

Abstract
Purpose:
DNA sequencing technology has unmasked a vast number of uncharacterized single nucleotide variants in disease-associated genes, and efficient methods are needed to determine pathogenicity and enable clinical care.
Methods:
We report herein an E.coli-based solubility assay for assessing the effects of variants on protein domain stability for three disease-associated proteins.
Results:
First, we examined variants in the Kv11.1 channel PAS domain (PASD) associated with inherited Long QT Syndrome type 2 and found that protein solubility correlated well with reported in vitro protein stabilities. A comprehensive solubility analysis of 56 Kv11.1 PASD variants revealed that disruption of membrane trafficking, the dominant loss-of-function disease mechanism, is largely determined by domain stability. We further validated this assay by using it to identify second-site suppressor PASD variants that improve domain stability and Kv11.1 protein trafficking. Finally, we applied this assay to several cancer-linked P53 tumor suppressor DNA-binding domain and myopathy-linked Lamin A/C Ig-like domain variants, which also correlated well with reported protein stabilities and functional analyses.
Conclusion:
This simple solubility assay can aid in determining the likelihood of pathogenicity for sequence variants due to protein misfolding in structured domains of disease-associated genes as well as provide insights into the structural basis of disease.
Keywords: sequence variant, protein misfolding, Kv11.1, LMNA, P53
Introduction
A pressing challenge in medicine is to understand the consequences of the enormous genetic variability in humans demonstrated by whole exome and genome sequencing projects and in clinical genetics.1 The classification of low frequency DNA coding variants as disease causing is of critical importance for the diagnosis, management, and prognostication of patients and families who harbor these variants. There is currently a dearth of functional data to support the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP)1,2 classification of variants as benign, likely benign, likely pathogenic, pathogenic, or uncertain significance. With sequence variants in about 3,000 genes underlying over 4,000 Mendelian phenotypes as well as the growing number of cancer-linked somatic variants, the biochemical and physiological characterization for all variants is impractical.3 Consequently, dozens of sequence and structure-based bioinformatics tools have been developed to predict their functional impact.1,4 In many proteins, the most common disease mechanism introduced by sequence variants is domain instability, which can lead to protein misfolding and aggregation (Fig S1).5,6 However, pathogenicity prediction tools such as FoldX7, a popular protein stability calculator, have limitations and are often unreliable8 highlighting the continued need for experimental characterization.
Assessment of protein stability using bacterial expression systems has been a focus of intense effort given applications to develop commercial proteins and biopharmaceuticals. Native and over-expressed proteins in bacteria can misfold and form insoluble protein aggregates (inclusion bodies). For the E.coli protein HypF, variants predicted to be destabilizing have increased aggregation compared to WT.9 Further, a high-throughput bacteria colony-based screen demonstrated a strong correlation between cellular aggregation and thermal stability for ten proteins from several different organisms.10 These observations focused on engineering more stable proteins for production, and few studies have examined the ability of such assays to evaluate human sequence variants recombinantly expressed in E.coli.11,12 Based on these studies and the simple methods available for screening protein solubility in bacteria, we set out to test on a larger scale and with several different proteins whether the presence of sequence variants causing protein misfolding could be efficiently assessed in E.coli by simply measuring the amount of soluble protein by immunoblot without the need for additional denaturation steps or bulky tags. We focused on missense (single nucleotide) variants since these are the most common variants and their detrimental effects are less predictable than the severe changes to protein structure and function such as insertions, frame-shifts, deletions, and premature truncations.
We chose three protein domains in human disease-associated genes with sequence variants reported to cause loss of function and have decreased thermal stabilities as benchmarks for testing the solubility assay. First, we tested variants in the Kv11.1 channel PASD responsible for a major component of cardiac action potential repolarization and associated with Long QT Syndrome type 2 (LQT2).13-16 The most common mechanism underlying LQT2 associated loss of Kv11.1 channel function is impaired protein trafficking to the surface membrane.16 Furthermore, trafficking for many of these variants can be improved with a variety of interventions including reduced culture temperature or culture with high affinity Kv11.1 channel blockers16, suggesting different degrees of protein stability. We took advantage of having trafficking data for over 60 PASD variants already reported to use for comparison with our solubility assay, toward providing a structural basis for different Kv11.1 trafficking phenotypes and in helping to identify LQT2-PASD suppressor variants. Next, we tested our solubility assay on several oncogenic variants in the DNA-binding domain (DBD) of the tumor-suppressor P53 protein. P53 accounts for half of all cancers with at least 1200 distinct variants identified.17-20 Finally, we tested variants in the Immunoglobulin D domain (IgD) of Lamin A/C, a major component of the nuclear envelope, associated with cardiomyopathy, muscular dystrophy, lipodystrophy and premature aging.21,22 Remarkably, we found solubility to be a good proxy for misfolding of structural domains in three different disease-associated proteins. This simple solubility assay should have broad utility towards rapid and efficient DNA sequence variant classification regarding pathogenicity via protein domain misfolding as well as gaining structural insights into disease and therapies.
