induced improvements in LV systolic function in the LZM mice from day 14 to day 56, with improvements in these parameters to the same degree as in WT mice. In fact, the degree of cardiac hypertrophy reduction with Sac/Val trended higher in the LZM compared with WT TAC mice, though this may relate to the abnormal cardiac phenotype of LZM mice discussed below. We interpret these findings to support that improvement in LV systolic function with Sac/Val in the setting of pressure overload does not require the PKGIα LZ interaction domain.
Importantly, though mutation of the PKGIα LZ domain did not affect the therapeutic benefit of sacubitril/valsartan on LV function and hypertrophy after TAC, the LZ mutation did inhibit PKGIα-mediated molecular signaling in the LV. Specifically, sacubitril/valsartan increased phosphorylation of two known PKGI substrates, cMyBP-C and VASP in LVs of WT, but not LZM, mice subjected to TAC These observations therefore support that chronic administration of sacubitril/valsartan induced PKGI activation in the LV, and that mutation of PKGIα blocked PKGI signaling. These combined findings provide further support that the therapeutic effects of sacubitril/valsartan did not require intact PKGIα.
These observations may have broader clinical implications. First, they raise the possibility that the outcome improvements with Sac/Val in HF1,2 do not require PKG, and thus may be independent of upstream NP modulation. While Sac/Val treatment in the PARADIGM trial correlated with increased urinary cGMP and with augmentation of BNP8, the degree to which these molecules mediated the improved outcomes remains unknown. Our results suggest that NEP inhibition may improve outcomes through mechanisms other than, or in addition to, PKG signaling.
We do note, however, that the Sac/Val effect on LV function could occur independently of PKGIα LZ-mediated signaling but could still require NP stimulation of cGMP synthesis. For example, besides PKGIα, other myocardial cGMP effectors include the less highly expressed PKGIβ isoform7, and cGMP-activated phosphodiesterase 2 which mediates cardiac protection through degradation of cAMP17. Additionally, though the PKG LZ mutation abolishes PKG interaction with and phosphorylation of important kinase substrates in the cardiovascular system7, 18, we did not directly test the possibility that non-LZ dependent substrates in the heart mediate the NP-cGMP effects.
This study has several additional limitations. First, we observed previously that LZM mice display baseline enhanced indices of LV systolic and diastolic function, compared with WT controls19. This baseline phenotype likely contributes to the relative reduction of these indices observed in LZM TAC mice, compared to sham13, and in the differential responses to TAC observed between genotypes in this study. We acknowledge that this may confound the comparison of shams between genotypes, and possibly makes it more difficult to compare differences in genotype response to therapies. For these reasons we present our echocardiographic data as a paired analysis, in order to compare the relative change in LV function within the same genotype, rather than comparing the genotypes to one another. Importantly, our prior work demonstrated a requirement of the PKGIα LZ domain for inhibition of cardiac remodeling and dysfunction after TAC, and further that mutation of the LZ domain completely abolished the therapeutic cardiac functional effects of cGMP modulating drugs such as sildenafil13. We therefore conclude that despite these baseline differences and differential response to pressure overload, the LZM mouse represents an adequate model for mechanistic studies of PKG activating therapies.
In summary, we have observed that NEP inhibition with sacubitril/valsartan improves cardiac hypertrophy and LV systolic function in a moderate, chronic mouse TAC model, and that this benefit does not require intact PKGIα LZM signaling. These findings suggest novel beneficial mechanisms modulated by NEP inhibition in addition to NP modulation of cGMP via PKG LZ interactions.
Supplementary Material
Acknowledgements
Sources of Funding
This study was funded by Novartis Pharmaceuticals through an investigator-initiated trial award (LCZ696BUSNC19T) to RMB and IZJ, and by the NIH R01HL131831 to RMB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- 1.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K and Zile MR. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993–1004. [DOI] [PubMed] [Google Scholar]
- 2.Solomon SD, Zile M, Pieske B, Voors A, Shah A, Kraigher-Krainer E, Shi V, Bransford T, Takeuchi M, Gong J, Lefkowitz M, Packer M and McMurray JJ. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet. 2012;380:1387–95. [DOI] [PubMed] [Google Scholar]
- 3.Lluri G, Lin J, Reardon L, Miner P, Whalen K and Aboulhosn J. Early Experience With Sacubitril/Valsartan in Adult Patients With Congenital Heart Disease. World J Pediatr Congenit Heart Surg. 2019;10:292–295. [DOI] [PubMed] [Google Scholar]
- 4.Singh JSS, Burrell LM, Cherif M, Squire IB, Clark AL and Lang CC. Sacubitril/valsartan: beyond natriuretic peptides. Heart (British Cardiac Society). 2017;103:1569–1577. [DOI] [PubMed] [Google Scholar]