Materials and Methods
Bioinformatics
Structural models were created in Pymol using Protein Data Bank (PDB) ID 5K7L for EAG1, PDB: 1BYW for the PASD,13 PDB: 1IFR for the IgD21 and PDB: 1TSR for the DBD.17 Three models (PDB 1BYW, 4HP9, 4HQA)13,23 were used as inputs for FoldX PASD calculations7 using the YASARA molecular graphics interface. PDB structures were first refined with the FoldX “RepairPDB” function and then predictions were made using default parameters. For the IgD and DBD, PDB 1IFR and 1TSR were used as inputs for FoldX calculations using the iRDP web server, respectively. Amino-acid conservation scores were calculated with ConSurf using default parameters. Scores are ranked from 1 (variable) to 9 (conserved). Relative solvent accessibilities were calculated using ASAview. Variants were also evaluated using the stability prediction web servers: I-Mutant, Eris, CUPSAT, MuPro, Polyphen, PoPMuSiC, and MutPred. Variants were binned using the PolyPhen descriptors ‘probably damaging’ (ΔG values ≥ 2.0), ‘possibly destabilizing’ (0<ΔΔG<2) or benign (ΔΔG ≤ 0). CUPSAT and I-Mutant gave results with opposite sign, so values were reversed to simplify comparisons. MutPred percentages ≥0.75 were assigned ‘probably destabilizing’, 0.5-0.75 possibly destabilizing, and ≤0.5 benign. References for all bioinformatics tools are in the supporting supplement.
Expression constructs
All variants were made using the QuikChange II XL kit from Agilent (Santa Clara, CA) using primers designed with the Agilent Primer Design Program. Primers were obtained from Integrated DNA Technologies (Coralville, IA). Templates for mutagenesis were pcDNA3-Kv11.1 previously published,16 pEGFP-p53 (Addgene # 12091),24 and pcDNA3-GFP-LaminA-N195K (Addgene #32708),25 which was mutated back to WT. Restriction digest analysis was used to test the integrity of all constructs and all variants were verified by sequencing at the UW-Biotechnology Center. For E.coli expression constructs, PCR was used to amplify the PASD (amino acids 2-135), IgD (amino acids 435-553), and DBD (amino acids amino acids 92-292) for ligation independent cloning (LIC) into a 6X-His tagged pET3 plasmid previously described.26
Protein expression and solubility assay
Single colonies of BL21 (DE3) cells transformed with each 6x His-tagged mutant construct were grown overnight (13-18hrs) at 37° in 2ml of auto-induction media (0.5% glycerol, 0.5% glucose, 0.2% α-lactose, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, and 2 mM MgSO4). An equal number of cells (1.5ml max) were harvested, washed (50mM Tris, 150mM NaCl, pH 7.5) once, resuspended in 100 μL of wash buffer and lysed for 10 min in Lysis buffer (1X Cell Lytic B ® (Sigma)) in wash buffer and 100 μM PMSF. 5μL of total cell lysate was diluted in 25μL of wash buffer and added to an equal volume of 2x sample buffer (125 mM Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 50mM DTT and 0.005% Bromophenol Blue) before western blot. Soluble protein was collected from the supernatant after a 15,000g spin for 10 min and diluted in equal amounts of 2x sample buffer for western blot or serially diluted 1:2 for dot blot (1μL). All samples were boiled for 1-2 minutes before 12% SDS-PAGE (50X more soluble protein was loaded by volume than total protein) and transferred to nitrocellulose paper. All blots were first blocked for 10min in blocking buffer (50mM Tris pH 7.5, 150mM NaCl, 0.05% Tween-20 and 10% dry milk) and detected with anti His-HRP antibodies (Santa Cruz Biotech for PASD and IgD or Rockland Immunochemicals Inc. for DBD). Densitometry was performed using ImageJ (NIH) to quantify immunoblots. For dot blots, one representative row from each serially diluted dot blot was quantified (n≥3) (see Fig S3 for representative examples).
Kv11.1 trafficking
HEK 293 cells were cultured at 37° and transfected with the Kv11.1 variants using Lipofectamine 2000 (Invitrogen; ThermoFisher). Immature and mature Kv11.1 protein bands were detected by immunoblot analysis of whole-cell lysates. Briefly, lysates were mixed with an equal amount of Laemmli sample buffer, separated by 7% SDS-PAGE, and detected with an antibody to the distal C-terminus as previously described.16
Kv11.1 Function
Stable cell lines were generated by transfecting HEK cells with mutant Kv11.1 pcDNA3 and selecting in G418 as previously described.16 Single colonies of G418 resistant cells were then tested for Kv11.1 expression by immunoblot. Cell lines that gave a robust 155kD band on immunoblot were used for electrophysiological analysis. Kv11.1 current was measured using the whole-cell patch clamp technique as previously described.16 Voltage protocols are described in Fig. 3 and data analysis was done using pCLAMP 8.0 (Axon Instruments) and Origin (6.0 Microcal).
Fig. 3. Rational design of PASD suppressor variants.

(a) Structure of the Kv11.1 PASD (PDB: 1BYW) showing the location and (b) conservation of each variant characterized. (c) FoldX calculated stabilities for increasingly larger hydrophobic substitutions and V41 and C64 mutations. Bars ± SD represent ΔΔG predictions from three different structures (PDB: 1BYW, 4HQA, 4HP9).23 (d) Representative western blot of each full-length Kv11.1 variant expressed in HEK cells under normal conditions (−) or at reduced temperature (27°). A lack of a 155kD bands indicates defective trafficking. (e) Representative immunoblots of recombinant PASD C64 and V41 hydrophobic substitutions expressed in E.coli. Bars ± SD represent relative solubility (% of WT) determined by dot blot (n≥3). (f) Changes in FoldX stabilities are shown for second-site mutations predicted to improve variant stability (smaller ΔΔG). Representative western blots of each full-length Kv11.1 variant expressed in HEK cells are also shown under normal conditions (−) or at reduced temperature (27°). (g) Representative immunoblots of recombinant PASD suppressor mutations expressed in E.coli. Bars ± SD represent relative solubility (% of WT) determined by dot blot (n≥3). (h) Current densities and western blots for full-length Kv11.1 variants expresses in HEK cells. Inset shows voltage-clamp protocol with representative current trace. Bars ± SD represent current density levels (n≥4 cells). Dashed line indicates WT current density level previously reported,16 which was performed at the same time and with the same intracellular and extracellular solutions as these variants. *P<0.05, a significant reduction in solubility of variants compared with WT (e), increase in solubility of the double variant compared to the single variant (g) or increase in current density of the double variant compared to the single variant (h).