- 5.Zois NE, Bartels ED, Hunter I, Kousholt BS, Olsen LH and Goetze JP. Natriuretic peptides in cardiometabolic regulation and disease. Nature reviews Cardiology. 2014;11:403–12. [DOI] [PubMed] [Google Scholar]
- 6.Ishii M, Kaikita K, Sato K, Sueta D, Fujisue K, Arima Y, Oimatsu Y, Mitsuse T, Onoue Y, Araki S, Yamamuro M, Nakamura T, Izumiya Y, Yamamoto E, Kojima S, Kim-Mitsuyama S, Ogawa H and Tsujita K. Cardioprotective Effects of LCZ696 (Sacubitril/Valsartan) After Experimental Acute Myocardial Infarction. JACC Basic to translational science. 2017;2:655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong Q and Blanton RM. Protein kinase G I and heart failure: Shifting focus from vascular unloading to direct myocardial antiremodeling effects. Circulation Heart failure. 2013;6:1268–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Packer M, McMurray JJ, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile M, Andersen K, Arango JL, Arnold JM, Belohlavek J, Bohm M, Boytsov S, Burgess LJ, Cabrera W, Calvo C, Chen CH, Dukat A, Duarte YC, Erglis A, Fu M, Gomez E, Gonzalez-Medina A, Hagege AA, Huang J, Katova T, Kiatchoosakun S, Kim KS, Kozan O, Llamas EB, Martinez F, Merkely B, Mendoza I, Mosterd A, Negrusz-Kawecka M, Peuhkurinen K, Ramires FJ, Refsgaard J, Rosenthal A, Senni M, Sibulo AS Jr, Silva-Cardoso J, Squire IB, Starling RC, Teerlink JR, Vanhaecke J, Vinereanu D and Wong RC. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation. 2015;131:54–61. [DOI] [PubMed] [Google Scholar]
- 9.Suematsu Y, Jing W, Nunes A, Kashyap ML, Khazaeli M, Vaziri ND and Moradi H. LCZ696 (Sacubitril/Valsartan), an Angiotensin-Receptor Neprilysin Inhibitor, Attenuates Cardiac Hypertrophy, Fibrosis, and Vasculopathy in a Rat Model of Chronic Kidney Disease. J Card Fail. 2018;24:266–275. [DOI] [PubMed] [Google Scholar]
- 10.Mochel JP, Teng CH, Peyrou M, Giraudel J, Danhof M and Rigel DF. Sacubitril/valsartan (LCZ696) significantly reduces aldosterone and increases cGMP circulating levels in a canine model of RAAS activation. Eur J Pharm Sci. 2019;128:103–111. [DOI] [PubMed] [Google Scholar]
- 11.Burke RM, Lighthouse JK, Mickelsen DM and Small EM. Sacubitril/Valsartan Decreases Cardiac Fibrosis in Left Ventricle Pressure Overload by Restoring PKG Signaling in Cardiac Fibroblasts. Circulation Heart failure. 2019;12:e005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michael SK, Surks HK, Wang Y, Zhu Y, Blanton R, Jamnongjit M, Aronovitz M, Baur W, Ohtani K, Wilkerson MK, Bonev AD, Nelson MT, Karas RH and Mendelsohn ME. High blood pressure arising from a defect in vascular function. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6702–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanton RM, Takimoto E, Lane AM, Aronovitz M, Piotrowski R, Karas RH, Kass DA and Mendelsohn ME. Protein kinase g ialpha inhibits pressure overload-induced cardiac remodeling and is required for the cardioprotective effect of sildenafil in vivo. J Am Heart Assoc. 2012;1:e003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards DA, Aronovitz MJ, Calamaras TD, Tam K, Martin GL, Liu P, Bowditch HK, Zhang P, Huggins GS and Blanton RM. Distinct Phenotypes Induced by Three Degrees of Transverse Aortic Constriction in Mice. Sci Rep. 2019;9:5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thoonen R, Cauwels A, Decaluwe K, Geschka S, Tainsh RE, Delanghe J, Hochepied T, De Cauwer L, Rogge E, Voet S, Sips P, Karas RH, Bloch KD, Vuylsteke M, Stasch JP, Van de Voorde J, Buys ES and Brouckaert P. Cardiovascular and pharmacological implications of haem-deficient NO-unresponsive soluble guanylate cyclase knock-in mice. Nat Commun. 2015;6:8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patrucco E, Domes K, Sbroggio M, Blaich A, Schlossmann J, Desch M, Rybalkin SD, Beavo JA, Lukowski R and Hofmann F. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:12925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vettel C, Lindner M, Dewenter M, Lorenz K, Schanbacher C, Riedel M, Lammle S, Meinecke S, Mason FE, Sossalla S, Geerts A, Hoffmann M, Wunder F, Brunner FJ, Wieland T, Mehel H, Karam S, Lechene P, Leroy J, Vandecasteele G, Wagner M, Fischmeister R and El-Armouche A. Phosphodiesterase 2 Protects Against Catecholamine-Induced Arrhythmia and Preserves Contractile Function After Myocardial Infarction. Circ Res. 2017;120:120–132. [DOI] [PubMed] [Google Scholar]
- 18.Thoonen R, Giovanni S, Govindan S, Lee DI, Wang GR, Calamaras TD, Takimoto E, Kass DA, Sadayappan S and Blanton RM. Molecular Screen Identifies Cardiac Myosin-Binding Protein-C as a Protein Kinase G-Ialpha Substrate. Circulation Heart failure. 2015;8:1115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blanton RM, Takimoto E, Aronovitz M, Thoonen R, Kass DA, Karas RH and Mendelsohn ME. Mutation of the protein kinase I alpha leucine zipper domain produces hypertension and progressive left ventricular hypertrophy: a novel mouse model of age-dependent hypertensive heart disease. J Gerontol A Biol Sci Med Sci. 2013;68:1351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.