Lamin A/C aggregation
HEK 293 cells of similar confluence were cultured at 37°C and transfected with the GFP-tagged constructs described above using Lipofectamine 2000 (Invitrogen; ThermoFisher). Cells were imaged the following day at 20X or 40X magnification using the EVOS FL Imaging System (ThermoFisher). The percentage of cells with aggregates was obtained by counting at least 100 cells from several different fields of view averaged over 4-6 transfections.
Statistics
All data are presented as mean ± SD. One-way analysis of variance (ANOVA) was used for statistical analysis followed by the Tukey post hoc test. P<0.05 was considered statistically significant.
Results
Assessing Kv11.1 PASD misfolding
Kv11.1 is a large multi-domain membrane protein containing a 110 amino acid N-terminal intracellular PASD harboring over 60 sequence variants putatively associated with LQT2 (Fig. 1a). We have previously used a membrane trafficking assay in HEK293 cells to show that LQT2-associated variants exhibit different trafficking efficiencies that can be grouped as follows: (a) trafficking deficient and uncorrectable, (b) trafficking deficient but correctable by culturing cells at reduced temperature, (c) trafficking deficient but correctable with reduced temperature and Kv11.1 channel blockers, and (d) variants that traffic similar to WT. Because domain misfolding leads to deficient trafficking, we first sought to see if computational models could predict the results of published trafficking assays for LQT2 variants in the PASD.16 Since destabilizing variants are typically overrepresented at evolutionarily conserved residues,27 in hydrophobic, buried regions in the core of the domain5 and are more chemically severe,28 we used the ConSurf and ASAView bioinformatics tools to determine their conservation score (scored 1-9) and relative solvent accessibility (RSA), respectively. Using ConSurf, LQT2 residues averaged 7.1 (9 being the highest) compared to 6.1 for all residues (Fig. 1a and Table S1). Most of the variants at highly variable residues involve proline - a unique amino acid that forces structural rigidity and backbone strain on PASD folding - or result in other large physicochemical changes (e.g. S26I, I30T). Using ASAView, LQT2 residues averaged 26% solvent accessibility compared to 32% for all residues (Table S1) with the most severe variants (i.e trafficking defective and uncorrectable) being buried (Fig. 1b).
Fig. 1. Bioinformatics analysis of Kv11.1 PASD variants.

(a) ConSurf calculated scores color-coded from orange (highly variable) to blue (highly conserved) for each residue. (b) ASAView calculated relative solvent accessibility percentages for variants grouped by trafficking phenotype. Circles outlined in black indicate the average. (c) FoldX calculated stabilities for color-coded by trafficking phenotype for each variant. Bars represent ± SD from three different structures (PDB: 1BYW, 4HQA, and 4HP9). Inset shows values grouped by trafficking phenotype. Circles outlined in black indicate the average. Reported changes in melting temperature (Tm) are shown below for comparison.14,15 Abbreviations: PAS domain (PASD), transmembrane domain (TMD), cyclic nucleotide-binding homology domain (cNBHD).
Using the structure-based stability prediction tools FoldX, PolyPhen, MUpro, CUPSAT, Eris, and I-Mutant 2.0, we found that ≥75% of LQT2-PASD variants are “possibly to probably destabilizing” (Fig. S2 and Table S1) but with a great deal of scatter. Overall, FoldX is the best predictor of stability when compared to trafficking phenotype but is unreliable when individual variants are compared to reported changes in melting temperatures underscoring the need for biochemical characterization (Fig. 1c).
To assess the stability of these variants, we expressed the PASD with extra N-terminal aliphatic helix region (amino acids 2-135) and tested their solubility as a proxy. To benchmark this method, we tested fourteen PASD missense variants reported in two different studies with thermal stabilities that correlate with their Kv11.1 trafficking properties (i.e. thermally unstable PASD variants are trafficking defective)14,15 and three variants we previously categorized as likely benign (E58D, V115M and F125C) (Fig. 2a).16 Fig. 2b shows the immunoblot results for nine of the reported proteins where total protein levels were all similar to WT but the amount of soluble protein varied using western blot (Fig. 2b and Table S1). Overall, destabilized PASD variants with lower melting temperatures and trafficking defects are less soluble than WT in contrast to trafficking competent but dysfunctional R56Q and N33T as well as WT-like trafficking and functional E58D, V115M and F125C (Fig. 2b-d).16 Further, the level of solubility largely trended with stability (i.e. the more destabilized the variant, the less soluble it is) (Fig. 2c). To simplify the misfolding assay further, we also assessed solubility by dot blot, which had a good correlation to Western blot analysis (R2=0.82) (Fig. 2b-d). We then used our solubility assay to perform a comprehensive analysis of all PASD variants previously characterized16 and found that trafficking phenotype largely correlates with PASD solubility (Fig. 2e). This result demonstrates a simple and rapid way of assessing misfolding for PASD variants and could replace Kv11.1 trafficking as an assay for misfolding and pathogenicity.
Fig. 2. Misfolding analysis of Kv11.1 PASD variants.

(a) Structure of the Kv11.1 PASD (PDB: 1BYW) with variants color-coded based on trafficking phenotype. (b) Representative immunoblots for recombinant PASD variants with reported melting temperatures expressed in E.coli. Bars ± SD represent relative solubility (% of WT) determined by dot blot (n≥3). (c) Comparison of solubility determined by western blot and reported melting temperatures.14,15 Inset compares relative solubilities of dot blot to western blot. (d) Representative immunoblots for recombinant PASD variants reported to be benign expressed in E.coli. Bars ± SD represent relative solubility (% of WT) determined by western blot (n≥3). (e) Bars ± SD represent relative solubility (% of WT) determined by western blot for each variant (n≥3). Inset shows relative solubility values grouped by trafficking phenotype. Circles outlined in black indicate the average. *P<0.05, a significant reduction in solubility of variants compared with WT.
Rational design of LQT2-PASD suppressor variants
Destabilization can result from loss of non-covalent interactions (Fig. S1). For example, the LQT2-linked variants V41F and C64Y/W introduce larger side-chains at conserved and buried residues and likely cause PASD destabilization through conformational strain (Fig. 3a, b). To test this, we used FoldX and the well-established doublet on immunoblot16 to assess PASD stability and Kv11.1 trafficking in HEK cells, respectively. Variants at V41 and C64 show larger ΔΔG (more destabilizing) and a more severe trafficking phenotype as the hydrophobic side-chain volume increases, respectively (Fig. 3c, d and Table S2).
To further support conformational strain of V41F and C64W, we assessed the solubility of each variant and found that as the hydrophobic side-chain volume increases, solubility tends to decrease (Fig. 3e). V41A likely decreases stability by causing a cavity in the core. However, C64A, C64M and C64I traffic normally but show decreased solubility revealing some limitations to this assay. One possible explanation is that PASD misfolding may be compensated for through domain-domain interactions for some variants. Overall, these results show that this assay can largely be used to assess the stability of engineered variants, which in turn can help to identify second-site suppressor variants. It could also help determine misfolding for different missense variants at disease-linked residues.
To mitigate conformational strain and improve PASD stability and consequently Kv11.1 trafficking, our strategy was to mutate nearby residues to help relieve clashes created by the larger side-chains. We chose the highly conserved residues C39 and C64 which would clash with larger side-chains at C64 and V41 as well as Q61, which is in a nearby flexible loop (Fig. 3a, b). Supporting this rationale, FoldX predicted that mutating these second-site variants to a smaller more flexible glycine or alanine should improve stability (Fig. 3f, Table S2). Indeed, Fig. 3g shows that these second-site variants improve the solubility for V41F and C64Y/W, respectively. Furthermore, immunoblot analysis of transiently transfected HEK cells showed that C39G, C64A and C64G corrected the trafficking of V41F at 27° while C64W and nearby I42N were correctable at 37° consistent with the solubility assay (Fig. 3f). To directly measure surface expression, current densities of stably transfected HEK cells showed that LQT2-C64W (4±2 pA/pF) can be significantly improved at 37° with C39G (38±6 pA/pF) and to levels not statistically different than WT (98±19 pA/pF) with Q61G (69±22 pA/pF). Likewise, LQT2-I42N (3±1 pA/pF) can also be significantly improved with Q61G (42±15 pA/pF) (Fig. 3h). These results show that our solubility assay can be used to design second-site suppressor variants to help understand the structural basis of disease as illustrated in (Fig. S4). In addition, many genes contain common single nucleotide polymorphisms (SNPs) in the same protein domain as rare sequence variants, and so this assay can study the effects of rare sequence variants with different genetic backgrounds which could impact protein folding and pathogenicity.
Assessing P53 DBD misfolding
P53 contains a DBD, which is a hotspot for missense variants including the six highest frequency cancer-associated P53 variants (Fig. 4a). Using the same ASAView, ConSurf, and FoldX analyses as above, we focused on 12 missense variants (V143A, R175H, S241F, C242S, G245S, R248Q, R248W, R249S, F270L, R273H, C277F, R282W); nine with reported ΔΔG values (Table S3). We found that several variants are not buried (S241F, R248Q/W, R273H, C277F) or highly conserved (V143A and F270L) and that the correlation between measured and FoldX ΔΔG was poor (R2 = 0.12) further underscoring the need for biochemical characterization to determine deleteriousness (Fig. S5 and Table S3).
Fig. 4. Properties of P53 DBD and Lamin A/C IgD variants.

(a) Structure of the P53 DBD (PDB: 1TSR) with cancer-associated variants in green. (b) Representative immunoblots of recombinant DBD variants expressed in E.coli. Bars ± SD represent relative solubility (% of WT) determined by dot blot (n≥3). Reported ΔΔG values are shown below for comparison.18-20 (c) Structure of the Lamin A/C IgD (PDB: 1IFR) with variants color-coded based on disease. (d) Representative immunoblots of recombinant IgD variants expressed in E.coli. Bars ± SD represent relative solubility (% of WT) determined by dot blot. (e) 40X images of HEK nuclei after GFP-tagged full-length Lamin A variant over-expression. (f) Bars ± SD represent the percentage of cells showing aggregation for each variant (n≥4 transfections). Abbreviations: transactivation domain (TAD), proline rich domain (PRD), DNA-binding domain (DBD), oligomerization domain (OD), C-term regulatory domain (RegD), coiled-coil “rod” domains (1A, 1B, 2A, and 2B), Immunoglobulin-like domain (IgD), n/a (not available). Disease classifications from Universal Mutation Database (UMD). *P<0.05, a significant reduction in solubility of variants compared with WT (b,d) or significant increase in aggregation (f).
We used our solubility assay to assess misfolding for 10 of the 12 missense variants analyzed above and found that while total protein expression levels are all similar to WT, the amount of soluble protein varied. Destabilized P53 variants with larger ΔΔG values were less soluble (Fig. 4b and Table S3). All variants have reduced solubility except for C277F, which is not predicted to be destabilizing (i.e. not buried and FoldX ΔΔG = −0.18).
Assessing Lamin A/C IgD misfolding
Lamin A/C contains an IgD, which is a hotspot for disease-associated missense variants (Fig. 4c). Using the same ASAView, ConSurf and FoldX analyses as above, we focused on four well-characterized variants (G449V, L489P, N456I, W514R)22 with decreased thermal stabilities reported and A529V; a homozygous variant not predicted to be destabilizing (i.e. not buried, highly variable residue, and FoldX ΔΔG = −1.01). All four variants are buried and predicted to be destabilizing by FoldX but only two are highly conserved (G449V and N456I) (Table S4). In contrast to the PASD and DBD, deleteriousness of these IgD variants was better predicted using these tools.
We used our solubility assay to assess misfolding of the IgD missense variants analyzed above and found that while total protein expression levels are all similar to WT, destabilized variants with reduced melting temperatures were less soluble than WT in contrast to A529V, which was similar to WT (Fig. 4d and Table S4).
Since some Lamin A variants aggregate upon ectopic expression,29 we transiently transfected HEK cells with each GFP-tagged Lamin A constructs and compared mutant aggregation to WT. Representative images are shown in Fig. 4e. Except for A529V (2±2%), all variants showed an increased percentage of cells with nuclear aggregation to varying degrees (W514R (9±10%), G449V (32±17%), N456I (44±16%), and L489P (40±22%) compared to WT (5±2%)) (Fig. 4f and Table S4). These results again validate our solubility assay and suggest that IgD misfolding can cause Lamin A/C aggregation in the nucleus.
Discussion
In this study, we developed a simple E.coli-based solubility assay to assess the damaging effects of sequence variants in disease-associated genes on protein domain stability. By analyzing over 50 LQT2-associated Kv11.1 PASD variants with reported thermal stabilities14,15 and functional consequences,16 we showed that this assay largely predicts whether a variant will be destabilizing and thus cause deficient Kv11.1 trafficking. We also extend these findings to several disease-associated LMNA IgD and P53 DBD variants where, again, the solubility assay largely correlates with reported thermal stability studies (i.e. destabilizing variants are less soluble). Combined, these results suggest that our assay may have widespread value as a new, simpler approach to assist with the complex interpretive process of deciding the clinical relevance of rare sequence variants,29-33
The broader application of this solubility assay across a range of Mendelian disorders will require further validation studies focused on missense variants in highly structured domains that have been studied with other established functional tests. Assuming the assay is validated for a particular domain, a strategy for incorporating this solubility assay into a workflow for determining the pathogenicity of sequence variants in disease-associated genes is proposed based on ACGM standards of evidence for pathogenicity (Fig. 5). Because of the simplicity and efficiency of the solubility assay, it can rapidly contribute key evidence for the classification of many missense variants which otherwise would typically require more cumbersome functional assessment. In addition, the solubility assay could provide more precision to certain pathogenicity criteria such as when a novel missense change occurs at an amino acid residue where a different missense change was previously documented to be pathogenic (PM5). Although we anticipate that many missense variants in a wide range of disease-associated genes will be amenable to this scheme, there will be a significant fraction of variants outside of highly structured protein domains or in domains in which the solubility assay is not possible.
Figure 5. Proposed strategy for characterizing pathogenicity of variants.
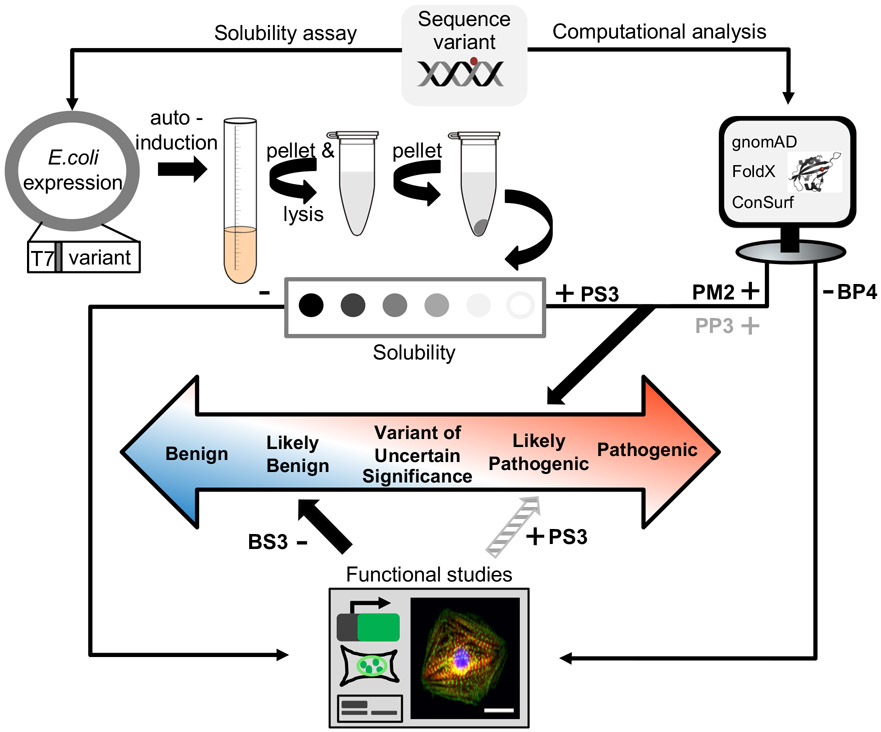
Missense variants of interest are initially evaluated using computational analysis and the solubility/misfolding assay followed by further functional analysis as needed. Computational analysis is performed to determine the minor allele frequency of the variant in the general population (e.g. gnomAD), to predict if the variant is destabilizing (e.g. FoldX), and to determine if the variant is evolutionarily conserved (e.g. ConSurf). The rapid solubility assay is used for functional assessment. In the case of a sequence variant that is absent or extremely rare in the population (PM2) and is also insoluble (PS3), then per ACMG guidelines1 this variant would be categorized as likely pathogenic and hence clinically actionable. Computational models could provide further evidence of pathogenicity (PP3). In cases where variants have WT-like solubilities, further functional studies (e.g. heterologous expression, patient-specific iPSCs) are needed to exclude other mechanisms of pathogenesis than domain misfolding. If a variant is soluble, functional studies are negative (BS3) and computational models are negative (BP4), then it will be classified as likely benign. Abbreviation: The Genome Aggregation Database (gnomAD).
Our solubility assay might also be useful for applications other than misfolding / pathogenicity assessment for individual domain variants by combining a second variant. Such second-site variants could be used to generate suppressor variants to understand the structural basis of disease,35 to assess the stability of engineered proteins important in biotechnology,10 and to study the important effect of background variants which may act as “genetic modifiers.” For example, disease-associated variants can occur together with more common sequence variants in the same domain, and the second-site variant can act as a suppressor by inhibiting misfolding and thus blocking the disease manifestation. The solubility assays could efficiently screen multiple combinations of a disease-associated variant with a range of relevant second-site variant for suppressor function for improved prediction of pathogenicity in a given genetic background. Additionally, correcting misfolding and aggregation is a promising therapeutic goal by directly targeting domains with small stabilizing molecules or indirectly by modulating the cell’s proteostasis network.36,37 By simply assessing solubility, our assay should help determine which variants are potential targets for these types of correction strategies.
In addition to describing a new method, we note several interesting biochemical observations. Kv11.1 misfolding, like many protein conformational diseases, leads to ER-associated degradation (Fig. S4).37 All trafficking defective PASD variants studied here have decreased solubility compared to WT making this assay a good predictor of defective Kv11.1 trafficking and could potentially be applied to other trafficking-related diseases. It also supports our previous model of Kv11.1 misfolding that proposed the level of defective trafficking correlates with domain stability,16 which we show here can be improved with stabilizing second-site mutations (Fig. S4). Our findings also support domain misfolding and aggregation as the likely mechanism underlying many Lamin A IgD and P53 DBD variants. Interestingly, destabilized variants within the same IgD can have variable effects on Lamin A aggregation in HEK cells with W514R being more aggregation prone than the other destabilizing variants. Finally, we provide new stability insights into three previously uncharacterized P53 variants. We observed that C277F is soluble (stable) in contrast to S241F, R248W and most other DBD variants characterized.18-20 Thus, C277F likely would not benefit from targeted therapeutics designed to stabilize the DBD.
Limitations
There are several limitations to this E.coli-based assay. Protein expression and solubility in bacteria can vary between different proteins and optimization may be needed (e.g. domain length, tag type and placement, growth and lysis conditions, etc.). Since protein expression levels can change between variants potentially confounding reduced solubility results, a second validation experiment should be performed to either test total expression or compare soluble and insoluble fractions12 to rule out false positives. Further, this assay might lead to ‘false negatives’ where the protein is soluble but loss of function (LOF) is through some other mechanism (e.g. altered gating16 or higher turnover38); however, when the assay is incorporated as part of a multistage variant classification strategy (Figure 5), such variants can be identified by other functional assays. Rare false positives were seen as with a few C64 variants that had decreased solubility, but normal Kv11.1 trafficking. This points to an important limitation of this assay in studying multidomain proteins in which domain-domain interactions can compensate for local domain misfolding or blunt quality control mechanisms.36,37 Finally, this assay addresses variants only in small protein domains that are easily expressed in E.coli and not variants in other regions of disease-associated proteins, and this constraint limits the number of clinically relevant variants that can be studied. However, protein misfolding and aggregation are major disease mechanisms,5,6 and so this assay should still be useful for characterizing a great number of targets and variants.
Conclusion
In summary, we demonstrate a simple solubility assay for quickly assessing protein misfolding, which should be applicable to most soluble protein domains that can be expressed in bacteria. Small culture volume and benchtop centrifugation also make this solubility assay amenable to higher throughput multi-well formats. Further, high throughput mutagenesis methods32 and E.coli protein aggregation protocols39 are available that could be adapted to study domain sequence variants. Finally, this method in conjunction with in silico analysis will aid in determining whether putative disease-associated sequence variants are actionable,40 possibly with strategies to correct misfolding or aggregation.36
Supplementary Material
Acknowledgements
We sincerely thank Caleb Hintz, Ryan Childs, Catherine Kuzmicki, and Madilyn Anderson for technical assistance and Robert Stroud, PhD (University of California-San Francisco) for providing the modified pET-based LIC plasmid. This study was supported by NIH R01 HL07887 (T.J.K.), NIH R01 HL060723 (C.T.J), AHA Competitive Catalyst Renewal Grant 17CCRG33700289 (B.P.D), AHA Midwest Affiliate Predoctoral Fellowship (C.L.A), Ruth L Kirschstein F32 HL128091 NRSA postdoctoral fellowship (C.L.A.) and NIH R01 HL139738-01 (L.L.E).
Footnotes
Confliction of Interest Notification Anderson et al.,
Disclosure: Dr. Kamp serves as a consultant for Fujifilm Cellular Dynamics, which is a stem cell technology company. Dr. Anderson, Mr. Routes, Dr. Eckhardt, Dr. Delisle, and Dr. January have nothing to disclose
References
- 1.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacArthur D, Manolio T, Dimmock D, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469–476. doi: 10.1038/nature13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chong JX, Buckingham KJ, Jhangiani SN, et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genetics. 2015;97(2):199–215. doi: 10.1016/j.ajhg.2015.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peterson TA, Doughty E, Kann MG. Towards Precision Medicine: Advances in Computational Approaches for the Analysis of Human Variants. J Mol Biol. 2013;425(21):4047–4063. doi: 10.1016/j.jmb.2013.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yue P, Li Z, Moult J. Loss of Protein Structure Stability as a Major Causative Factor in Monogenic Disease. J Mol Biol. 2005;353(2):459–473. doi: 10.1016/j.jmb.2005.08.020 [DOI] [PubMed] [Google Scholar]
- 6.Baets G, Doorn L, Rousseau F, Schymkowitz J. Increased Aggregation Is More Frequently Associated to Human Disease-Associated Mutations Than to Neutral Polymorphisms. Plos Comput Biol. 2015;11(9):e1004374. doi: 10.1371/journal.pcbi.1004374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(suppl_2):W382–W388. doi: 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan S, Vihinen M. Performance of protein stability predictors. Hum Mutat. 2010;31(6):675–684. doi: 10.1002/humu.21242 [DOI] [PubMed] [Google Scholar]
- 9.Calloni G, Zoffoli S, Stefani M, Dobson CM, Chiti F. Investigating the Effects of Mutations on Protein Aggregation in the Cell. J Biol Chem. 2005;280(11):10607–10613. doi: 10.1074/jbc.m412951200 [DOI] [PubMed] [Google Scholar]
- 10.Asial I, Cheng Y, Engman H, et al. Engineering protein thermostability using a generic activity-independent biophysical screen inside the cell. Nat Commun. 2013;4(1):2901. doi: 10.1038/ncomms3901 [DOI] [PubMed] [Google Scholar]
- 11.Mayer S, Rüdiger S, Ang H, Joerger AC, Fersht AR. Correlation of Levels of Folded Recombinant p53 in Escherichia coli with Thermodynamic Stability in Vitro. J Mol Biol. 2007;372(1):268–276. doi: 10.1016/j.jmb.2007.06.044 [DOI] [PubMed] [Google Scholar]
- 12.Lee Y, Stiers KM, Kain BN, Beamer LJ. Compromised Catalysis and Potential Folding Defects in in Vitro Studies of Missense Mutants Associated with Hereditary Phosphoglucomutase 1 Deficiency. J Biol Chem. 2014;289(46):32010–32019. doi: 10.1074/jbc.M114.597914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Crystal Structure and Functional Analysis of the HERG Potassium Channel N Terminus. Cell. 1998;95(5):649–655. doi: 10.1016/s0092-8674(00)81635-9 [DOI] [PubMed] [Google Scholar]
- 14.Harley CA, Jesus CS, Carvalho R, Brito RM, Morais-Cabral JH. Changes in Channel Trafficking and Protein Stability Caused by LQT2 Mutations in the PAS Domain of the HERG Channel. Plos One. 2012;7(3):e32654. doi: 10.1371/journal.pone.0032654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ke Y, Ng CA, Hunter MJ, Mann SA, Heide J, Hill AP, Vandenberg JI. Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain-domain interactions. Biochem J. 2013;454:69–77. doi: 10.1042/BJ20130328 [DOI] [PubMed] [Google Scholar]
- 16.Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP, January CT. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat Commun. 2014;5(1):5535. doi: 10.1038/ncomms6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho Y, Gorina S, Jeffrey P, Pavletich N. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265(5170):346–355. doi: 10.1126/science.8023157 [DOI] [PubMed] [Google Scholar]
- 18.Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19(10):1245–1256. doi: 10.1038/sj.onc.1203434 [DOI] [PubMed] [Google Scholar]
- 19.Ang H, Joerger AC, Mayer S, Fersht AR. Effects of Common Cancer Mutations on Stability and DNA Binding of Full-length p53 Compared with Isolated Core Domains. J Biol Chem. 2006;281(31):21934–21941. doi: 10.1074/jbc.m604209200 [DOI] [PubMed] [Google Scholar]
- 20.Joerger A, Ang H, Fersht A. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc National Acad Sci. 2006;103(41):15056–15061. doi: 10.1073/pnas.0607286103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhe-Paganon S, Werner ED, Chi Y-I, Shoelson SE. Structure of the Globular Tail of Nuclear Lamin. J Biol Chem. 2002;277(20):17381–17384. doi: 10.1074/jbc.c200038200 [DOI] [PubMed] [Google Scholar]
- 22.Dialynas G, Shrestha OK, Ponce JM, et al. Myopathic Lamin Mutations Cause Reductive Stress and Activate the Nrf2/Keap-1 Pathway. Plos Genet. 2015;11(5):e1005231. doi: 10.1371/journal.pgen.1005231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adaixo R, Harley CA, Castro-Rodrigues AF, Morais-Cabral JH. Structural Properties of PAS Domains from the KCNH Potassium Channels. Plos One. 2013;8(3):e59265. doi: 10.1371/journal.pone.0059265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyd SD, Tsai KY, Jacks T. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat Cell Biol. 2000;2(9):563–568. doi: 10.1038/35023500 [DOI] [PubMed] [Google Scholar]
- 25.Gilchrist S, Gilbert N, Perry P, Östlund C, Worman HJ, Bickmore WA. Altered protein dynamics of disease-associated lamin A mutants. Bmc Cell Biol. 2004;5(1):46. doi: 10.1186/1471-2121-5-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savage DF, Anderson CL, Robles-Colmenares Y, Newby ZE, Stroud RM. Cell-free complements in vivo expression of the E. coli membrane proteome. Protein Sci. 2007;16(5):966–976. doi: 10.1110/ps.062696307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vitkup D, Sander C, Church GM. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003;4(11):R72. doi: 10.1186/gb-2003-4-11-r72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson HA, Accili EA. Evolutionary analyses of KCNQ1 and HERG voltage-gated potassium channel sequences reveal location-specific susceptibility and augmented chemical severities of arrhythmogenic mutations. Bmc Evol Biol. 2008;8(1):188. doi: 10.1186/1471-2148-8-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hübner S, Eam JE, Wagstaff KM, Jans DA. Quantitative analysis of localization and nuclear aggregate formation induced by GFP-lamin A mutant proteins in living HeLa cells. J Cell Biochem. 2006;98(4):810–826. doi: 10.1002/jcb.20791 [DOI] [PubMed] [Google Scholar]
- 30.Pricolo MR, Herrero-Galán E, Mazzaccara C et al. Protein Thermodynamic Destabilization in the Assessment of Pathogenicity of a Variant of Uncertain Significance in Cardiac Myosin Binding Protein C. J Cardiovasc Transl Res. 2020;[Epub ahead of print]. doi: 10.1007/s12265-020-09959-6 [DOI] [PubMed] [Google Scholar]
- 31.Woods NT, Baskin R, Golubeva V, et al. Functional assays provide a robust tool for the clinical annotation of genetic variants of uncertain significance. Npj Genom Medicine. 2016;1(1):16001. doi: 10.1038/npjgenmed.2016.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glazer AM, Kroncke BM, Matreyek KA, Yang T, Wada Y, Shields T, Salem JE, Fowler DM, Roden DM. Deep Mutational Scan of an SCN5A Voltage Sensor. Circ: Genom and Precis Med. 2020;13e002786. doi: 10.1161/CIRCGEN.119.002786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiltshire T, Ducy M, Foo TK, et al. Functional characterization of 84 PALB2 variants of uncertain significance. Genetics in Medicine. 2019;[Epub ahead of print]. doi: 10.1038/s41436-019-0682-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma N, Zhang J, Itzhaki I, et al. Determining the Pathogenicity of a Genomic Variant of Uncertain Significance Using CRISPR/Cas9 and Human-Induced Pluripotent Stem Cells. Circulation. 2018;138(23). doi: 10.1161/circulationaha.117.032273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otsuka K, Kato S, Kakudo Y, Mashiko S, Shibata H, Ishioka C. The screening of the second-site suppressor mutations of the common p53 mutants. Int J Cancer. 2007;121(3):559–566. doi: 10.1002/ijc.22724 [DOI] [PubMed] [Google Scholar]
- 36.Tao Y-X, Conn MP. Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiol Rev. 2018;98:697–725. doi: 10.1152/physrev.00029.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foo B, Williamson B, Young JC, Lukacs G, Shrier A. hERG quality control and the long QT syndrome. J Physiol. 2016;594(9):2469–2481. doi; 10.113/JP270531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanner SA, Jain A, Colecraft HM. Development of a High-Throughput Flow Cytometry Assay to Monitor Defective Trafficking and Rescue of Long QT2 Mutant hERG Channels. Fron Physiol. 2018;9:397 Doi: 10.3389/fphys.2018.00397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cornvik T, Dahlroth SL, Magnusdottir A, Herman MD, Knaust R, Ekberg M, Nordlund P. Colony filtration blot: a new screening method for soluble protein expression in Escherichia coli. Nature Methods. 2005;2:507–509. doi: 10.1038/nmeth767 [DOI] [PubMed] [Google Scholar]
- 40.Hunter J, Irving SA, Biesecker LG, et al. A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet Med. 2016;18(12):1258–1268. doi: 10.1038/gim.2016.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.